

Summary
The Forkhead transcription factor, FoxO3a, is a known suppressor of primary tumor growth through transcriptional regulation of key genes regulating cell cycle arrest and apoptosis. In many types of cancer, in response to growth factor signaling, FoxO3a is phosphorylated by Akt, resulting in its exclusion from the nucleus. Here we show that FoxO3a remains nuclear in anaplastic thyroid carcinoma (ATC). This correlates with lack of Akt phosphorylation at serine473 in ATC cell lines and tissues of ATC patients, providing a potential explanation for nuclear FoxO3a. Mechanistically, nuclear FoxO3a promotes cell cycle progression by transcriptional upregulation of cyclin A1, promoting proliferation of human ATC cells. Silencing FoxO3a with a reverse genetics approach leads to downregulation of CCNA1 mRNA and protein. These combined data suggest an entirely novel function for FoxO3a in ATC promotion by enhancing cell cycle progression and tumor growth through transcriptional upregulation of cyclin A1. This is clinically relevant since we detected highly elevated CCNA1 mRNA and protein levels in tumor tissues of ATC patients. Our data indicate therapeutic inactivation of FoxO3a may lead to attenuation of tumor expansion in ATC. This new paradigm also suggests caution in relation to current dogma focused upon reactivation of FoxO3a as a therapeutic strategy against cancers harboring active PI3-K and Akt signaling pathways.
Key words: FoxO3a, Anaplastic thyroid carcinoma, Cyclin A1, Transcriptional regulation
Introduction
Thyroid cancer incidence in the United States has doubled in the past decade with deaths rising in parallel by one-third, making it the fifth most diagnosed cancer in women in 2012 and an emerging public health concern. Thyroid cancers account for 5% of all human malignancies in the U.S. with 56,460 new cases estimated in 2012 disproportionately affecting over 43,000 women (Siegel et al., 2012). One subtype, anaplastic thyroid carcinoma (ATC), is characterized by its loss of differentiation and is one of the most aggressive tumors in humans (Green et al., 2006). Even though ATC is rare and represents only 1.7% of all malignant thyroid diseases, it accounts for approximately 40% of thyroid carcinoma related deaths in the US (Green et al., 2006; Hundahl et al., 1998). Thus, the diagnosis of ATC is essentially a death sentence, with the five year survival rate of approximately seven percent; patients survive, on average, less than five months after diagnosis (Edge and Compton, 2010; Smallridge and Copland, 2010). Such a bleak prognosis is due to ATC aggressive growth, invasiveness, metastatic behavior of the cancer and lack of effective therapies. Due to high mortality rates, further preclinical studies of ATC pathogenesis and molecular signaling are critically needed for developing effective therapeutic interventions.
Tumor enlargement is, in part, a result of persistent cell cycle progression due to aberrant signaling along with the lack of tumor suppressors and regulators. ATC has been shown to have multiple pathways overexpressed and actively signaling including: EGFR, phosphorylated Akt (p-Akt), p-ERK and JAK/STAT pathways (Neff et al., 2008; Sherman, 2006; Smallridge et al., 2009; Woyach and Shah, 2009). Cell cycle progression is coordinated by cyclins and specifically cyclins D1, D3 and E have all been shown to be overexpressed in ATC. Growth arrest is facilitated by the cyclin-dependent kinase inhibitors and tumor suppressors and in ATC it has been shown that genes encoding p16, p21, p27, p53, Rb and PTEN are either mutated or less expressed (Smallridge and Copland, 2010; Smallridge et al., 2009).
Forkhead homeobox type O3a (FoxO3a, FKHRL1) is a transcription factor with known tumor suppressor activity. Foxo3a targets and activates the pro-apoptotic genes Bim and FasL and induces cyclin kinase inhibitors such as p21CIP1/WAF1 and p27Kip1, but also represses cyclin D1 (Burgering, 2008; Carter and Brunet, 2007; Fu and Tindall, 2008; Furukawa-Hibi et al., 2005). FoxO3a is a member of the FoxO superfamily that all share a conserved 110 amino acid DNA-binding domain (Vogt et al., 2005) and all recognize two consensus DNA-binding sequences: 5′-TTGTTTAC-3′ and 5′-(C/A)(A/C)AAA(C/T)AA-3′ (Furuyama et al., 2000; Obsil and Obsilova, 2011). FoxO3a is tightly controlled post-translationally by acetylation (p300/CBP, SIRT1), ubiquitylation (Skp2), and phosphorylation [Akt, serum and glucocorticoid inducible kinase (SGK1), AMP-activated protein kinase (AMPK), dual-specificity tyrosine phosphorylation-regulated kinase 1a (DYRK1a), IκB kinase β (IKKβ), c-Jun N-terminal kinase (JNK), and mammalian ortholog of Ste20-like protein kinase (Mst1)] in order to regulate metabolism, homeostasis, cell cycle, DNA repair, oxidative stress resistance and apoptosis (Huang and Tindall, 2007; Storz, 2011). Regulation of FoxO3a by phosphorylation is important for nuclear–cytoplasmic shuttling. Phosphorylation of FoxO3a by JNK, AMPK or Mst1 leads to activation and nuclear localization, while phosphorylations mediated by Akt, DYRK1a, IKKβ or SGK1 lead to nuclear exclusion and inactivation (Brunet et al., 1999; Brunet et al., 2001; Carter and Brunet, 2007; Shukla et al., 2009; Tzivion et al., 2011; van den Berg and Burgering, 2011; Vogt et al., 2005). Akt phosphorylates FoxO3a at 3 residues T32, S253 and S315 (Finnberg and El-Deiry, 2004).
Akt activity correlates with its phosphorylation at T308 and S473. Akt activity has been reported to be highly elevated in ATC (Santarpia et al., 2008). Therefore, our original hypothesis was that by suppressing Akt either by shRNA or novel Akt inhibitors, its target FoxO3a would relocalize to the nucleus and induce its antitumor activity. However, the contrary was discovered with high nuclear FoxO3a in the presence of biologically inactive Akt in ATC patient tissue samples and cell lines. Moreover, biologically active FoxO3a in ATC cell lines transcriptionally upregulated cyclin A1 gene expression leading to enhanced cell cycle progression and cell proliferation. Thus, we unexpectedly identified a novel mechanism and paradigm shift leading to a new tumor promoting role of FoxO3a in a highly aggressive and invasive cancer.
Results
FoxO3a localization and Akt activity in ATC cells
Immunocytochemistry (ICC) showed that FoxO3a was localized primarily in the nucleus in four ATC cell lines (Fig. 1A; supplementary material Fig. S1A). KTC2 and KTC3 cells were lentiviral infected with either nontarget or FoxO3a shRNA to demonstrate antibody specificity (verification of shRNA knockdown in Fig. 3A). Seven ATC cell lines were screened for FoxO3a expression and FoxO3a was expressed in all, with a lesser degree in FRO and FF1 cells (Fig. 1B). However, FoxO3a is thought of as a tumor suppressor that becomes inactivated and shuttled out of the nucleus as a result of phosphorylation by activated Akt (Carter and Brunet, 2007; Shukla et al., 2009; Vogt et al., 2005). Thus, phosphorylated FoxO3a (p-FoxO3a; Ser253, The32, S318) levels were examined (Fig. 1B) and phosphorylated S253 was detected in four of the seven cell lines. Generally, it is accepted that ATC has aberrant and elevated Akt signaling (García-Rostán et al., 2005; Santarpia et al., 2008). However in all ATC cell lines (except SW1736) p-Akt S473 was surprisingly absent, while p-Akt T308 (phosphorylated activation loop of Akt) was consistently elevated to varying degrees (Fig. 1B). Since S473 is known to be phosphorylated by the mTORC2 complex, we next examined mTORC2 activity in ATC cells. Very low levels of active mTORC2 [p-mTOR(2) S2481 staining] were observed indicative of low pAkt S473 phosphorylation, previously shown to be required for phosphorylation of FoxO3a leading to inactivation (Jacinto et al., 2006). We next examined Akt activity by analyzing the phosphorylation status of glycogen synthase kinase 3 beta (GSK3β) at S9, a residue that is directly targeted by Akt (Stambolic and Woodgett, 1994). In all ATC cell lines examined, p-GSK3β S9 levels were low to nonexistent suggesting that Akt is not active. We also analyzed if nuclear accumulation of FoxO3a could be due to increased activity of kinases that phosphorylate and promote nuclear FoxO3a [i.e. Mst1 and JNK (Fig. 1B)]. Mst-1 was expressed and active (as judged by its phosphorylation at T183) in all cell lines, indicating that it may also contribute to nuclear FoxO3a activity. Total JNK was expressed, however, p-JNK T183/Y185 was expressed at low levels except in KTC3 cells. These cumulative data indicate that nuclear FoxO3a in ATC cells may be a result of joint action of two regulatory mechanisms, including inactive Akt allowing active FoxO3a to remain nuclear and active Mst-1 driving FoxO3a into the nucleus.
