

Abstract
Systemic administration of nicotine increases dopaminergic (DA) neuron firing in the ventral tegmental area (VTA), which is thought to underlie nicotine reward. Here, we report that the medial prefrontal cortex (mPFC) plays a critical role in nicotine-induced excitation of VTA DA neurons. In chloral hydrate-anesthetized rats, extracellular single-unit recordings showed that VTA DA neurons exhibited two types of firing responses to systemic nicotine. After nicotine injection, the neurons with type-I response showed a biphasic early inhibition and later excitation, whereas the neurons with type-II response showed a monophasic excitation. The neurons with type-I, but not type-II, response exhibited pronounced slow oscillations (SOs) in firing. Pharmacological or structural mPFC inactivation abolished SOs and prevented systemic nicotine-induced excitation in the neurons with type-I, but not type-II, response, suggesting that these VTA DA neurons are functionally coupled to the mPFC and nicotine increases firing rate in these neurons in part through the mPFC. Systemic nicotine also increased the firing rate and SOs in mPFC pyramidal neurons. mPFC infusion of a non-α7 nicotinic acetylcholine receptor (nAChR) antagonist mecamylamine blocked the excitatory effect of systemic nicotine on the VTA DA neurons with type-I response, but mPFC infusion of nicotine failed to excite these neurons. These results suggest that nAChR activation in the mPFC is necessary, but not sufficient, for systemic nicotine-induced excitation of VTA neurons. Finally, systemic injection of bicuculline prevented nicotine-induced firing alterations in the neurons with type-I response. We propose that the mPFC plays a critical role in systemic nicotine-induced excitation of VTA DA neurons.
Introduction
The mammalian ventral tegmental area (VTA) (A10) is a midbrain region long implicated as an integrative center mediating incentive and motivational effects for almost all addictive drugs including nicotine. Within the VTA, dopamine (DA) neurons and their associated ascending projections to the nucleus accumbens (NAc) and medial prefrontal cortex (mPFC) comprise the well-characterized mesolimbic and mesocortical pathways, respectively. It has been reported that VTA DA neurons receive glutamatergic inputs directly or indirectly from the PFC (Kalivas, 1993; Charara et al., 1996; Carr and Sesack, 2000; Omelchenko and Sesack, 2007) and other brain regions (Geisler and Wise, 2008). In addition, the VTA receives cholinergic projections from the laterodorsal tegmental nucleus (Garzón et al., 1999). Although effects must occur via different molecular mechanisms of action and perhaps through different neuronal circuits, increases in VTA DA neuronal firing and in DA release in the NAc and PFC have been proposed to be a common mechanism underlying dependence on addictive drugs (Mathon et al., 2003).
Our current understanding of the mechanisms underlying nicotine-induced excitation of VTA DA neurons is based on studies done using brain slices (Dani and Heinemann, 1996; Mansvelder and McGehee, 2000; Mansvelder et al., 2002). Because the interaction between the VTA and other brain regions is interrupted in brain slices, studies in a more intact in vivo system are needed. Emerging evidence demonstrates that the mPFC plays an important role in controlling VTA DA neuronal function (Mansvelder et al., 2009). The mPFC contributes to cognitive processes, such as attention, spatial learning, behavioral planning, and working memory (Fuster, 2000). Nicotine has been reported to act through nicotinic acetylcholine receptors (nAChRs) in the PFC to enhance working memory and attention (Levin, 1992; Granon et al., 1995; Levin et al., 2006; Counotte et al., 2011; Guillem et al., 2011). In addition, the mPFC has been suggested to be a key brain region underlying neural mechanisms of drug addiction and craving (Tzschentke, 2001).
Functionally, the mPFC and VTA have been shown to be closely coupled (Peters et al., 2004; Gao et al., 2007). Stimulation of the mPFC increases burst firing in VTA DA neurons, whereas disruption of signaling from the PFC induces the opposite effect (Gariano and Groves, 1988; Svensson and Tung, 1989; Murase et al., 1993; Overton et al., 1996a; Tong et al., 1996). We reported previously that, under nonstimulation conditions, VTA DA neurons exhibit robust oscillatory activity (0.5–1.5 Hz) in anesthetized animals defined as slow oscillations (SOs), which may reflect coupling between PFC and VTA neurons (Shi, 2005). We found that the activity of VTA DA neurons covaried with mPFC neuronal activity, consistent with mPFC–VTA functional coupling (Gao et al., 2007).
In the present study, we tested whether the mPFC plays a role in nicotine-induced VTA DA neuron excitation by using single-unit recordings in anesthetized rats. Our results demonstrate that systemic nicotine excites a subset of VTA DA neurons through a perturbation of mPFC–VTA coupling. This nicotinic action may represent a novel mechanism through which nicotine produces its rewarding effect in vivo.
Materials and Methods
The protocol for single-unit recording from VTA DA neurons in anesthetized rats was approved by the Institutional Animal Care and Use Committee of the Barrow Neurological Institute.
Single-unit recordings in vivo
VTA neurons. Male Wistar rats (200–220 g) were anesthetized with chloral hydrate (400 mg/kg, i.p., with supplemental doses administrated via a lateral tail vein) and mounted in a stereotaxic apparatus (Narishige). Body temperature was maintained at 36–37°C with a homeothermic blanket system (Harvard Apparatus). DA neurons were identified and recorded extracellularly as described previously (Gao et al., 2007, 2011; Zhang et al., 2008). Glass microelectrodes with resistance ranging from 6 to 8 MΩ were filled with 2 m NaCl and 2% pontamine sky blue dye. The latter was used to confirm the location of recording site within the VTA at the end of each experiment by visual inspection (Fig. 1A,B). The recording electrode was placed into the VTA through a small burr hole (located 3.0 mm anterior to lambda and 0.5–0.9 mm lateral to the midline) and positioned using a stepping motor micromanipulator. DA neurons were usually recorded between 6.5 and 8.5 mm below the cortical surface and identified according to published criteria (Grace and Onn, 1989; Ungless et al., 2004; Zhang et al., 2008), including presence of a long action potential (positive/negative) duration (2–5 ms), a relatively slow firing rate (FR; 1–10 Hz) characterized by an irregular or burst firing pattern, low pitch sounds produced on an audio amplifier, and a ≥1.1 ms duration from the start of the action potential to the negative trough (Ungless et al., 2004). Although we identified each recorded VTA DA neuron based on well-established electrophysiological criteria (Bunney et al., 1973; Grace and Bunney, 1983; Ungless et al., 2004), we do acknowledge that there are the technical limitations in the determination of the identities of our recorded neurons using extracellular single-unit recording from anesthetized rats. Additionally, we also realize that chloral hydrate has an inhibitory effect on GABAergic function. However, in the present study, we have evaluated the effects of systemic nicotine on VTA DA neuronal firing, and our data show that, under chloral hydrate anesthesia, systemic nicotine exposure excites all recorded VTA DA neurons, suggesting that the chloral-hydrate-anesthetized rat is an appropriate in vivo model for studying the mechanisms of systemic nicotine-induced VTA DA neuron excitation.
Figure 1.

The recording sites of VTA DA neurons and mPFC pyramidal neurons. A, A phase-contrast image illustrates electrode entry and track sites (black arrows) and the general recording site within the VTA (red arrow). B, Schematic diagram of coronal sections through the VTA shows the recording sites of 24 neurons labeled after electrophysiological and pharmacological experimentation. Black dots are type-I neurons (n = 17 from 17 rats), and white circles are type-II neurons (n = 7 from 7 rats). IF, Intrafascicular nucleus; PN, paranigral nucleus; PIF, paraintrafascicular nucleus; PBP, parabrachial pigmented nucleus; RN, red nucleus; tth, trigeminothalamic tract; IP, interpeduncular nucleus; ml, medial lemniscus; SN, substantia nigra. C, A phase-contrast image illustrates electrode general recording site within the mPFC (red arrow). D, A coronal section through the rat brain illustrating the mPFC (shaded area). Neurons (n = 6 from 6 rats) were filled in coronal sections at AP, 3.0 mm; ML, 0.7 mm; DV, 3.5 mm from bregma. ACd, Dorsal anterior cingulated cortex; PL, prelimbic cortex; IL, infralimbic cortex; OFC, orbitofrontal cortex.
mPFC neurons.
In 11 Wistar rats, the pyramidal neurons within mPFC [from bregma, anteroposterior (AP), 3.0 mm; mediolateral (ML), 0.7 mm; dorsoventral (DV), 3.5 mm] were recorded extracellularly. At the end of each experiment, the recording site within the mPFC was verified by visual inspection (Fig. 1B,C).
Drug applications
For systemic nicotine studies, nicotine bitartrate (Sigma-Aldrich) was dissolved in saline to a final concentration of 0.5 mg/ml (all nicotine concentrations refer to the concentration of the salt, but 0.5 mg/ml salt is equivalent to a free base concentration of 0.17 mg/ml). After obtaining a single-unit recording with stable firing for ∼5 min, nicotine bitartrate was injected intravenously (0.5 mg/kg, i.v.) through the tail-vein in a volume based on rat body weight and using a regulated syringe pump (Harvard Apparatus). The injection time was set to 30 s. Bicuculline methiodide (BMI; Sigma-Aldrich) was similarly delivered intravenously at a concentration of 2.5 mg/kg. For in situ nicotine experiments, silica capillary tubing (outer diameter, 160 μm; inner diameter, 100 μm) was used to deliver drugs into the mPFC (from bregma, AP, 3.0 mm; ML, 0.7 mm; DV, 3.5 mm). Drugs tested included 10 ng/μl tetrodotoxin (TTX), 100 μm mecamylamine (MEC), or 0.5 μm nicotine in PBS. After a stable baseline recording was established, drugs were infused at 0.75 μl/min for 2 min into both sides of the mPFC (controlled by a syringe pump; Harvard Apparatus). Ten minutes after the infusion, nicotine was injected systemically. Only one cell was recorded per rat. At the end of each experiment, the injection site within the mPFC was verified by visual inspection (Fig. 1B,C).
Acute PFC transection
To confirm the role of the PFC in nicotine-induced increase of VTA DA neuronal firing, bilateral transections were made immediately caudal to the PFC to interrupt PFC inputs to VTA DA neurons. A slit was drilled in the skull 2.0 mm anterior to bregma. The sharp pointed tip of a 3-mm-wide scalpel blade was lowered to the base of skull and passed along the slit to completely interrupt connections between the PFC and the rest of the brain.
Data analyses
Data were collected and analyzed using a data acquisition system (Fudan University, Shanghai, China) and stored in a computer for offline analysis. All data analyses were performed using programs written in Visual Basic for Applications in Microsoft Excel (Shi et al., 2004). Bursting was identified according to the “80/160 ms” definition proposed by Grace and Bunney. Thus, the onset of a burst was identified as the concurrence of two spikes with an interspike interval <80 ms, and the termination of a burst was defined as an interspike interval >160 ms (Grace and Bunney, 1984). Firing periodicity was examined using methods similar to those described previously (Shi et al., 2004). Briefly, rate histograms (bandwidth, 50 ms) and autocorrelograms (using an autocovariance function with a bandwidth of 50 ms over 2048 bins) were constructed based on 2 min recordings selected from each cell. After tapering using the Hanning–Tukey window function (15 windows with 50% overlap) and removal of the linear trend, fast Fourier transforms (FFTs) were performed on both the rate histograms and autocorrelograms, yielding spectra with a resolution of 0.078 Hz. Spectra obtained from rate histograms and autocorrelograms were qualitatively identical. The frequency band of 0.5–1.5 Hz was set to determine the power of SOs. We used interstimulus intervals to make autocorrelogram curves, and we used these curves to do FFTs.
Drug effects were determined by comparing measures before and after drug injection, and statistical significance of the comparison between two groups of matched datasets was assessed using two-tailed Student's t test. Spectral data were log-transformed before being subjected to statistical comparisons. The numerical data were expressed as means ± SEM, and p and t values were presented as the accurate number (unless the number <0.001 was presented as 0.001), and the p values <0.05 were considered significant.
Results
Systemic exposure to nicotine induces biphasic and monophasic firing activity in VTA DA neurons
Of 24 DA neurons tested (from 24 rats), 17 neurons exhibited a biphasic (initial decrease followed by long-lasting increase) change in FR after systemic injection of nicotine (defined as type-I response; Fig. 2Aa,Ba). The remaining seven neurons had a monophasic increase in FR (defined as type-II response; Fig. 2Ab,Bb). Additional analysis (using paired t test) showed that, in 17 VTA DA neurons with type-I response, 1 min after systemic administration of nicotine, FR, fraction of bursting (BF), and power of slow oscillations (PSO) were 77.6 ± 5.3% (t(16) = 6.49, p < 0.001), 57.9 ± 10.9% (t(16) = 3.06, p=0.007), and 33.3 ± 7.4% (t(16) = 5.11, p < 0.001) of baseline levels (Fig. 2Ca), whereas in seven VTA neurons with type-II response, they were 121.6 ± 4.8% (t(6) = −3.13, p = 0.020), 180.3 ± 46.4% (t(4) = −1.52, p = 0.20), and 170.7 ± 42.1% of baseline (t(6) = −2.13, p = 0.077) (Fig. 2Cb). Five minutes after nicotine administration, the FR, BF, and PSO for the neurons with type-I response (n = 17) were 124.2 ± 7.7% (t(16) = −4.71, p < 0.001), 142.2 ± 28.9% (t(16) = −2.49, p = 0.024), and 36.2 ± 8.2% (t(16) = 5.73, p < 0.001) of baseline levels (Fig. 2Ca); for the neurons with type-II response (n = 7), they were 108.8 ± 5.8% (t(6) = 3.84, p = 0.054), 203.3.0 ± 60.0% (t(4) = 2.36, p = 0.078), and 139.1 ± 32.8% (t(6) = −3.15, p = 0.201) of baseline, respectively (Fig. 2Cb). Thus, although nicotine exposure can alter the FR and BF for the VTA DA neurons with both types of responses, the time course for its differential effects across response types on FR is notable, and it has significant and polar opposite effects on PSO between the neurons with type-I and type-II responses.
Figure 2.

Nicotine alters the firing of VTA DA neurons. A, Systemic injection of nicotine (0.5 mg/kg, i.v., duration is indicated by the solid horizontal bar labeled “Nic”) induced different responses in VTA DA neurons. B, One group of neurons showed a biphasic response (type-I response, n = 17 from 17 rats) in which the FR was initially reduced (∼1 min after nicotine) and then increased (peak increase ∼5 min after nicotine; Ba). In the second group of neurons, nicotine-induced monophasic response (type-II response, n = 7 from 7 rats), in which FR was transiently increased, peaking ∼1 min after nicotine, and returns to baseline levels ∼3 min after nicotine (Bb). C, Statistical analysis indicated that, 1 min after nicotine injection, VTA DA neurons with type-I (Ca) and type-II (Cb) responses showed opposite direction in FR, BF, and PSO (at ∼1 Hz). However, 5 min after injection of nicotine, FR and BF were increased, whereas PSO remained significantly reduced in the neurons with type-I (Ca) but not type-II (Cb) response. In this and the following figures, error bars indicate SEM, and *p < 0.05 and **p < 0.01.
These results indicate that VTA DA neurons exhibit two types of firing responses based on their differing electrophysiological patterns to systemic exposure to nicotine at a concentration (0.5 mg/kg) relevant to human cigarette smoking (Benowitz et al., 1982; Henningfield et al., 1993; Corrigall et al., 1994; Pidoplichko et al., 1997).