Fig. 1.

FoxO3a is predominantly nuclear in ATC cell lines. (A) ICC shows nuclear localization of FoxO3a in KTC2 and KTC3 cells. Cells were lentiviral-infected with either nontarget or FoxO3a 1566 shRNA. Less fluorescence is seen in the FoxO3a-silenced cells, indicating that the nuclear expression is specific. DAPI was used to stain the nuclei. (B) Western blot of ATC cell lines for total and phosphorylated forms of FoxO3a, Akt, mTOR, Mst-1 and JNK expression. MDA231 was used as a positive control (+) that was run on the same blot (another control was removed). β-actin was used as a loading control. (C) QPCR of unmatched normal and ATC patient tissues shows that there is no significant difference between FoxO3a mRNA levels. Data are plotted as average fold change compared with unmatched normal ± s.d. (n = 10). (D) IHC in adjacent normal thyroid and ATC patient tissues demonstrates FoxO3a is nuclear and expression is similar for both as shown by H scores. Normal patient pancreatic tissue was used as a positive control, and shows cytoplasmic staining of FoxO3a, further demonstrating the specificity of the antibody for nuclear staining in thyroid tissue (top left panel). Breast tumor tissue was used as a positive control for the remaining control panels.
Fig. 3.
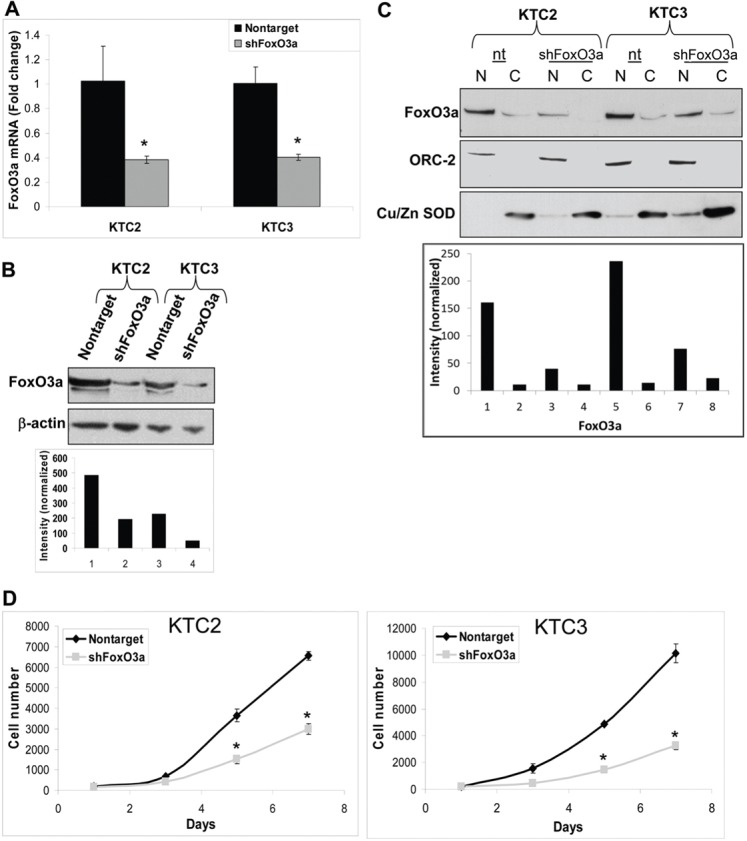
Silencing FoxO3a reduces proliferation. (A) QPCR verifies that FoxO3a has been silenced in KTC2 and KTC3 cells that have been infected with lentiviral FoxO3a 1566 shRNA but not in nontarget control cells. Data are plotted as fold change ± s.d. as compared with nontarget control; *P<0.05. (B) Western blot of the same cells confirms that FoxO3a is reduced in the FoxO3a shRNA cells compared with nontarget control. β-actin was used as a loading control and was used to normalize data for densitometric analysis. (C) The same cells were then fractionated to demonstrate that FoxO3a is predominantly in the nuclear fraction and its expression can be attenuated by shRNA. ORC-2 and Cu/Zn SOD were used to normalize data for densitometric analysis. N, nuclear; C, cytoplasmic. (D) Proliferation curve of the lentiviral-infected cells showing decreased proliferation over 7 days when FoxO3a is silenced. Data are plotted as cell number ± s.d. and *P<0.05, compared with nontarget control.
When FoxO3a was examined in patient samples, FoxO3a mRNA demonstrated no significant difference in expression levels in ATC patient tissues when compared to unmatched normal samples (Fig. 1C). This was also true by immunohistochemistry (IHC) where total FoxO3a protein expression and localization were not different between adjacent normal thyroid and tumor tissues with FoxO3a localized predominantly to the nucleus (Fig. 1D). To demonstrate antibody specificity, normal human pancreatic tissue (Fig. 1D, top left panel), human breast tissues and BT474 breast cancer cells (supplementary material Fig. S1B) were used as positive controls showing cytoplasmic staining of FoxO3a protein. Ten normal and tumor breast tissue slides were also stained for FoxO3a as a control and it showed more nuclear than cytoplasmic staining in the normal tissue versus the tumor tissue (supplementary material Fig. S1B). These data are corroborated by IHC data from other laboratories for normal breast and tumor tissues (Chen et al., 2010; Habashy et al., 2011; Hu et al., 2004). p-FoxO3a T32, indicative of inactive FoxO3a, was elevated ∼50% above that of adjacent normal thyroid tissue and was cytoplasmic (supplementary material Fig. S1C). p-FoxO3a S253/S318 were essentially nonexistent in patient normal thyroid and ATC tissues (Fig. 1D; supplementary material Fig. S1C). Interestingly, p-Akt T308 was elevated in ATC patient tissues while p-Akt S473 appeared absent for the most part (Fig. 1D). Thus, there was a strong concordance between patient and cell line data indicative that FoxO3a was predominantly nuclear and S473 Akt phosphorylation was absent.