Firing patterns of VTA DA neurons with type-I or type-II response to systemic nicotine
On closer examination, we found that the VTA DA neurons with type-I and type-II responses to systemic nicotine have distinct baseline firing patterns. The neurons with type-I response (n = 17) showed relatively irregular spontaneous spike activity (Fig. 3Aa) and a high level of PSO (Fig. 3B, black line). In contrast, the neurons with type-II response exhibited regular spiking (Fig. 2Ab) and a low level of PSO (Fig. 3B, gray line). Statistical analyses demonstrated that the baseline values for FR, BF, and PSO in type-I responded DA neurons (n = 17) were 4.5 ± 0.5 Hz, 38.3 ± 7.5%, and 1.7 ± 0.3, whereas in type-II responded DA neurons (n = 7), the baseline values were 4.1 ± 0.8 Hz, 16.1 ± 7.2%, and 0.3 ± 0.1. Statistical comparisons between type-I and type-II responded neurons showed that, although there were no significant differences in their FRs (t(22) = 0.54, p = 0.592), the BF was significantly higher for type-I responded neurons (t(22) = 1.33, p = 0.048), and the values for PSO of the neurons with type-I response were significantly higher than the neurons with type-II response (t(22) = 5.29, p < 0.001) (Fig. 3C). These results again suggest that VTA DA neurons have two types of electrophysiological responses not only to systemic nicotine but also exhibit two types of intrinsic firing patterns and different SO power.
Figure 3.
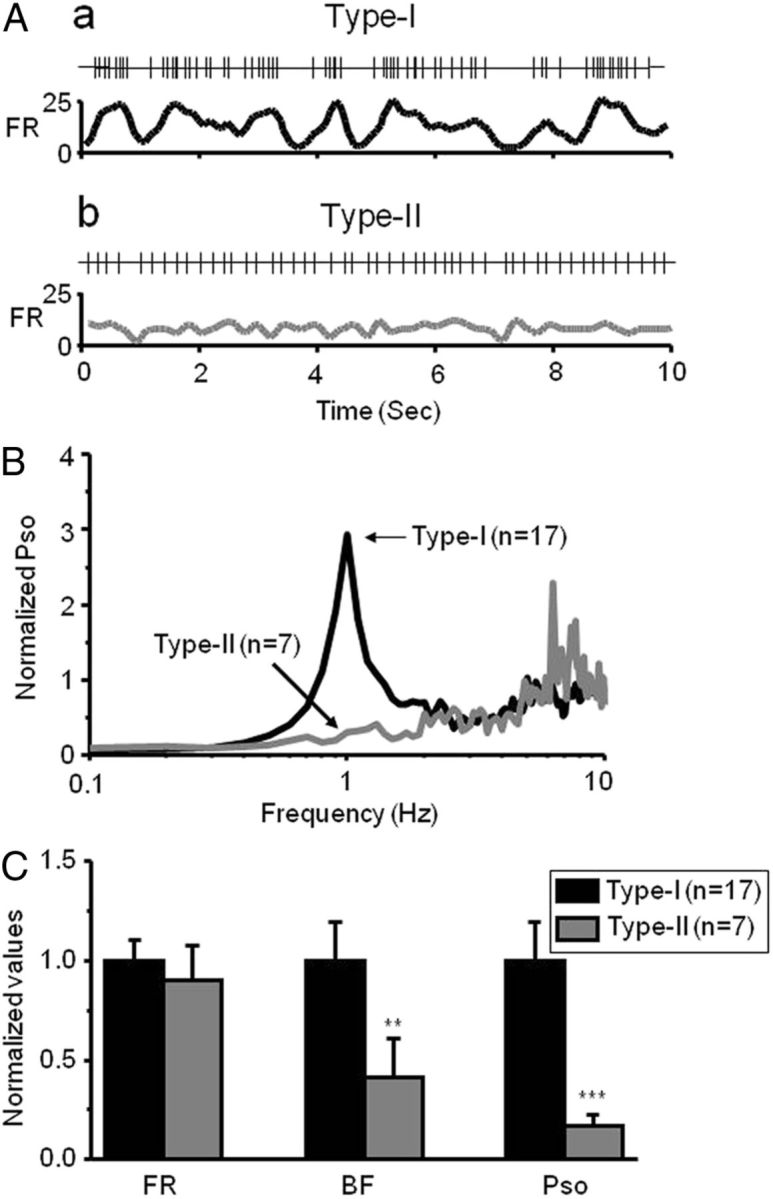
Firing patterns of VTA DA neurons. A, VTA DA neurons with two types of responses to systemic nicotine exhibited different firing patterns. Segments of spike trains recorded and corresponding, smoothed rate histograms from a type-I(Aa) and a type-II(Ab) responded neuron, respectively. B, Comparison of PSO between type-I (n = 17 from 17 rats) and type-II (n = 7 from 7 rats) responded neurons. SOs with a peak frequency close to 1 Hz were observed in the VTA DA neurons with type-I (indicated by a horizontal arrow), but not type-II, response. C, Bar graph summarizes the pool data to compare neuronal firing patterns between type-I and type-II neurons.
Effects of TTX infusion into the PFC on nicotine-induced firing of VTA DA neurons
Because VTA DA neuron SOs have been found to be an important sign of functional coupling between the PFC and the VTA (Shi, 2005; Gao et al., 2007), we hypothesized that VTA DA neurons with type-I, but not type-II, response to systemic nicotine may be coupled via cortico-meso innervations. To test this, we ablated mPFC–VTA coupling by a local infusion of the sodium channel blocker TTX (10 ng/μl in PBS) bilaterally (0.75 μl/min for 2 min through a syringe pump) into the mPFC (from bregma, AP, 3.0 mm; ML, ± 0.7 mm; DV, 3.5 mm) (Gao et al., 2007). We then examined the effects of systemic nicotine on VTA DA neuron firing. The results showed that local infusion of TTX into the mPFC for 4 min prevented the systemic nicotine-induced excitatory effect on the VTA DA neurons with type-I response (Fig. 4Aa, n = 5) but not the excitatory effect on the neurons with type-II response (Fig. 4Ba, n = 5).
Figure 4.

Effects of mPFC inactivation on nicotine-induced responses in VTA DA neurons. A, Local infusion of TTX into the mPFC (over the duration indicated by the larger, horizontal, solid bar) prevented the increase in FR induced by nicotine in type-I (Aa, n = 5 from 5 rats) but not type-II (Ba, n = 5 from 5 rats) responded DA neurons. The insets indicate PSO levels and differences also help to distinguish VTA DA neurons with type-I (Aa) and type-II (Ba) responses. Statistical analysis showed that TTX infusion into the mPFC significantly reduced BF and PSO in type-I (Ab) but not type-II (Bb) neurons. Subsequent injection of nicotine failed to increase, but persistently decreases, FRs in type-I responded neurons (Ab), while increasing the FR in type-II responded neurons.
Additional analysis (using paired t test) quantified the effects of systemic nicotine after TTX inhibition of mPFC function. These studies indicated that the effect of systemic nicotine injection on the FR, BF, and PSO (relative to baseline at 100%) for VTA type-I responded neurons was reduced to 74.5 ± 6.1% (t(4) = 3.53, p = 0.024), 37.2 ± 5.4% (t(4) = 2.84, p = 0.047), and 26.1 ± 7.8% (t(4) = 2.09, p = 0.009) when measured 5 min after nicotine exposure (Fig. 4Ab). However, for type-II responded neurons, 5 min after systemic nicotine injection, there still were increases in FR to 144.3 ± 11.5% (t(4) = −3.40, p = 0.027), and no significant effects were observed on BF (148.8 ± 43.6%, t(4) = −1.08, p = 0.341), and PSO (93.2 ± 7.4%, t(4) = −0.03, p = 979) (Fig. 4B). These results support the hypothesis that the VTA DA neurons with type-I, rather than type-II, response to nicotine are functionally coupled to the mPFC.
Effects of mPFC–VTA transection on nicotine-induced increases in FR of VTA DA neurons
To complement the preceding experiments, we then bilaterally transected the mPFC immediately caudal to the PFC (2 mm anterior to bregma) (Gao et al., 2007), to physically disconnect mPFC inputs to the VTA DA neurons. Based on our previous experiments involving local TTX application, we predicted that the transection of mPFC inputs (1) should eliminate SOs in the VTA DA neurons with type-I response and (2) would interfere with nicotinic excitation of these neurons but have no effect on the nicotinic excitation in the VTA DA neurons with type-II response.
VTA type-I responded DA neurons (n = 12 from 12 rats) were identified in part because they failed to show the late-phase increase in FR attributable to nicotine injection after transection of mPFC inputs to the VTA (Fig. 5Aa). Presumed type-II responded DA neurons still displayed an increase in FR by nicotine exposure after disruption of mPFC–VTA connections (Fig. 5Ab, n = 6 from 6 rats). When measured 5 min after nicotine exposure (i.e., once responses had stabilized), FR was reduced to 84.9 ± 0.9% in type-I responded DA neurons (t(11) = 4.57, p = 0.001; Fig. 5Ba) but increased to 146.3 ± 13.3% in type-II DA neurons (t(5) = −5.77, p=0.002; Fig. 5Bb) compared with baseline levels. These changes in FR are similar to those observed after local TTX infusion and then systemic nicotine exposure. Note that the type-I responded neurons no longer showed the biphasic pattern in FR response to systemic nicotine as seen in intact animals (Fig. 2Ba), whereas the type-II responded neurons remained qualitatively similar if not slightly prolonged compared with what was seen in intact controls (Fig. 2Bb). Conversely, BF and PSO in type-I responded neurons were not significantly altered after nicotine exposure (t(11) = 1.11, p = 0.292; t(11) = 0.176, p = 0.864), partly because both levels were very low after mPFC transection (Fig. 5Ca,Cb) but consistent with disruption of the otherwise important role for the mPFC in the generation of bursting and SO in a group of DA neurons (Shi, 2005; Gao et al., 2007).
Figure 5.

Effects of mPFC transection on nicotine-induced responses in VTA DA neurons. A, In animals with mPFC transections, nicotine (0.5 mg/kg, i.v.) decreased the FR in type-I responded neurons (Aa), but it increased the FR in type-II responded neurons (Ab). B, Bar graphs show the effects of mPFC transection on nicotine-induced changes of firing patterns in type-I (Ba) and type-II (Bb) neurons. C, After mPFC transaction, the SO was very low in both type-I and type-I responded neurons (n = 18 from 18 rats, Ca). Nicotine had no significant effects on BF and PSO in these VTA cells (Cb).
Effects of local infusion of an nAChR antagonist or agonist into the mPFC on VTA DA neurons with type-I response to systemic nicotine
Thus far, we have demonstrated that, in the VTA, type-I (but not type-II) responded DA neurons are functionally coupled to the PFC. We further elucidated how systemic nicotine affected VTA DA neurons with type-I response through the alteration of this PFC–VTA coupling. One hypothesis was that systemic nicotine exposure might primarily act on the nAChRs in mPFC neurons, in turn altering PFC–VTA coupling and leading to excitation of these VTA DA neurons. We tested this hypothesis in two experiments.
First, we infused the nAChR antagonist MEC (100 μm, 0.75 μl/2 min) locally into the mPFC and determined the subsequent effects of systemic nicotine injection on VTA type-I responded neuron firing. The results showed that there was no effect of MEC infusion into the PFC alone on FR, BF, or PSO in VTA type-I responded neurons. However, 4 min of exposure to MEC abolished the systemic nicotine-induced firing increase in these neurons (Fig. 6Aa). In nine VTA DA neurons with type-I response, after local infusion of MEC into the mPFC, systemic nicotine-induced alterations in FR, BF, and SO were 98.8 ± 3.7% (t(8) = 0.07, p = 0.949), 114.8 ± 11.4% (t(8) = −1.36, p = 0.211), and 160.4 ± 23.5% (t(8) = −1.05, p = 0.327) of baseline levels when measured 5 min later (Fig. 6Ab). These results implicated a role for nAChRs in the mPFC and of mPFC–VTA coupling in this group of VTA DA neurons.
Figure 6.

Effects of PFC infusion of a nicotinic antagonist or agonist on DA neuron firing. A, Local infusion of MEC (100 μm; duration of infusion shown by open horizontal bar) into the mPFC prevented the increase in firing in the VTA neurons with type-I response (Aa, n = 9 from 9 rats) usually seen after systemic nicotine exposure. Statistical analysis showed that the infusion inhibited all effects of nicotine on FR, BF, and PSO in these neurons (Ab). B, Local infusion of nicotine (500 nm) into the mPFC did not alter the FR (Ba), but it significantly enhances PSO in the VTA DA neurons with type-I response (Bb, n = 6 from 6 rats, *p < 0.05).
Second, we locally infused nicotine itself (0.5 μm, 0.75 μl/2 min) into the mPFC, and, 1 or 5 min later, we examined its effects on VTA type-I responded neuron firing. There was no effect on neuronal FR measured 1 or 5 min after local infusion of nicotine into the mPFC (Fig. 6B, n = 6, t(5) = 0.99, p = 0.365 vs baseline). However, additional analysis showed a significant increase in PSO in these neurons (Fig. 6Bb, t(5) = −2.74, p = 0.041). These experiments suggest that local infusion of nicotine into the mPFC increases mPFC descending coupling to VTA DA neurons with type-I response to systemic nicotine, as exemplified by increased PSO. However, this method of nicotine administration (0.5 mg/kg nicotine bitartrate) is insufficient to mimic all of the effects of systemic nicotine on VTA type-I DA neuron firing, suggesting that additional target sites might be involved in systemic nicotine-induced excitation of VTA DA neurons, although actions in the mPFC might gate such effects.
Systemic injection of nicotine acutely increases mPFC pyramidal neuron firing
To test the hypothesis that systemic nicotine can act on mPFC nAChRs to excite mPFC pyramidal neurons, we recorded mPFC pyramidal neuron firing after systemic nicotine injection. Nicotine (0.5 mg/kg, i.v.) induced a biphasic change in FR, BF, and PSO (initial increase followed by long-lasting decrease) of mPFC pyramidal neurons (Fig. 7, n = 6), which interestingly is the inverse of the pattern observed for nicotine-induced changes in the FR for VTA DA neurons with type-I response (Fig. 2Ba).
Figure 7.