The conundrum of nuclear FoxO3a and elevated p-Akt T308 levels in the absence of p-Akt S473 led us to overexpress constitutively active Akt (CA Akt) to further analyze whether p-Akt T308 was defective in phosphorylating FoxO3a thereby stimulating FoxO3a shuttling out of the nucleus. One report showed that if Akt was phosphorylated only on T308, FoxO3a could not be phosphorylated by Akt and thus no relocalization followed by inactivation in the cytoplasm (Jacinto et al., 2006). This report suggested that p-Akt T308 may be unable to phosphorylate FoxO3a thereby providing a mechanistic explanation for nuclear accumulation of FoxO3a in ATC. To test this, CA Akt was overexpressed in KTC3 and KTC2 cells (Fig. 2A). The phosphoinositide 3-kinase (PI3-K) inhibitor, LY294002, attenuated phosphorylation of Akt at S473 and T308 (Fig. 2A). While total FoxO3a in each of the two cell lines showed little change in the presence of CA Akt and/or LY294002, phosphorylation of T32 on FoxO3a was enhanced upon LY294002 treatment (Fig. 2A). Unexpectedly, there was no difference in proliferation rates of the CA-Akt-expressing cells as compared to the pcDNA3 control in either of the two ATC cell lines (Fig. 2B). Treatment with the PI3-K inhibitors, LY294002 and wortmannin (data not shown), showed a small but statistically significant decrease in proliferation in the pcDNA3 controls (Fig. 2B) while the CA Akt cells in the presence of LY294002 demonstrated slight but statistical growth inhibition when compared to pcDNA3 plus LY294002 (Fig. 2B). We noted that KTC3 and KTC2 cells transiently transfected with overexpressed CA Akt were more sensitive to growth inhibition upon LY294002 treatment compared to that of empty vector (Fig. 2A). This was also true for the biochemical readouts where phosphorylation of T32 on FoxO3a were enhanced upon LY294002 treatment (Fig. 2B). This was quite puzzling since loss of S473 and T308 Akt phosphorylation indicated that the LY294002 was effective. Collectively, these data indicated that LY294002 may have a growth inhibitory effect independent of active Akt. Subcellular localization was next examined for FoxO3a and Akt to determine if CA Akt was nuclear and whether CA Akt phosphorylated FoxO3a (Fig. 2C,D). ICC for total Akt demonstrated enhanced nuclear staining in the CA-Akt-overexpressing cells when compared to pcDNA3 controls (Fig. 2C, panel 1). When ICC was examined, endogenous total FoxO3a remained nuclear with some enhanced cytoplasmic FoxO3a in the CA-Akt-expressing cells (Fig. 2C, panel 2). When phosphorylation sites on FoxO3a were examined, p-FoxO3a S253 showed that some FoxO3a was shuttled into the cytoplasm in the presence of CA Akt (Fig. 2C, panel 3). Interestingly, p-FoxO3a T32 was present in the nucleus in pcDNA3 control cells and unchanged with CA Akt overexpression (Fig. 2C, panel 4). Furthermore, in cells undergoing mitosis, p-FoxO3a T32 was localized at the centrioles (Fig. 2C, panel 4). Our ICC findings were validated utilizing cytosolic and nuclear fractionation followed by western blot analysis to examine FoxO3a and Akt subcellular localization (Fig. 2D). FoxO3a was predominantly in the nuclear fraction and when CA Akt was overexpressed the cytoplasmic fraction doubled (Fig. 2D). These cumulative data demonstrated a novel finding in that FoxO3a remained predominantly in the nucleus of ATC cells and tumor tissues even when exposed to exogenous CA Akt.
Fig. 2.

Overexpression of constitutively active Akt has no effect on proliferation. (A) Western blot of KTC3 and KTC2 cells transfected with pcDNA3 control or a constitutively active (CA) Akt expression plasmid demonstrates overexpression of CA Akt versus pcDNA3 control. When cells were untreated or treated with 1 µM LY294002 (LY), it was shown that LY attenuates p-Akt levels. Total FoxO3a and p-FoxO3a protein levels were also examined for each of these conditions. (B) Proliferation curves of transfected KTC3 and KTC2 cells over 7 days, show that there is no difference in proliferation rates between the pcDNA3 control and the CA Akt cells. Treatment with the PI3-K inhibitor, LY294002, resulted in only slight growth inhibition in the pcDNA3 controls (gray arrow) compared with pcDNA3-DMSO-treated cells, whereas the CA Akt cells showed modest but statistically significant growth inhibition (black arrow) when compared with the LY-treated pcDNA3 cells of both cell lines. Data are plotted as cell number ± s.d. (C) ICC of transfected KTC3 reveals that total Akt is both nuclear and cytoplasmic (panel 1). Total FoxO3a (panel 2) remains predominantly nuclear in the CA Akt cells with minimal shuttling of p-FoxO3a S253 to the cytoplasm (panel 3), whereas p-FoxO3a T32 remains cytoplasmic (panel 4). DAPI was used to stain the nuclei. (D) Subcellular fractionation followed by western blotting and densitometric analysis confirmed that FoxO3a is predominantly in the nuclear fraction and when CA Akt is overexpressed, very little if any FoxO3a is shuttled into the cytoplasm. ORC-2 was used to normalize for the nuclear fraction and Cu/Zn SOD for the cytoplasmic fraction. N, nuclear, C, cytoplasmic.
FoxO3a promotes ATC cell proliferation
We previously identified FoxO3a shRNA 1566 to be selective in silencing FoxO3a mRNA expression (Storz et al., 2009). KTC2 and KTC3 (high FoxO3a expression) cells were infected with lentiviral nontarget or FoxO3a 1566 shRNA. Silencing of FoxO3a mRNA (∼60%) was shown by Quantitative PCR (QPCR; Fig. 3A) and this transferred to reduction of protein expression as confirmed by western blot (Fig. 3B). To further verify that FoxO3a was predominantly nuclear, the same cells were then fractionated to demonstrate that the majority of FoxO3a is in the nuclear fraction and its expression can be attenuated by shRNA (Fig. 3C). Data were normalized to ORC-2 for the nuclear fraction and Cu/Zn superoxide dismutase (SOD) for the cytoplasmic fraction. When proliferation rates were examined, there was decreased growth in the FoxO3a-silenced cells (Fig. 3D). These data were verified using additional siRNA targeting FoxO3a in four independent ATC cell lines that expressed high levels of FoxO3a (supplementary material Fig. S2A). FoxO3a siRNA #1 inhibited proliferation by 61–99% while #6 inhibited by 28–90% (supplementary material Fig. S2B). Due to this intriguing finding, overexpression of FoxO3a was next examined in low expressing FoxO3a ATC cell lines (see Fig. 1B). FRO and FF1 cells were transiently transfected with pECE empty vector control or wild-type (wt) FoxO3a and shown by QPCR that wt FoxO3a mRNA was elevated by ∼3-fold in FRO cells and ∼80-fold in FF1 cells (Fig. 4A). FoxO3a overexpression was confirmed by western blot analysis for both day one and day seven after transient transfection (Fig. 4B). When ICC was examined, transfected wt FoxO3a shuttled into the nucleus (Fig. 4C). Thus, nuclear localization of FoxO3a in ATC cells holds true in these transfected cells in the presence of Akt (see Fig. 1B). The proliferation rates were increased in the transiently overexpressing wt FoxO3a cells (Fig. 4D). Thus, silencing or overexpressing FoxO3a resulted in the novel finding that FoxO3a promoted cell proliferation, a paradigm shift.
Fig. 4.

Overexpression of FoxO3a increases proliferation. (A) QPCR verifies that wild-type (wt) FoxO3a has been overexpressed in transiently transfected FRO and FF1 cells compared with pECE control (24 hours post-transfection). Data are plotted as fold change ± s.d. compared with empty vector control, *P<0.05. (B) Western blots of the same transiently transfected cells confirm that wt FoxO3a is overexpressed compared with its empty control, 1 and 7 days after transfection. β-actin was used as a loading control. (C) ICC shows nuclear localization of transiently transfected wt FoxO3a in both ATC cell lines. DAPI was used to stain the nuclei. (D) Proliferation curves of the transiently transfected cells show that overexpressed wt FoxO3a increases proliferation over 7 days. Data are plotted as cell number ± s.d.; *P<0.05, compared with empty vector control.
FoxO3a regulates cyclin A1 in ATC cells
In order to further demonstrate that FoxO3a was not functioning as a tumor suppressor in ATC cells, cell cycle regulation was examined. First, flow cytometry in KTC3 cells with silenced FoxO3a, demonstrated increased G0/G1 (∼8.9%) and decreased G2/M (∼14%) phase (Fig. 5A). In the transiently overexpressing wt FoxO3a FRO cells, G0/G1 was decreased by ∼7.8% with a slight increase in the G2/M (∼2%) phase (Fig. 5A). Because of these changes in G0/G1 and G2/M phases of the cell cycle, all the major cyclins (D, E, A, and B) and cyclin-dependent kinase inhibitors (CKIs) of the INK4 and CIP/KIP families were examined (Fig. 5B, and data not shown). Only cyclin A and B expression levels were consistently affected by altered FoxO3a in both models (Fig. 5B). QPCR showed that CCNA1 and CCNA2 mRNA were decreased by ∼30% in cells when FoxO3a is silenced in KTC3 cells (Fig. 5C, top panel). However, only CCNA1 mRNA was elevated by ∼2-fold when wt FoxO3a was overexpressed (Fig. 5C, bottom panel). Despite changes in cyclin B protein expression by western blot (Fig. 5B); no change occurred in either mRNA levels as detected by QPCR (Fig. 5C) or promoter activity of cyclin B (data not shown).