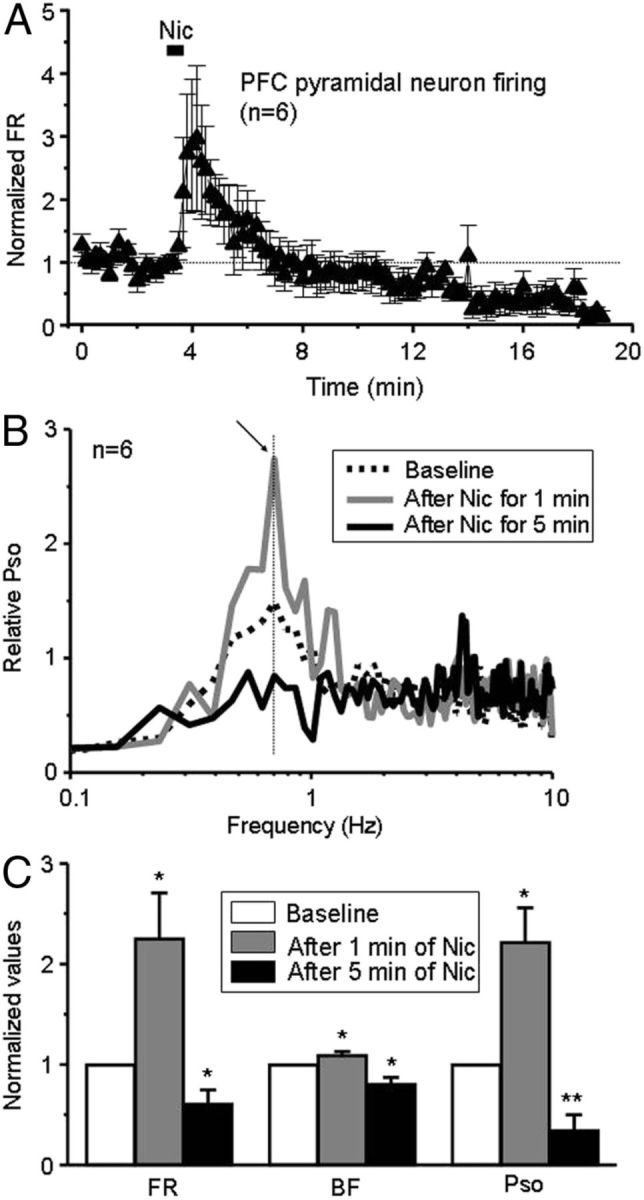
Nicotine alters mPFC neuron firing. A, Systemic nicotine exposure produces a biphasic change in mPFC pyramidal neuron firing, an increase in FR, followed by a decrease 5 min after nicotine (n = 6 from 6 rats). B, PSO is elevated 1 min after nicotine exposure and then reduced 5 min after the injection (left). C, Analysis suggests that all changes induced by nicotine are statistically significant.
Statistical analyses (using paired t test) showed that, after the injection of nicotine, the FR, BF, and PSO were 225.0 ± 45.6%, (t(5) = −2.95, p=0.032), 109.2 ± 3.8%, (t(5) = −2.75, p = 0.040), and 221.5 ± 34.3%, (t(5) = −2.01, p = 0.021) of baseline levels in mPFC pyramidal neurons when measured 1 min after nicotine injection but were 60.8 ± 4.0% (t(5) = 2.535, p = 0.048), 80.5 ± 6.7% (t(5) = 2.87, p = 0.035), and 34.6 ± 15.2% (t(5) = 2.76, p = 0.008) of baseline levels when measured 5 min after nicotine injection (Fig. 7C).
These results provide direct evidence that systemic nicotine rapidly and significantly excites mPFC pyramidal neuronal firing, bursting activity, and SO, but these effects do not persist and, in fact, reverse 5 min after nicotine administration.
Effects of BMI on the nicotine-induced increase in the VTA DA neurons with type-I response to nicotine
The apparent inverse relationship between firing patterns of mPFC and VTA DA neurons (with type-I response) suggested that there may be an intervening, inhibitory connection, likely involving GABA, in the descending mPFC–VTA pathway (Gao et al., 2007). To test this possibility, we systemically injected the GABAA receptor antagonist BMI (2.5 mg/kg, i.v.) and then evaluated nicotine-induced effects on both mPFC pyramidal neurons and VTA type-I DA neurons.
There was no change in baseline firing of mPFC pyramidal neurons (n = 6) in rats exposed to BMI (Fig. 8Aa). However, subsequent systemic administration of nicotine still excited mPFC neurons as reflected in the significant increase measured 1 min after nicotine exposure relative to baseline levels in FR (160.2 ± 30.3%, t(5) = −4.07, p = 0.009), BF (150.2 ± 15.1%, t(5) = −3.37, p = 0.014), and PSO (149.2 ± 35.3%, t(5) = −2.08, p = 0.009; Fig. 8Ab). However, BMI preexposure did abolish the nicotine-induced late-phase reduction in mPFC neuron firing, otherwise (Fig. 7) seen 5 min after nicotine injection (FR = 60.8 ± 4.0% of baseline, t(5) = 2.54, p = 0.048 for nicotine alone and FR = 101.8 ± 11.0% for nicotine after BMI injection, t(5) = −1.76, p = 0.139, n = 6; Fig. 8Ab).
Figure 8.

Effects of BMI. Aa, Systemic injection of the GABAA receptor antagonist BMI (2.5 mg/kg, i.v.; delivery duration indicated by first, solid horizontal bar) had no effect on the early excitation, but it suppressed the late inhibition of mPFC neurons induced by subsequent nicotine (Nic) injection. Ab, Bar graph summarized effects of BMI. BMI had no effect alone and did not affect the early, nicotine-induced increase in FR and BF in mPFC neurons, but it abolished the later inhibition of mPFC neurons (n = 6 from 6 rats). There was a reduction in PSO after BMI exposure alone and 5 min after subsequent delivery of nicotine, but BMI did not alter the early increase in PSO induced by nicotine. Ba, After BMI injection (2.5 mg/kg, i.v.), which had no effect on FR alone, systemic injection of nicotine did not significantly alter FR in DA neurons with type-I response (n = 6 from 6 rats). Bb, Statistical analysis showed that BMI exposure alone had no effect on FR and BF but prevented effects of subsequently delivered nicotine on FR and BF measured at 1 or 5 min after nicotine injection. In addition, BMI delivery with or without nicotine reduced PSO in the VTA DA neurons with type-I response (Bb).
We next determined the effects of BMI preexposure on nicotine-induced effects on type-I responded VTA DA neurons (n = 6 from 6 rats). The results show that, although BMI exposure did not alter the FR and BF, it did diminish PSO significantly (Fig. 8Bb). Statistical analysis showed that, in six VTA type-I responded DA neurons tested after BMI treatment, nicotine-induced alterations in FR, BF, and PSO were 96.0 ± 5.0% (t(5) = 0.78, p = 0.47), 92.4 ± 9.4% (t(5) = 0.99, p = 0.37), and 69.3 ± 4.2% (t(5) = 1.86, p = 0.007) of baseline levels when measured 1 min after nicotine injection, and they were 97.9 ± 7.5% (t(5) = 0.56, p = 0.597), 107.9 ± 10.2% (t(5) = −0.23, p = 0.824), and 70.1 ± 3.8% (t(5) = 1.75, p = 0.005; p = 0.974 compared with the baseline after BMI) when measured 5 min after nicotine injection. This indicates that BMI eliminates SO and both the early inhibitory and the late excitatory effects of systemic nicotine on the VTA DA neurons with type-I response.
These results also are consistent with the hypothesis that systemic nicotine exposure has its effects on VTA type-I responded DA neuronal firing by altering the activity of mPFC neurons, which ordinarily are inversely coupled to those VTA DA neurons through intervening GABAergic synapses.
Discussion
The release of DA into the NAc and the mPFC after the excitation of VTA DA neurons is considered to be an important mechanism underlying drug reward and addiction (Wise, 2004). Patch-clamp recordings in VTA slices have shown that nicotine at concentrations comparable with those found in human cigarette smokers excites VTA DA neurons (Pidoplichko et al., 1997). Nicotine also induced desensitization of α4β2–nAChRs on GABAergic neurons in the VTA, thereby reducing endogenous cholinergic excitation of those neurons and release of GABA onto DA neurons, which thus become disinhibited (Pidoplichko et al., 1997, 2004; Mansvelder et al., 2002).