Fig. 5.

FoxO3a alters cyclin A expression. (A) Flow cytometry of lentiviral-infected KTC3 cells to examine silencing of FoxO3a, and transiently transfected FRO cells to examine overexpression of wt FoxO3a. Prior to analysis, cells were synchronized for 24 hours and then released for 72 hours. DNA content was analyzed using propidium iodide fluorescence and histograms were plotted of the percentage of cells in each phase of the cell cycle. (B) Western blot of the same cells indicate that when FoxO3a is silenced in KTC3 cells, cyclin A and B expression decreases, whereas overexpressing wt FoxO3a in FRO cells increases cyclin A and B expression. β-actin was used as a loading control. (C) QPCR confirms that only CCNA1 and A2 mRNA levels are decreased in FoxO3a-silenced KTC3 cells and only CCNA1 mRNA levels are increased in wt FoxO3a-overexpressing FRO cells. There is no change in CCNB1 and CCNB2 mRNA levels. Data are plotted as fold change ± s.d. compared with control, *P<0.05.
Analysis of the cyclin A1 promoter region identified a single putative FoxO3a binding site in the first 1181 bases upstream of the transcription start site (Fig. 6A). Knockdown of FoxO3a in the KTC3 cells significantly inhibited the transcriptional activity of the cyclin A1 promoter by over 50%. Conversely, overexpression of wt FoxO3a significantly induced the transcriptional activity of cyclin A1 in transiently transfected FRO cells by ∼2-fold (Fig. 6B, *). Mutation of the putative FoxO3A binding site in the cyclin A1 promoter (Fig. 6A) significantly decreased reporter activity in both KTC3 nontarget and FRO/pECE controls by ∼50% compared to wild-type cyclin A1 promoter (Fig. 6B, +). In addition, the luciferase activity of cyclin A1 decreased by ∼85% in FRO/wt FoxO3a cells when comparing wt versus the mutant cyclin A1 promoter (Fig. 6B, ++). While we found no other consensus FoxO3a response elements, a 50% decrease of luciferase of the mutant cyclin A1 promoter when compared to the cyclin A1 promoter indicates that another functional FoxO3a response element exists. We further confirmed our novel finding by performing chromatin immunoprecipitation (CHiP) and demonstrate FoxO3a bound to the cyclin A1 promoter (Fig. 6C). Taken together, these findings indicated that FoxO3a transcriptionally regulated cyclin A1 to promote cell cycle progression and enhanced cell proliferation in ATC.
Fig. 6.

FoxO3a regulates the cyclin A promoter. (A) Cyclin A1 promoter region contains a putative FoxO3a binding site. The response element was mutated as described in Materials and Methods for a mutant cyclin A1 promoter lacking a functional FoxO3a consensus sequence. (B) Comparison of luciferase activity of the wild-type cyclin A1 promoter with the mutant cyclin A1 promoter in KTC3 shFoxO3a and FRO wt FoxO3a cells. Cells were transiently transfected as described in Materials and Methods. The results given as average relative luminescent units (firefly activity/renilla luciferase vector activity) ± s.d. *Comparison between control and FoxO3a shRNA or wt FoxO3a within a promoter, +comparison of activities across wild-type and mutant cyclin A1 promoters. (C) FRO cells were transfected with vector control or FLAG-tagged FoxO3a. FoxO3a/DNA complexes were immunoprecipitated (anti-FLAG) after crosslinking. An immunoprecipitation with mouse IgG served as a negative control (control). Precipitates were analyzed by PCR for the FoxO3a-bound cyclin A1 promoter. A PCR for the cyclin A1 promoter using the input DNA served as an additional control.
We next examined CCNA1 mRNA and cyclin A1 protein levels in adjacent normal and ATC patient tissues finding elevated cyclin A1 mRNA and protein levels (Fig. 7A,B). Using two different CCNA1 lentiviral shRNAs, QPCR shows 70–80% silencing of CCNA1 mRNA in KTC3 and FRO/wt FoxO3a cells (Fig. 7C). When examining proliferation of these same cells, there was a 2- to 4-fold reduction in cell proliferation (Fig. 7D). Thus, our cumulative data mapped a new signaling pathway whereby FoxO3a transcriptionally upregulated cyclin A1 which in turn promoted cell cycle progression clearly linking FoxO3a to cyclin A1-driven cell proliferation.
Fig. 7.

Cyclin A1 is overexpressed in ATC patient tumor tissues. (A) QPCR of unmatched normal and ATC patient tissues show there is a significant increase in CCNA1 mRNA levels. Data are plotted as average fold change compared with unmatched normal ± s.d., *P<0.05. (B) IHC in adjacent normal and ATC patient tissues demonstrates cyclin A1 is overexpressed as shown by H scores. (C) QPCR verifies that CCNA1 mRNA has been silenced in KTC3 and FRO/wt FoxO3a cells that have been infected with lentiviral CCNA1 387 and 1018 shRNA compared with levels in nontarget control. Data are plotted as fold change ± s.d. compared with nontarget control, *P<0.05 (D) Proliferation analysis of lentiviral-infected cells shows that when CCNA1 is silenced in KTC3 or FRO/wt FoxO3a-overexpressing cells, decreased cell proliferation occurs. Data are plotted as cell number ± s.d.; *P<0.05, compared with nontarget control.
Discussion
Our data go against the dogma that FoxO3a is a tumor suppressor that becomes inactivated by growth factor signaling and localized to the cytoplasm (Carter and Brunet, 2007; Shukla et al., 2009; Vogt et al., 2005). For example, cytosolic FoxO3a has been correlated with tumor grade in prostate cancer (Shukla et al., 2009). Generally, it is accepted that nuclear (active) FoxO3a induces apoptosis through Bim1 and FasL, and cell cycle arrest through GADD45, p21 and p27 while repressing cyclins D1 and D2. FoxO3a has also been shown to promote DNA repair through GADD45 and DBB1 (Carter and Brunet, 2007; Vogt et al., 2005). Limited data are available in thyroid tumors, but nuclear FoxO3a was reduced in follicular thyroid carcinoma (FTC) tissues and unchanged in papillary thyroid carcinoma (PTC), while cytoplasmic FoxO3a was increased in both FTC and PTC (Karger et al., 2009). Shin et al. showed in a follicular cell line (FTC 133) that nuclear FoxO3a induced anti-proliferative (Shin et al., 2010) and pro-apoptotic genes, while Weidinger et al. found similar results (Weidinger et al., 2010). These data contrast with our data showing nuclear localization of FoxO3a in ATC. This is quite interesting in light of current thought that FTC and PTC may progress to ATC versus that of ATC arising de novo, suggesting again a switch in FoxO3a function.
On the other hand, there are currently only two publications that support the notion that FoxO3a possesses oncogenic functions. Storz and colleagues were the first, showing that nuclear FoxO3a promotes invasion through induced expression of MMP9 and MMP13 in breast cancer cells (Storz et al., 2009). Chen et al. have shown that nuclear FoxO3a correlates with lymph node metastasis and poor survival as well as elevated p-Akt T308 in breast cancer (Chen et al., 2010). They further showed an uncoupling of the Akt:FoxO3a signaling axis in drug-resistant breast cancer cells in culture. Now, our laboratory recapitulates this finding correlating nuclear FoxO3a and elevated p-Akt T308 in another aggressive cancer, ATC. For the first time, we show that nuclear FoxO3a transcriptionally induces cyclin A1 to promote cell proliferation in ATC cell lines (Figs 5 and 6). We also demonstrated that overexpression of a constitutively active Akt mutant is not sufficient to inactivate and shuttle FoxO3a into the cytoplasm (Fig. 2). Akt is phosphorylated at two key residues located at the catalytic site (T308) and the C-terminal hydrophobic motif (S473) site. Our data support previous work (Guertin et al., 2006) demonstrating that p-Akt T308 is not sufficient to phosphorylate FoxO3a. Work by the Sabatini laboratory demonstrated that disruption of the mTORC2 complex leads to loss of p-Akt S473 phosphorylation and loss of Akt-induced phosphorylation of FoxO proteins. This is consistent with the lack of S473 phosphorylation seen in six of seven ATC cell lines and patient ATC tumors that we screened (Fig. 1). Jacinto and colleagues further showed that dual phosphorylations of Akt at the hydrophobic motif and catalytic sites synergistically activate Akt but are not required for all Akt functions. Importantly, they also observed that phosphorylation of FoxO1 or FoxO3a did not occur in defective p-Akt S473 phosphorylation cells, while singly T308-phosphorylated Akt was sufficient to phosphorylate other Akt targets such as TSC2 and GSK3, S6K and 4E-BP1 (Jacinto et al., 2006). The mTORC2 complex appeared to have minimal activating phosphorylation [p-mTOR(2) S2481] in our ATC cell lines indicative of an inability to phosphorylate Akt at S473 (Fig. 2B). These observations most likely explain our data whereby FoxO3a remains nuclear in ATC in the presence of highly elevated p-Akt T308 and is functional. Our data also suggest that FoxO3a is somehow resistant to CA Akt remaining in the nucleus even with overexpression of CA Akt (Fig. 2). This may be due to active Mst1 known to drive FoxO3a into the nucleus (Fig. 1B).
In addition, overexpression of FoxO3a results in nuclear accumulation and leads to increased proliferative rates (Fig. 4). We speculate that as thyroid tumors progress from benign to well-differentiated to metastatic and undifferentiated, tumor alterations occur in the FoxO3a signaling pathway, shifting from tumor suppressor to that of oncogenic. Changes may occur in the nuclear/cytoplasmic distribution, the phosphorylation and/or acetylation status of FoxO3a, or in proteins that regulate, are regulated by, or bind to FoxO3a. The mechanism by which FoxO3a now acts as a positive regulator at the level of the cyclin A1 promoter remains to be discovered.
Cyclin A2, also known as cyclin A, is the major A-type cyclin in mammals while cyclin A1 is an alternative CDK2 associated A-type cyclin (Joshi et al., 2009). Cyclin A1 differs from other cyclins in its highly restricted expression pattern. Besides its expression during spermatogenesis, cyclin A1 is also expressed in hematopoietic progenitor cells and in acute myeloid leukemia. Cyclin A1 is expressed at low levels in most other tissues, but no phenotype other than male infertility has been reported for mice lacking the cyclin A1 gene (Ji et al., 2004). Cyclin A1 mRNA and protein are present at very low levels in cells at the G0 phase. It increases during the progression of the cell cycle and reaches the highest levels in the S and G2/M phases. This cyclin binds both cyclin dependent kinase 2 (CDK2) and CDC2 kinases, which give two distinct kinase activities, one appearing in S phase, the other in G2, and thus regulate separate functions in the cell cycle. CDK2–cyclin A1 complex has kinase activities for histone H1, E2F-1, and the Rb family of proteins. Transcriptional control of cyclin A1 has been demonstrated for Sp1 and Sp3, which bind to four GC boxes between nucleotides −130 and −80 upstream of the transcriptional start site. c-Myc has also been shown to directly transactivate the cyclin A1 promoter and may be involved in the high-level expression of cyclin A1 observed in acute myeloid leukemia (Chan et al., 2009). We now add FoxO3a as a new positive transcriptional regulator of cyclin A1 in ATC. In contrast, FoxO3a has been shown to suppress cyclin A1 expression in follicular thyroid carcinoma (FTC) cell lines, suggestive that FoxO3a is also transcriptionally active but with opposite results in regard to cell cycle regulation in another thyroid cancer histotype (Karger et al., 2009; Shin et al., 2010).
One could envision cyclin A1 as a novel molecular target for therapy against ATC, especially since cyclin A1 demonstrates very selective tissue expression (Ji et al., 2004; Sharma et al., 2001). Beside human leukemia, cyclin A1 has been shown to play a role in enhanced cell proliferation in non-small cell lung cancer (Chan et al., 2009; Cho et al., 2006). Because of its selective interaction with CDK and CDC kinases as well as important cell cycle regulators such as E2F1, retinoblastoma (Rb) and the p21 family proteins, screening small compound libraries may identify selective inhibitors of cyclin A1. Sharma et al. identified moderately active antagonists based upon E2F1/cyclin A interactions and further showed that attenuation of cyclin A/CDK2 activity leads to apoptosis (Sharma et al., 2001). In testing our ATC cell lines with and without FoxO3a, we discovered that FoxO3a profoundly accelerates proliferation and enhances the G2/M phase of the cell cycle. Thus, we identified a novel mechanism and paradigm shift in the role of FoxO3a in a highly aggressive and invasive cancer. This new finding is highly suggestive that targeting cyclin A1 may lead to tumor suppression in ATC.
Even though FoxO3a is considered a tumor suppressor, no other lab, to our knowledge, has directly silenced FoxO3a and examined proliferative effects. The paper by Lin et al. was the only manuscript that examined proliferation when FoxO3a was silenced using miR-96. However, they showed increased proliferation in breast cancer cell lines with increased cyclin D1 and downregulation of p21CIP1/WAF1 and p27Kip1 (Lin et al., 2010). Our laboratory has mechanistically linked FoxO3a to enhanced proliferation rates (Figs 3 and 4) through FoxO3a transcriptional regulation of cyclin A1 (Figs 6 and 7). This paradigm shift, where a good gene turns bad in an aggressive cancer, provokes many more questions especially related to the FoxO3a nuclear complex positively regulating cyclin A1 in ATC cells; as well as, the prevalence of this phenomenon in aggressive cancers. It also brings into question the notion of targeting FoxO3a for re-expression in cancers as a therapeutic strategy for inhibiting tumor growth (Singh et al., 2011; Yang and Hung, 2011; Yang and Hung, 2009).
Materials and Methods
Immunohistochemistry
IHC was performed on patient-matched thyroid samples and pancreatic and breast tissue as a control to examine FoxO3a and Akt expression and localization. Tissues were mounted on slides from paraffin-embedded blocks and blocked with Diluent that contained Background Reducing Components (Dakocytomation, Denmark) for 30 minutes and probed for FoxO3a, p-Akt T308 (Cell Signaling, Beverly, MA); p-Akt S473 (Dakocytomation) and cyclin A1 (Labvision, Fremont, CA). Negative sections were prepared by incubating the slides in the absence of the primary antibody. Images were obtained at 20× using Scanscope XT and Imagescope software (Aperio Technologies, Vista, CA). The staining of the tissue sections was scored using an algorithm in the Imagescope software created by a histologist based upon signal intensity (0–3+). H scores were calculated using the formula: [(1+%×1)+(2+%×2)+(3+%×3)]. Cases were excluded from the study if a section could not be assigned a score due to insufficient tumor tissue. This study was approved by the Mayo Institutional Review Board.
Cell lines and proliferation curves
The following anaplastic thyroid carcinoma cell lines used in these studies were kindly provided as follows: KTC2 and KTC3 by Junichi Kurebayashi (Kawasaki Medical School, Japan); FRO by G. J. Juillard (University of California-Los Angeles); FF1 by Franco Frasca (University of Catania, Italy); BHT101 by Istvan Palyi (National Institute of Oncology, Hungary); SW1736 by Leibowitz and McCombs III (Scott and White Memorial Hospital, Texas); OCUT1 by Naoyoshi Onoda (Osaka City University Graduate School of Medicine, Japan); THJ-11T, THJ-16T, THJ-21T and THJ-29T were originated in our laboratory (Marlow et al., 2010). MDA231, BT474 and HEK293 were purchased from ATCC (Manassas, VA). All cell lines are short-tandem repeat (STR) verified (Marlow et al., 2010; Schweppe et al., 2008) and were maintained in RPMI 1640 medium (Cellgro, Herndon VA) supplemented with 10% charcoal-stripped fetal bovine serum (Gemini Bioproducts, West Sacramento, CA), non-essential amino acids (Cellgro), sodium pyruvate (Cellgro), HEPES (Cellgro) and penicillin–streptomycin–amphotericin B (Cellgro) at 37°C in a humidified atmosphere with 5% CO2. For all proliferation curves, cells were plated in 12-well plates (Midwest Scientific, St. Louis, MO) in triplicate at a concentration of 2×104 cells/well. As indicated, LY294002 (Sigma-Aldrich, St. Louis, MO) was added on day 1 and again on day 4 when medium was changed. After 1, 3, 5, and 7 days, cells were trypsinized and counted on a Coulter Particle Counter (Beckman, Brea, CA).