However, systemic administration of nicotine affects the activity of DA neurons through mechanisms that are more complex, involving the VTA and other brain regions (Mameli-Engvall et al., 2006; Avale et al., 2008), such as the mPFC (Erhardt et al., 2002; Ikemoto et al., 2006). Several studies have consistently shown a functional coupling between the mPFC and the VTA. For example, stimulation of the mPFC increases burst firing in VTA DA neurons, whereas inactivating the mPFC decreases VTA DA neuronal activity (Gariano and Groves, 1988; Svensson and Tung, 1989; Murase et al., 1993; Overton et al., 1996a; Tong et al., 1996). In addition, mPFC-induced increase in DA neuronal bursting is often preceded by an inhibition of DA neuronal firing that lasts up to several hundred milliseconds (Nakamura et al., 1979; Thierry et al., 1979; Gariano and Groves, 1988; Overton et al., 1996b; Tong et al., 1996; Overton and Clark, 1997), suggesting that initial excitation of mPFC neurons inhibits VTA DA neurons via activation of inhibitory GABAergic neurons. Furthermore, stimulating mPFC excites VTA GABAergic neurons in either a monophasic or biphasic pattern (Geisler and Wise, 2008). More importantly, the mPFC produces a relatively long-lasting GABAergic inhibitory action on VTA DA neurons, which is thought to underlie disinhibition-induced DA neuron excitation, especially during exposure to addictive drugs (Tong et al., 1996). We previously reported a covariance of spontaneous activity of VTA DA neurons and mPFC neurons suggestive of functional coupling (Gao et al., 2007). However, simultaneous dual-field potential recordings demonstrated that field SOs in the mPFC preceded those in the VTA by ∼30 ms, and transection of, or local injection of TTX into, the mPFC abolished VTA neuronal SOs. Together, this suggests a negative coupling between mPFC and VTA DA neurons (Gao et al., 2007). Finally, pharmacological MRI studies also showed that systemic nicotine exposure in humans caused a prominent increase in cerebral blood flow to the mPFC and the VTA (Choi et al., 2006; Gozzi et al., 2006), consistent with the concept that mPFC–VTA circuits are important targets for nicotinic action.
In the present study, we analyzed neuronal PSO as an index to reflect PFC–VTA coupling before and after nicotine exposure because it is known that VTA DA neurons exhibit robust oscillatory activity in anesthetized rats (Shi, 2005). In this study (Fig. 5Ca), inactivation of PFC abolished VTA SOs, suggesting that SOs in VTA DA neurons are related to activity in the mPFC. This idea is consistent with the dual-field potential recordings in the PFC and the VTA that display highly synchronized SOs between mPFC and the VTA (Peters et al., 2004).
VTA DA neurons exhibit two types of spiking profiles in response to systemic nicotine. The high PSO only seen in the neurons with type-I response, and the inverse relationships between the changes of their FRs and the FRs of mPFC neurons after nicotine exposure, provide clear indications of mPFC–VTA negative coupling, as well as a useful means for identifying those cells experimentally. The new findings based on pharmacological disruption or a physical severance of connections to the mPFC underscores their pivotal role in mPFC mediation of nicotine-induced excitation of those VTA DA neurons with type-I response. Whether or not these VTA DA neurons with differential responses to nicotine are correlated to a defined subgroup of DA neurons in the VTA (based on neuronal locations, projections, or response to reward/aversive stimulations) are unclear. It will be highly interesting to evaluate this in a future study.
Systemic nicotine administration significantly reduces PSO in type-I responded DA neurons while increasing their FR. This is consistent with nicotine acting to functionally uncouple the mPFC–VTA circuit. BMI exposure also lowers PSO and completely blocks initial inhibitory and late excitatory effects of nicotine on the VTA DA neurons with type-I response. This is consistent with nicotine having its effects and with mPFC–VTA DA neuronal inverse coupling being mediated via an inhibitory, GABAergic connection. It has been reported that GABAergic neurons in the VTA exhibit oscillations at 1–2 Hz (Steffensen et al., 1998) and that VTA GABAergic neurons receive glutamatergic innervations from the PFC (Carr and Sesack, 2000; Geisler and Wise, 2008). These GABAergic neurons may play a role in the transfer of SOs from mPFC to the VTA neurons with type-I response. A role for mPFC nAChRs in the excitatory response of these DA neurons to nicotine is evident from the sensitivity of that response to MEC applied locally into the mPFC. Moreover, the ability of BMI administration to prevent the late inhibitory phase of the actions of nicotine on mPFC neuronal firing suggests that GABAA receptors, at least partly, mediate that effect. However, the insensitivity to BMI of the initial, excitatory effect of nicotine exposure on mPFC neuronal firing is consistent with an effect via nAChR-mediated activation of presynaptic glutamate release in the mPFC (Dickinson et al., 2008; Konradsson-Geuken et al., 2009; Livingstone et al., 2009), but this idea requires additional investigation. These results suggest that the nicotine-induced initial inhibition on the DA neurons with type-I response may be mediated by GABAergic neurons that may not be controlled by the PFC.
In the mPFC, nicotine exposure culminates in a delayed but longer-lasting reduction in mPFC neuron firing, likely through activation of GABAergic mechanisms (Aracri et al., 2010). Such a process, which would lead to lasting activation of VTA DA neurons, has been implicated in morphine-, muscimol-, and benzodiazepine-induced VTA DA neuron excitation (Gysling and Wang, 1983; Lokwan et al., 2000). However, our studies suggest that consideration should be given to the nicotine exposure-induced impairment of inverse coupling between the mPFC and the VTA via the GABAergic neurons as defined by diminished PSO as a novel, alternate mechanism for nicotine-induced DA neuron excitation in vivo.
The major feature of drug addiction is a reduced ability to regulate control over the desire to procure drugs regardless of the risks involved. Traditional models implicated the neural “reward” (i.e., VTA) system as the key brain region in providing a neurobiological model of addiction. Computational models of the local VTA circuitry suggested that a biphasic pattern of response to nicotine could be mediated by nicotine action on the nAChRs at local GABAergic neurons and subsequent disinhibition of the DA neurons (Ahmed et al., 2009; Graupner and Gutkin, 2009). However, ideas that are more recent have expanded on this circuitry to include two separate but interconnecting systems: the VTA in the incentive sensitization of drugs, and the PFC in regulating inhibitory control over drug use.
The neurophysiological results we show here provide experimental evidence to support this new model. This is particularly significant when put into the larger context of drug addiction theory. The development of drug addiction has been likened to a progressive habitualization of behavior and a loss of cognitive control (Everitt and Robbins, 2005; Feil et al., 2010), possibly through a disconnection of higher cortical influence over reward circuitry (Kalivas, 2009). Consistent with the notion that the mPFC plays a role in drug addiction through the modulation of descending inhibitory system (Feil et al., 2010) and in line with the concept that the mPFC mediates cognitive control (executive functions), the inverse coupling that we show here suggests that the mPFC exerts this control through multisynaptic gating of inhibition and excitation of DA transmission and that nicotine selectively disrupts this inhibitory control, leading to a dysfunctional modulation of the VTA DA neurons with type-I response. Furthermore, recent computational theories propose an opposition of cognitive control versus reward-driven habitual action (Daw et al., 2005), suggesting that a model-based cortical system can ensure flexible achievement of long-term goals, whereas a model-free, reward-based, DA system promotes habitual seeking of immediate rewards. The theory proposes that an optimal integration between the two systems for control of behavior is the key and may be mediated by cholinergic signals encoding the uncertainty of the behavioral context (Yu and Dayan, 2005). Thus, we speculate that nicotine would pathologically remove the influence of the cortical model-based cognitive control system over the DA circuitry and promote habitual immediate-reward-seeking behavior. Our results, by pointing to a nicotine-induced uncoupling of the mPFC-mediated inhibitory control over reward circuitry, provide a potential neural basis for the bias toward impulsivity and loss of cognitive control seen in nicotine-dependent individuals and possibly other types of drug dependencies.
Footnotes
This work was supported by the Barrow Neurological Foundation, Arizona Biomedical Research Commission Grants 0028 and 0057, the Institute for Mental Health Research, Philip Morris International through their External Research Program, and the National Institutes of Health Grants R01 NS040417 and R01 DA015389. We thank Dr. A. Oster, Dr. M. Graupner, Harrison Stratton, and Dr. E. Siagain for their careful reading of this manuscript as well as their discussions and comments.