siRNA screening
ATC cells were reverse-transfected with siRNA as previously described (Azorsa et al., 2009). Screening with siRNA was performed using a subset of the Apoptosis siRNA library (Qiagen, Valencia, CA). Negative control non-Silencing siRNA and positive control lethal siRNA were purchased from Qiagen. Library and control siRNA were diluted in siRNA buffer (Qiagen) and 9.3 ng of siRNA was printed onto white 384-well plates. The targeting sequences for FOXO3A siRNA #1 and #6 were 5′-CTGAATGATGGGCTGACTGAA-3′ and 5′-TCGATTCATGCGGGTCCAGAA-3′, respectively. Diluted Lipofectamine RNAiMAX reagent (Invitrogen, Carlsbad, CA) in OptiMEM (Invitrogen) was added to the wells and allowed to complex with siRNA for 30 min at room temperature. ATC cells were resuspended in growth media without antibiotics at a final concentration of 1000 cells/well. Plates were incubated at 37°C with 5% CO2. After 96 hours total cell number was determined by the addition of Cell Titer Glo (Promega, Madison, WI) and relative luminescence units (RLU) were measured. Raw RLU data were used to calculate viability relative to untreated wells.
Plasmids and transfections
The constitutively active Akt (pcDNA3/Myr CA AKT) plasmid # 9008 was purchased from Addgene (Cambridge, MA). The control (pECE) and pECE/wt FoxO3a plasmids were a gift from Anne Brunet (Stanford University). The FoxO3a insert was confirmed by DNA sequencing. For stable transfection, KTC3 cells were transfected for 24 hours using Lipofectamine 2000 (Invitrogen) followed by selection with 500 µg/ml neomycin (MP Biomedicals, Solon, OH). For transient transfections, FRO and FF1 cells were first transfected for 24 hours using Lipofectamine 2000 (Invitrogen) and then seeded for the experiment. The pRL-CMV-renilla luciferase plasmid was purchased from Promega (Madison, WI). The cyclin A1 promoter luciferase reporter plasmid (pGL/cyclin A1 luciferase) was a gift from Michael Brattain (University of Nebraska, Omaha, NE). Mutation of the FoxO3A binding site within the cyclin A1 promoter was created using the QuikChange II Site Directed Mutagenesis Kit (Stratagene, La Jolla, CA) as per manufacturer's protocol using the primers 5′-CAGTTTCCTTTGGTCCACCCTTCACTCGCCTGC-3′ and 5′-CGAGGCGAGTGAAGGGTGGACCAAAGGAAACTG-3′. The two base pair mutation was verified by DNA sequencing before purification with PureLink HiPure Plasmid Maxiprep Kit (Invitrogen).
Lentivirus and infections
Self-inactivating shRNA lentiviruses were generated using MISSION shRNA pLKO.1 constructs (Sigma-Aldrich, St. Louis, MO). The nontarget control was a random scrambled sequence (SHC002) and the target sequence for FoxO3a was 5′-GTCACTGCATAGTCGATTCAT-3′ (clone NM_001455.1-1566s1c1) (Sigma-Aldrich). For cyclin A1, the target sequences were 5′-GCTAACTGCAAATGGGCAGTA-3′ (clone NM_003914.2-387s1c1) and 5′-GCTTCGAAATATGAAGAGATA-3′ (clone NM_003914.2-1018s1c1). Lentiviruses were packaged using HEK293FT cells by transient transfection of the pLKO.1 constructs along with ViraPower (Invitrogen) using Lipofectamine 2000 (Invitrogen). Supernatants were collected 72 hours post-transfection, passed through a 0.45 µm PVDF syringe filter (Millipore, Bedford, MA) and applied to KTC2 and KTC3 cells for infection along with 5 µg/ml polybrene (American Bioanalytical, Natick, MA) for 24 hours. Cells were then selected with 2 µg/ml puromycin (Fisher Scientific, Houston TX).
Cell lysis and western blot
Cells were plated in 10-cm plates (Midwest Scientific) and grown to ∼70% confluence prior to cell lysis in M-PER extraction buffer (Pierce, Rockford, IL) containing protease inhibitor cocktail (Roche, Mannheim, Germany) and phosphatase inhibitor (Pierce). For tissues, 50 mM Tris, pH 8.0 + 1% SDS was used. After centrifugation, supernatant protein concentrations were measured by bicinchoninic acid (BCA) assay (Pierce). Nuclear and cytoplasmic fractions from two million cells were collected using NE-PER kit as per manufacturer's protocol (Pierce). All samples were loaded on Novex 4–12% Bis-Tris gels in MES buffer (Invitrogen) followed by transfer to 0.2 µm Immobilon Psq membranes (Millipore). For western blot analysis, primary antibodies [FoxO3a, p-FoxO3a S253, p-FoxO3a S318, p-JNK T183/Y185, JNK, p-Mst1 T183, Mst1, HA tag, total cyclin A, cyclin B, p-Akt T308 and Akt (Cell Signaling, Beverly, MA); p-Akt S473 (R&D Biosystems, Minneapolis, MN); p-FoxO3a T32 (Abcam, Cambridge, MA); ORC-2 (BD Pharmingen, San Diego, CA); Cu/Zn SOD (Enzo Life Sciences, Farmingdale, NY); β-actin (Sigma-Aldrich)] were incubated overnight at 4°C. Secondary species-specific horseradish-peroxidase-labeled antibodies (Jackson Immunoresearch, West Grove, PA) were applied in 3% milk/TBS for 45 minutes at room temperature. Detection was performed using Supersignal chemiluminescence kit (Pierce). Protein expression from western analysis was quantitated using Image Quant 5.0 (Molecular Dynamics, GE Healthcare, Piscataway, NJ). Blots were background corrected and normalized to loading controls.
Immunocytochemistry
Cells were plated in 2-well chamber slides (Fisher Scientific) and grown to ∼70% confluence. For ICC analysis, cells were fixed with 2% paraformaldehyde followed by permeabilization with ice-cold 100% methanol. Samples were blocked with diluent containing background reducing components (Dako, Carpenteria, CA) and then probed with either FoxO3a, p-FoxO3a S253, Akt (Cell Signaling), or p-FoxO3a T32 (Abcam) in diluent for 2 hours at room temperature. Negative controls were generated by incubating slides in the absence of primary antibody. Secondary anti-rabbit FITC- or TRITC-conjugated antibodies (Sigma-Aldrich) were applied in diluent for 45 minutes at room temperature. DAPI staining (Fisher Scientific) was added for 3 minutes to stain cell nuclei followed by washing with PBS. ICC images were captured under white light and fluorescence filters using an Olympus microscope (Olympus IX71, C Squared Corporation, Pittsburgh, PA).
RNA isolation and quantitative PCR
Total mRNA was isolated from cells using Purelink RNA isolation kit (Invitrogen) per the manufacturer's protocol. The OD 260/280 ratio of the mRNA was at least 1.8 and the 18 s/28 s bands were verified on a 1% agarose gel. Two-step quantitative reverse transcriptase-mediated real-time PCR (QPCR) was used to measure changes in mRNA in response to changes in FoxO3a expression. The RT step was achieved by synthesizing cDNA from 3 µg RNA using the High Capacity Reverse Transcription kit as per the manufacturer's protocol (Applied Biosystems, Foster City, CA). The PCR step was done using Taqman® Fast Universal PCR Master Mix (Applied Biosystems) and TaqMan FAMTM dye-labeled probes for FoxO3a (Hs00818121_m1), CCNA1 (Hs00171105_m1), CCNA2 (Hs00996788_m1), CCNB1 (Hs01030103_m1), CCNB2 (Hs00270424_m1) and GAPDH (Hs99999905_m1). Data were normalized to GAPDH for each sample. Fold change values between FoxO3a shRNA and wt FoxO3a and control samples were calculated using the ΔΔCt method (Schmittgen and Livak, 2008).
Flow cytometry
Cells were plated in 6-cm plates (Midwest Scientific) and grown to ∼50% confluence prior to overnight serum starvation. Cells were then incubated in regular media for 72 hours followed by collection using 0.05% trypsin (Cellgro). Cells were then fixed and stained with propidium iodide (BD Pharmingen, San Jose, CA) per the manufacturer's protocol. FACS analysis was performed on Accuri C6 flow cytometer (Accuri, Ann Arbor, MI). Unstained cells were used as controls for setting the cell population parameters and overlay of histograms show no deviation or drift of channels.
Luciferase reporter gene analysis
Cells were plated in 12-well culture plates (Midwest Scientific) in triplicate at 1×105 cells/well and incubated overnight. Cells were transfected with 0.4 µg pGL/cyclin A Luc and 25 ng pRL-CMV-renilla in either KTC3 shRNA cells or FRO cells along with 0.4 µg pECE or pECE/wt FoxO3a using Lipofectamine 2000 (Invitrogen). After 24 hours, cells were washed with DPBS (Cellgro), and lysed using Promega's Dual Luciferase assay kit per the manufacturer's protocol. Luciferase activity was measured using a Veritas luminometer (Promega) and the enzyme activity was normalized for efficiency of transfection on the basis of renilla activity levels and reported as relative luminescent units.
Chromatin immunoprecipitation assay
ChIP assays were performed using the EZ-ChIPTM Chromatin Immunoprecipitation (ChIP) KIT from Millipore (Bedford, MA) according to the manufacturer's protocol. 4 µg primary antibody (anti-FLAG, Sigma) or mouse IgG control was used for chromatin immunoprecipitations. Immunoprecipitates were analyzed by PCR using the primer set 5′-TAGAGTCAGCCTTCGGACAG-3′ and 5′-ATCCCGCGACTATTGAAAT-3′ to amplify a 253 bp fragment of the human cyclin A1 promoter corresponding to the FoxO3a binding site.
Statistical analysis
Data are presented as the mean ± s.d. and comparisons were analyzed by two-tailed paired Student's t-tests. Data for comparison of multiple groups are presented as mean ± s.d. and were analyzed by ANOVA. P<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Alex Toker (Harvard Medical School) for helpful comments related to Akt signaling.
Footnotes
Funding
This work was supported by the National Institutes of Health Cancer Center Support Grant [grant number P30CA15083 to R.C.S.]; the Mayo Clinic Research Committee [to R.C.S.], Florida Department of Health Bankhead-Coley Cancer Research Program [grant number FL09BW202 to J.A.C. and R.C.S.; FLA07BN-08 to P.S.]; National Institutes of Health [grant number R01CA136665 to J.A.C. and R.C.S.; R01CA140182 to P.S.]; a generous gift from Alfred D. and Audrey M. Petersen to R.C.S.; a grant for rare cancers from Ellis and Dona Brunton (to J.A.C.); and the Translational Genomics Research Institute institutional research funds [to D.O.A.]. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.097428/-/DC1
References
- Azorsa D. O., Gonzales I. M., Basu G. D., Choudhary A., Arora S., Bisanz K. M., Kiefer J. A., Henderson M. C., Trent J. M., Von Hoff D. D., et al. (2009). Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. J. Transl. Med. 7, 43 10.1186/1479-5876-7-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A., Bonni A., Zigmond M. J., Lin M. Z., Juo P., Hu L. S., Anderson M. J., Arden K. C., Blenis J., Greenberg M. E. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868 10.1016/S0092-8674(00)80595-4 [DOI] [PubMed] [Google Scholar]
- Brunet A., Park J., Tran H., Hu L. S., Hemmings B. A., Greenberg M. E. (2001). Protein kinase SGK mediates survival signals by phosphorylating the forkhead transcription factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 21, 952–965 10.1128/MCB.21.3.952-965.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgering B. M. (2008). A brief introduction to FOXOlogy. Oncogene 27, 2258–2262 10.1038/onc.2008.29 [DOI] [PubMed] [Google Scholar]
- Carter M. E., Brunet A. (2007). FOXO transcription factors. Curr. Biol. 17, R113–R114 10.1016/j.cub.2007.01.008 [DOI] [PubMed] [Google Scholar]
- Chan C. B., Liu X., Jang S. W., Hsu S. I. H., Williams I., Kang S., Chen J., Ye K. (2009). NGF inhibits human leukemia proliferation by downregulating cyclin A1 expression through promoting acinus/CtBP2 association. Oncogene 28, 3825–3836 10.1038/onc.2009.236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J., Gomes A. R., Monteiro L. J., Wong S. Y., Wu L. H., Ng T. T., Karadedou C. T., Millour J., Ip Y. C., Cheung Y. N., et al. (2010). Constitutively nuclear FOXO3a localization predicts poor survival and promotes Akt phosphorylation in breast cancer. PLoS ONE 5, e12293 10.1371/journal.pone.0012293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho N. H., Choi Y. P., Moon D. S., Kim H., Kang S., Ding O., Rha S. Y., Yang Y. J., Cho S. H. (2006). Induction of cell apoptosis in non-small cell lung cancer cells by cyclin A1 small interfering RNA. Cancer Sci. 97, 1082–1092 10.1111/j.1349-7006.2006.00292.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edge S. B., Compton C. C. (2010). The American Joint Committee on Cancer: the 7th edition of the AJCC cancer staging manual and the future of TNM. Ann. Surg. Oncol. 17, 1471–1474 [DOI] [PubMed] [Google Scholar]
- Finnberg N., El–Deiry W. S. (2004). Activating FOXO3a, NF-kappaB and p53 by targeting IKKs: an effective multi-faceted targeting of the tumor-cell phenotype? Cancer Biol. Ther. 3, 614–616 10.4161/cbt.3.7.1057 [DOI] [PubMed] [Google Scholar]
- Fu Z., Tindall D. J. (2008). FOXOs, cancer and regulation of apoptosis. Oncogene 27, 2312–2319 10.1038/onc.2008.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa–Hibi Y., Kobayashi Y., Chen C., Motoyama N. (2005). FOXO transcription factors in cell-cycle regulation and the response to oxidative stress. Antioxid. Redox Signal. 7, 752–760 10.1089/ars.2005.7.752 [DOI] [PubMed] [Google Scholar]
- Furuyama T., Nakazawa T., Nakano I., Mori N. (2000). Identification of the differential distribution patterns of mRNAs and consensus binding sequences for mouse DAF-16 homologues. Biochem. J. 349, 629–634 10.1042/0264-6021:3490629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García–Rostán G., Costa A. M., Pereira–Castro I., Salvatore G., Hernandez R., Hermsem M. J., Herrero A., Fusco A., Cameselle–Teijeiro J., Santoro M. (2005). Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 65, 10199–10207 10.1158/0008-5472.CAN-04-4259 [DOI] [PubMed] [Google Scholar]
- Green L. D., Mack L., Pasieka J. L. (2006). Anaplastic thyroid cancer and primary thyroid lymphoma: a review of these rare thyroid malignancies. J. Surg. Oncol. 94, 725–736 10.1002/jso.20691 [DOI] [PubMed] [Google Scholar]
- Guertin D. A., Stevens D. M., Thoreen C. C., Burds A. A., Kalaany N. Y., Moffat J., Brown M., Fitzgerald K. J., Sabatini D. M. (2006). Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev. Cell 11, 859–871 10.1016/j.devcel.2006.10.007 [DOI] [PubMed] [Google Scholar]
- Habashy H. O., Rakha E. A., Aleskandarany M., Ahmed M. A., Green A. R., Ellis I. O., Powe D. G. (2011). FOXO3a nuclear localisation is associated with good prognosis in luminal-like breast cancer. Breast Cancer Res. Treat. 129, 11–21 10.1007/s10549-010-1161-z [DOI] [PubMed] [Google Scholar]