References
- Ahmed SH, Graupner M, Gutkin B. Computational approaches to the neurobiology of drug addiction. Pharmacopsychiatry. 2009;42(Suppl 1):S144–S152. doi: 10.1055/s-0029-1216345. [DOI] [PubMed] [Google Scholar]
- Aracri P, Consonni S, Morini R, Perrella M, Rodighiero S, Amadeo A, Becchetti A. Tonic modulation of GABA release by nicotinic acetylcholine receptors in layer V of the murine prefrontal cortex. Cereb Cortex. 2010;20:1539–1555. doi: 10.1093/cercor/bhp214. [DOI] [PubMed] [Google Scholar]
- Avale ME, Faure P, Pons S, Robledo P, Deltheil T, David DJ, Gardier AM, Maldonado R, Granon S, Changeux JP, Maskos U. Interplay of beta2* nicotinic receptors and dopamine pathways in the control of spontaneous locomotion. Proc Natl Acad Sci U S A. 2008;105:15991–15996. doi: 10.1073/pnas.0807635105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benowitz NL, Kuyt F, Jacob P., 3rd Circadian blood nicotine concentrations during cigarette smoking. Clin Pharmacol Ther. 1982;32:758–764. doi: 10.1038/clpt.1982.233. [DOI] [PubMed] [Google Scholar]
- Bunney BS, Aghajanian GK, Roth RH. Comparison of effects of l-dopa, amphetamine and apomorphine on firing rate of rat dopaminergic neurones. Nat New Biol. 1973;245:123–125. doi: 10.1038/newbio245123a0. [DOI] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Projections from the rat prefrontal cortex to the ventral tegmental area: target specificity in the synaptic associations with mesoaccumbens and mesocortical neurons. J Neurosci. 2000;20:3864–3873. doi: 10.1523/JNEUROSCI.20-10-03864.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charara A, Smith Y, Parent A. Glutamatergic inputs from the pedunculopontine nucleus to midbrain dopaminergic neurons in primates: Phaseolus vulgaris-leucoagglutinin anterograde labeling combined with postembedding glutamate and GABA immunohistochemistry. J Comp Neurol. 1996;364:254–266. doi: 10.1002/(SICI)1096-9861(19960108)364:2<254::AID-CNE5>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Choi JK, Mandeville JB, Chen YI, Kim YR, Jenkins BG. High resolution spatial mapping of nicotine action using pharmacologic magnetic resonance imaging. Synapse. 2006;60:152–157. doi: 10.1002/syn.20284. [DOI] [PubMed] [Google Scholar]
- Corrigall WA, Coen KM, Adamson KL. Self-administered nicotine activates the mesolimbic dopamine system through the ventral tegmental area. Brain Res. 1994;653:278–284. doi: 10.1016/0006-8993(94)90401-4. [DOI] [PubMed] [Google Scholar]
- Counotte DS, Goriounova NA, Li KW, Loos M, van der Schors RC, Schetters D, Schoffelmeer AN, Smit AB, Mansvelder HD, Pattij T, Spijker S. Lasting synaptic changes underlie attention deficits caused by nicotine exposure during adolescence. Nat Neurosci. 2011;14:417–419. doi: 10.1038/nn.2770. [DOI] [PubMed] [Google Scholar]
- Dani JA, Heinemann S. Molecular and cellular aspects of nicotine abuse. Neuron. 1996;16:905–908. doi: 10.1016/s0896-6273(00)80112-9. [DOI] [PubMed] [Google Scholar]
- Daw ND, Niv Y, Dayan P. Uncertainty-based competition between prefrontal and dorsolateral striatal systems for behavioral control. Nat Neurosci. 2005;8:1704–1711. doi: 10.1038/nn1560. [DOI] [PubMed] [Google Scholar]
- Dickinson JA, Kew JN, Wonnacott S. Presynaptic alpha 7- and beta 2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol. 2008;74:348–359. doi: 10.1124/mol.108.046623. [DOI] [PubMed] [Google Scholar]
- Erhardt S, Schwieler L, Engberg G. Excitatory and inhibitory responses of dopamine neurons in the ventral tegmental area to nicotine. Synapse. 2002;43:227–237. doi: 10.1002/syn.10044. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Feil J, Sheppard D, Fitzgerald PB, Yücel M, Lubman DI, Bradshaw JL. Addiction, compulsive drug seeking, and the role of frontostriatal mechanisms in regulating inhibitory control. Neurosci Biobehav Rev. 2010;35:248–275. doi: 10.1016/j.neubiorev.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Fuster JM. Executive frontal functions. Exp Brain Res. 2000;133:66–70. doi: 10.1007/s002210000401. [DOI] [PubMed] [Google Scholar]
- Gao M, Liu CL, Yang S, Jin GZ, Bunney BS, Shi WX. Functional coupling between the prefrontal cortex and dopamine neurons in the ventral tegmental area. J Neurosci. 2007;27:5414–5421. doi: 10.1523/JNEUROSCI.5347-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao M, Chu HY, Jin GZ, Zhang ZJ, Wu J, Zhen XC. l-Stepholidine-induced excitation of dopamine neurons in rat ventral tegmental area is associated with its 5-HT(1A) receptor partial agonistic activity. Synapse. 2011;65:379–387. doi: 10.1002/syn.20855. [DOI] [PubMed] [Google Scholar]
- Gariano RF, Groves PM. Burst firing induced in midbrain dopamine neurons by stimulation of the medial prefrontal and anterior cingulate cortices. Brain Res. 1988;462:194–198. doi: 10.1016/0006-8993(88)90606-3. [DOI] [PubMed] [Google Scholar]
- Garzón M, Vaughan RA, Uhl GR, Kuhar MJ, Pickel VM. Cholinergic axon terminals in the ventral tegmental area target a subpopulation of neurons expressing low levels of the dopamine transporter. J Comp Neurol. 1999;410:197–210. doi: 10.1002/(sici)1096-9861(19990726)410:2<197::aid-cne3>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Geisler S, Wise RA. Functional implications of glutamatergic projections to the ventral tegmental area. Rev Neurosci. 2008;19:227–244. doi: 10.1515/revneuro.2008.19.4-5.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozzi A, Schwarz A, Reese T, Bertani S, Crestan V, Bifone A. Region-specific effects of nicotine on brain activity: a pharmacological MRI study in the drug-naive rat. Neuropsychopharmacology. 2006;31:1690–1703. doi: 10.1038/sj.npp.1300955. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons. 1. Identification and characterization. Neuroscience. 1983;10:301–315. doi: 10.1016/0306-4522(83)90135-5. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci. 1984;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Onn SP. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J Neurosci. 1989;9:3463–3481. doi: 10.1523/JNEUROSCI.09-10-03463.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granon S, Poucet B, Thinus-Blanc C, Changeux JP, Vidal C. Nicotinic and muscarinic receptors in the rat prefrontal cortex: differential roles in working memory, response selection and effortful processing. Psychopharmacology. 1995;119:139–144. doi: 10.1007/BF02246154. [DOI] [PubMed] [Google Scholar]
- Graupner M, Gutkin B. Modeling nicotinic neuromodulation from global functional and network levels to nAChR based mechanisms. Acta Pharmacol Sin. 2009;30:681–693. doi: 10.1038/aps.2009.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillem K, Bloem B, Poorthuis RB, Loos M, Smit AB, Maskos U, Spijker S, Mansvelder HD. Nicotinic acetylcholine receptor beta2 subunits in the medial prefrontal cortex control attention. Science. 2011;333:888–891. doi: 10.1126/science.1207079. [DOI] [PubMed] [Google Scholar]
- Gysling K, Wang RY. Morphine-induced activation of A10 dopamine neurons in the rat. Brain Res. 1983;277:119–127. doi: 10.1016/0006-8993(83)90913-7. [DOI] [PubMed] [Google Scholar]