- Hu M. C., Lee D. F., Xia W., Golfman L. S., Ou–Yang F., Yang J. Y., Zou Y., Bao S., Hanada N., Saso H., et al. (2004). IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell 117, 225–237 10.1016/S0092-8674(04)00302-2 [DOI] [PubMed] [Google Scholar]
- Huang H., Tindall D. J. (2007). Dynamic FoxO transcription factors. J. Cell Sci. 120, 2479–2487 10.1242/jcs.001222 [DOI] [PubMed] [Google Scholar]
- Hundahl S. A., Fleming I. D., Fremgen A. M., Menck H. R. (1998). A National Cancer Data Base report on 53,856 cases of thyroid carcinoma treated in the U.S., 1985-1995. Cancer 83, 2638–2648 10.1016/j.cell.2006.08.033 [DOI] [PubMed] [Google Scholar]
- Jacinto E., Facchinetti V., Liu D., Soto N., Wei S., Jung S. Y., Huang Q., Qin J., Su B. (2006). SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127, 125–137 10.1016/j.cell.2006.08.033 [DOI] [PubMed] [Google Scholar]
- Ji P., Agrawal S., Diederichs S., Bäumer N., Becker A., Cauvet T., Kowski S., Beger C., Welte K., Berdel W E. (2004). Cyclin A1, the alternative A-type cyclin, contributes to G1/S cell cycle progression in somatic cells. Oncogene 24, 2739–2744 [DOI] [PubMed] [Google Scholar]
- Joshi A. R., Jobanputra V., Lele K. M., Wolgemuth D. J. (2009). Distinct properties of cyclin-dependent kinase complexes containing cyclin A1 and cyclin A2. Biochem. Biophys. Res. Commun. 378, 595–599 10.1016/j.bbrc.2008.11.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karger S., Weidinger C., Krause K., Sheu S. Y., Aigner T., Gimm O., Schmid K. W., Dralle H., Fuhrer D. (2009). FOXO3a: a novel player in thyroid carcinogenesis? Endocr. Relat. Cancer 16, 189–199 10.1677/ERC-07-0283 [DOI] [PubMed] [Google Scholar]
- Lin H., Dai T., Xiong H., Zhao X., Chen X., Yu C., Li J., Wang X., Song L. (2010). Unregulated miR-96 induces cell proliferation in human breast cancer by downregulating transcriptional factor FOXO3a. PLoS ONE 5, e15797 10.1371/journal.pone.0015797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow L. A., D'Innocenzi J., Zhang Y., Rohl S. D., Cooper S. J., Sebo T., Grant C., McIver B., Kasperbauer J. L., Wadsworth J. T., et al. (2010). Detailed molecular fingerprinting of four new anaplastic thyroid carcinoma cell lines and their use for verification of RhoB as a molecular therapeutic target. J. Clin. Endocrinol. Metab. 95, 5338–5347 10.1210/jc.2010-1421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neff R. L., Farrar W. B., Kloos R. T., Burman K. D. (2008). Anaplastic thyroid cancer. Endocrinol. Metab. Clin. North Am. 37, 525–538 xi 10.1016/j.ecl.2008.02.003 [DOI] [PubMed] [Google Scholar]
- Obsil T., Obsilova V. (2011). Structural basis for DNA recognition by FOXO proteins. Biochim. Biophys. Acta 18131946–1953 10.1016/j.bbamcr.2010.11.025 [DOI] [PubMed] [Google Scholar]
- Santarpia L., El–Naggar A. K., Cote G. J., Myers J. N., Sherman S. I. (2008). Phosphatidylinositol 3-kinase/akt and ras/raf-mitogen-activated protein kinase pathway mutations in anaplastic thyroid cancer. J. Clin. Endocrinol. Metab. 93, 278–284 10.1210/jc.2007-1076 [DOI] [PubMed] [Google Scholar]
- Schmittgen T. D., Livak K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 10.1038/nprot.2008.73 [DOI] [PubMed] [Google Scholar]
- Schweppe R. E., Klopper J. P., Korch C., Pugazhenthi U., Benezra M., Knauf J. A., Fagin J. A., Marlow L. A., Copland J. A., Smallridge R. C., et al. (2008). Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. J. Clin. Endocrinol. Metab. 93, 4331–4341 10.1210/jc.2008-1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. K., Ramsey T. M., Chen Y-N. P., Chen W., Martin M. S., Clune K., Sabio M., Bair K. W. (2001). Identification of E2F-1/Cyclin A antagonists. Bioorg. Med. Chem. Lett. 11, 2449–2452 10.1016/S0960-894X(01)00486-3 [DOI] [PubMed] [Google Scholar]
- Sherman S. I. (2006). Anaplastic Carcinoma. Humana Press, Totowa, NJ [Google Scholar]
- Shin D., Lee M., Hong Z., Lee E. (2010). The Role of Akt/PKB/FOXO3a Signaling in Growth of Follicular Thyroid Cancer. The 14th International Thyroid Congress. 14, P–0854 [Google Scholar]
- Shukla S., Shukla M., Maclennan G. T., Fu P., Gupta S. (2009). Deregulation of FOXO3A during prostate cancer progression. Int. J. Oncol. 34, 1613–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R., Naishadham D., Jemal A. (2012). Cancer statistics, 2012. CA Cancer J. Clin. 62, 10–29 10.3322/caac.20138 [DOI] [PubMed] [Google Scholar]
- Singh A., Plati J., Khosravi–Far R. (2011). Harnessing the tumor suppressor function of FOXO as an alternative therapeutic approach in cancer. Curr. Drug Targets 12, 1311–1321 10.2174/138945011796150271 [DOI] [PubMed] [Google Scholar]
- Smallridge R. C., Copland J. A. (2010). Anaplastic thyroid carcinoma: pathogenesis and emerging therapies. Clin. Oncol. (R. Coll. Radiol.) 22, 486–497 10.1016/j.clon.2010.03.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallridge R. C., Marlow L. A., Copland J. A. (2009). Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocr. Relat. Cancer 16, 17–44 10.1677/ERC-08-0154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V., Woodgett J. R. (1994). Mitogen inactivation of glycogen synthase kinase-3 beta in intact cells via serine 9 phosphorylation. Biochem. J. 303, 701–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P. (2011). Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid. Redox Signal. 14, 593–605 10.1089/ars.2010.3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storz P., Döppler H., Copland J. A., Simpson K. J., Toker A. (2009). FOXO3a promotes tumor cell invasion through the induction of matrix metalloproteinases. Mol. Cell. Biol. 29, 4906–4917 10.1128/MCB.00077-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzivion G., Dobson M., Ramakrishnan G.2011). FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 1813, 1938–1945 [DOI] [PubMed] [Google Scholar]
- van den Berg M. C. W., Burgering B. M. T. (2011). Integrating opposing signals toward Forkhead box O. Antioxid. Redox Signal. 14, 607–621 10.1089/ars.2010.3415 [DOI] [PubMed] [Google Scholar]
- Vogt P. K., Jiang H., Aoki M. (2005). Triple layer control: phosphorylation, acetylation and ubiquitination of FOXO proteins. Cell Cycle 4, 908–913 10.4161/cc.4.7.1796 [DOI] [PubMed] [Google Scholar]
- Weidinger C., Krause K., Klagge A L., Fuhrer D. (2010). FOXO3 is inhibited by the oncogenic p110a mutant H1074R in thyreocytes, but can be reactivated by NSAIDS. The 14th International Thyroid Congress Abstract Booklet. 14, OC–012 [Google Scholar]
- Woyach J. A., Shah M. H. (2009). New therapeutic advances in the management of progressive thyroid cancer. Endocr. Relat. Cancer 16, 715–731 10.1677/ERC-08-0335 [DOI] [PubMed] [Google Scholar]
- Yang J. Y., Hung M. C. (2009). A new fork for clinical application: targeting forkhead transcription factors in cancer. Clin. Cancer Res. 15, 752–757 10.1158/1078-0432.CCR-08-0124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. Y., Hung M. C. (2011). Deciphering the role of forkhead transcription factors in cancer therapy. Curr. Drug Targets 12, 1284–1290 10.2174/138945011796150299 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.