- Henningfield JE, Stapleton JM, Benowitz NL, Grayson RF, London ED. Higher levels of nicotine in arterial than in venous blood after cigarette smoking. Drug Alcohol Depend. 1993;33:23–29. doi: 10.1016/0376-8716(93)90030-t. [DOI] [PubMed] [Google Scholar]
- Ikemoto S, Qin M, Liu ZH. Primary reinforcing effects of nicotine are triggered from multiple regions both inside and outside the ventral tegmental area. J Neurosci. 2006;26:723–730. doi: 10.1523/JNEUROSCI.4542-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalivas PW. Neurotransmitter regulation of dopamine neurons in the ventral tegmental area. Brain Res Brain Res Rev. 1993;18:75–113. doi: 10.1016/0165-0173(93)90008-n. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Konradsson-Geuken A, Gash CR, Alexander K, Pomerleau F, Huettl P, Gerhardt GA, Bruno JP. Second-by-second analysis of alpha 7 nicotine receptor regulation of glutamate release in the prefrontal cortex of awake rats. Synapse. 2009;63:1069–1082. doi: 10.1002/syn.20693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED. Nicotinic systems and cognitive function. Psychopharmacology. 1992;108:417–431. doi: 10.1007/BF02247415. [DOI] [PubMed] [Google Scholar]
- Levin ED, McClernon FJ, Rezvani AH. Nicotinic effects on cognitive function: behavioral characterization, pharmacological specification, and anatomic localization. Psychopharmacology. 2006;184:523–539. doi: 10.1007/s00213-005-0164-7. [DOI] [PubMed] [Google Scholar]
- Livingstone PD, Srinivasan J, Kew JN, Dawson LA, Gotti C, Moretti M, Shoaib M, Wonnacott S. alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate dopamine release in vitro and in vivo in the rat prefrontal cortex. Eur J Neurosci. 2009;29:539–550. doi: 10.1111/j.1460-9568.2009.06613.x. [DOI] [PubMed] [Google Scholar]
- Lokwan SJ, Overton PG, Berry MS, Clark D. The medial prefrontal cortex plays an important role in the excitation of A10 dopaminergic neurons following intravenous muscimol administration. Neuroscience. 2000;95:647–656. doi: 10.1016/s0306-4522(99)00467-4. [DOI] [PubMed] [Google Scholar]
- Mameli-Engvall M, Evrard A, Pons S, Maskos U, Svensson TH, Changeux JP, Faure P. Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron. 2006;50:911–921. doi: 10.1016/j.neuron.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS. Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron. 2000;27:349–357. doi: 10.1016/s0896-6273(00)00042-8. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905–919. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, Mertz M, Role LW. Nicotinic modulation of synaptic transmission and plasticity in cortico-limbic circuits. Semin Cell Dev Biol. 2009;20:432–440. doi: 10.1016/j.semcdb.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathon DS, Kamal A, Smidt MP, Ramakers GM. Modulation of cellular activity and synaptic transmission in the ventral tegmental area. Eur J Pharmacol. 2003;480:97–115. doi: 10.1016/j.ejphar.2003.08.097. [DOI] [PubMed] [Google Scholar]
- Murase S, Grenhoff J, Chouvet G, Gonon FG, Svensson TH. Prefrontal cortex regulates burst firing and transmitter release in rat mesolimbic dopamine neurons studied in vivo. Neurosci Lett. 1993;157:53–56. doi: 10.1016/0304-3940(93)90641-w. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Iwatsubo K, Tsai CT, Iwama K. Cortically induced inhibition of neurons of rat substantia nigra (pars compacta) Jpn J Physiol. 1979;29:353–357. doi: 10.2170/jjphysiol.29.353. [DOI] [PubMed] [Google Scholar]
- Omelchenko N, Sesack SR. Glutamate synaptic inputs to ventral tegmental area neurons in the rat derive primarily from subcortical sources. Neuroscience. 2007;146:1259–1274. doi: 10.1016/j.neuroscience.2007.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overton PG, Clark D. Burst firing in midbrain dopaminergic neurons. Brain Res. 1997;25:312–334. doi: 10.1016/s0165-0173(97)00039-8. [DOI] [PubMed] [Google Scholar]
- Overton PG, Tong ZY, Brain PF, Clark D. Preferential occupation of mineralocorticoid receptors by corticosterone enhances glutamate-induced burst firing in rat midbrain dopaminergic neurons. Brain Res. 1996a;737:146–154. doi: 10.1016/0006-8993(96)00722-6. [DOI] [PubMed] [Google Scholar]
- Overton PG, Tong ZY, Clark D. A pharmacological analysis of the burst events induced in midbrain dopaminergic neurons by electrical stimulation of the prefrontal cortex in the rat. J Neural Transm. 1996b;103:523–540. doi: 10.1007/BF01273151. [DOI] [PubMed] [Google Scholar]
- Peters Y, Barnhardt NE, O'Donnell P. Prefrontal cortical up states are synchronized with ventral tegmental area activity. Synapse. 2004;52:143–152. doi: 10.1002/syn.20015. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, DeBiasi M, Williams JT, Dani JA. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- Pidoplichko VI, Noguchi J, Areola OO, Liang Y, Peterson J, Zhang T, Dani JA. Nicotinic cholinergic synaptic mechanisms in the ventral tegmental area contribute to nicotine addiction. Learn Mem. 2004;11:60–69. doi: 10.1101/lm.70004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi WX. Slow oscillatory firing: a major firing pattern of dopamine neurons in the ventral tegmental area. J Neurophysiol. 2005;94:3516–3522. doi: 10.1152/jn.00317.2005. [DOI] [PubMed] [Google Scholar]
- Shi WX, Pun CL, Zhou Y. Psychostimulants induce low-frequency oscillations in the firing activity of dopamine neurons. Neuropsychopharmacology. 2004;29:2160–2167. doi: 10.1038/sj.npp.1300534. [DOI] [PubMed] [Google Scholar]
- Steffensen SC, Svingos AL, Pickel VM, Henriksen SJ. Electrophysiological characterization of GABAergic neurons in the ventral tegmental area. J Neurosci. 1998;18:8003–8015. doi: 10.1523/JNEUROSCI.18-19-08003.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson TH, Tung CS. Local cooling of pre-frontal cortex induces pacemaker-like firing of dopamine neurons in rat ventral tegmental area in vivo. Acta Physiol Scand. 1989;136:135–136. doi: 10.1111/j.1748-1716.1989.tb08640.x. [DOI] [PubMed] [Google Scholar]
- Thierry AM, Deniau JM, Feger J. Effects of stimulation of the frontal cortex on identified output VMT cells in the rat. Neurosci Lett. 1979;15:102–107. doi: 10.1016/0304-3940(79)96097-x. [DOI] [PubMed] [Google Scholar]
- Tong ZY, Overton PG, Clark D. Stimulation of the prefrontal cortex in the rat induces patterns of activity in midbrain dopaminergic neurons which resemble natural burst events. Synapse. 1996;22:195–208. doi: 10.1002/(SICI)1098-2396(199603)22:3<195::AID-SYN1>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM. Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol. 2001;63:241–320. doi: 10.1016/s0301-0082(00)00033-2. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2042. doi: 10.1126/science.1093360. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nat Rev Neurosci. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Yu AJ, Dayan P. Uncertainty, neuromodulation, and attention. Neuron. 2005;46:681–692. doi: 10.1016/j.neuron.2005.04.026. [DOI] [PubMed] [Google Scholar]
- Zhang D, Yang S, Jin GZ, Bunney BS, Shi WX. Oscillatory firing of dopamine neurons: differences between cells in the substantia nigra and ventral tegmental area. Synapse. 2008;62:169–175. doi: 10.1002/syn.20479. [DOI] [PubMed] [Google Scholar]