

Abstract
Background:
Fibrodysplasia ossificans progressiva is a rare autosomal dominant disorder characterized by congenital malformation of the great toes and by progressive heterotopic ossification of skeletal muscle and soft connective tissues. The disorder is caused by a recurrent missense mutation in the glycine-serine activation domain of activin A receptor type I, a bone morphogenetic protein (BMP) type-I receptor, in all classically affected individuals. Osteochondromas of the proximal part of the tibia are benign osteochondral neoplasms or orthotopic lesions of skeletal remodeling associated with dysregulated BMP signaling and have been considered an atypical feature of fibrodysplasia ossificans progressiva, but they may be underdiagnosed because of their often asymptomatic nature. The purpose of the present study was to determine the prevalence and characteristics of proximal tibial osteochondromas in individuals who have fibrodysplasia ossificans progressiva.
Methods:
Over a period of thirty months, we evaluated all patients with new and established fibrodysplasia ossificans progressiva for the presence of proximal tibial osteochondromas on the basis of medical history, physical examination, and radiographic studies. We quantified the prevalence of osteochondromas and characterized the types of osteochondromas to identify relevant trends.
Results:
Ninety-six patients (including fifty-two female patients and forty-four male patients) with fibrodysplasia ossificans progressiva were evaluated on the basis of a history and physical examination. Plain radiographs were available for sixty-seven patients. Ninety percent of all patients had osteochondroma of the proximal part of the tibia. These lesions usually were asymptomatic, most commonly were bilateral, and typically were located at the pes anserinus. Seventy-five percent of the lesions were pedunculated, and 25% were sessile.
Conclusions:
Proximal tibial osteochondromas are a common phenotypic feature of fibrodysplasia ossificans progressiva, a finding that expands the recognized consequences of recurrent activating mutations in activin A receptor type I to include not only congenital skeletal malformations and heterotopic skeletogenesis but also benign osteochondral neoplasms or orthotopic lesions of skeletal modeling. The present study provides insight into the genetic basis of osteochondroma formation in patients with fibrodysplasia ossificans progressiva and possibly into that of more common conditions in which these lesions occur.
Fibrodysplasia ossificans progressiva (Online Mendelian Inheritance in Man #135100) is a rare autosomal dominant disorder characterized by congenital malformation of the great toes and by progressive heterotopic ossification of skeletal muscle and soft connective tissue in specific anatomic patterns1-6. It initially affects patients during the first decade of life with the episodic development of inflammatory fibroproliferative masses that arise in skeletal muscles and aponeuroses, predominantly in the axial region. These masses typically progress to form mature heterotopic bone through an endochondral process2. Common misdiagnoses for these early-stage lesions include aggressive juvenile fibromatosis and soft-tissue sarcoma1,7,8. During childhood and adolescence, heterotopic ossification continues in predictable anatomic patterns, both spontaneously and in response to soft-tissue injury and viral infections4,6,9-11.
As fibrodysplasia ossificans progressiva advances, the inflammatory fibroproliferative masses are replaced by heterotopic bone that forms through an endochondral process. Patients become progressively disabled as a result of ankylosis of the major joints of the axial and appendicular skeleton and, as a result, are often confined to a wheelchair by the age of thirty years4,5,7. Many patients have a shortened lifespan due to thoracic insufficiency syndrome resulting from severe restrictive disease of the chest wall12. Although nonsteroidal anti-inflammatory drugs and glucocorticoids can be used for palliative care and to reduce the early inflammatory response, a definitive medical treatment for the disease does not exist9.
At a molecular level, fibrodysplasia ossificans progressiva involves dysregulation of the bone morphogenetic protein (BMP) signaling pathway13-18. All patients with classic features of the disease possess the same heterozygous missense mutation in the glycine-serine activation domain of activin A receptor type I (ACVR1), a BMP type-I receptor19. Dysregulated BMP signaling is associated with the formation of osteochondromas20, classified by some as benign osteochondral neoplasms and by others as disorders of orthotopic skeletal modeling21. Osteochondromas have been recognized as a feature associated with fibrodysplasia ossificans progressiva2.
The radiographic appearance of an osteochondroma is diagnostic and reflects the pathologic characteristics of the tumor. Osteochondromas are benign orthotopic osteocartilaginous lesions composed of cortical and trabecular bone with an overlying hyaline cartilage cap. The bone marrow cavity of an osteochondroma is continuous with that of the underlying bone. The cartilaginous cap is covered with a thin layer of fibrous tissue that is continuous with the perichondrium22.
Osteochondromas of the proximal part of the tibia, and less commonly of the humerus, femur, and coronoid process of the mandible, have been reported in patients with fibrodysplasia ossificans progressiva; such occurrences have been reported as an unusual and variable feature of the disease2,23,24. However, osteochondromas could be easily overlooked in patients who have fibrodysplasia ossificans progressiva because of their often asymptomatic nature and because of the disabling nature of heterotopic bone formation in these patients. The purpose of the present study was to determine the prevalence and characteristics of proximal tibial osteochondromas in patients who have fibrodysplasia ossificans progressiva.
Materials and Methods
During a thirty-month time-period from January 2001 through July 2003, all patients with new and established fibrodysplasia ossificans progressiva who were seen and examined by the senior author (F.S.K.) were evaluated for the presence of osteochondromas of the proximal part of the tibia as part of their routine physical examination. The proximal part of the tibia was prospectively chosen as the preferred anatomic site for evaluation because of sporadic reports that have cited its association with fibrodysplasia ossificans progressiva, the ease of physical examination of the site, and the anecdotal observations of osteochondromas in skeletal surveys of our patients who have this disease. In all cases, the diagnosis of fibrodysplasia ossificans progressiva was confirmed by the presence of congenital malformation of the great toes and by progressive heterotopic ossification of soft tissues in characteristic anatomic patterns5-7. Investigational studies were approved by the institutional review board, and all patients gave informed consent for participation.
As part of their routine history, patients and/or their parents were questioned about lower limb symptoms, including pain, swelling, irritation, numbness, paresthesias, and any history of antecedent trauma. Additionally, patients were questioned to identify a history of heterotopic bone formation at the proximal part of the tibia. As part of the physical examination, the proximal parts of the tibiae of all patients were carefully examined and palpated for masses.
Although plain radiographs of the proximal part of the tibia were not available for all patients, most patients have skeletal surveys at the time of diagnosis, during a flare-up, or as part of routine monitoring of the disease. We reviewed all available skeletal radiographs for each patient whom we examined. The presence of unilateral or bilateral proximal tibial osteochondromas on plain radiographs was recorded. Osteochondromas at other anatomic sites also were noted when the relevant radiographs were available. For all skeletal radiographs, the dimensions of each osteochondroma were measured and the lesions were classified as sessile (with the dimension of the base of the tumor exceeding that of its length) or pedunculated (with the dimension of the length of the tumor exceeding that of its base)22.
All patients and/or their parents were questioned with regard to previous treatment for osteochondromas and any previous episodes of disease flare-up at the proximal part of the tibia. In cases in which an osteochondroma had been surgically resected prior to the definitive diagnosis of fibrodysplasia ossificans progressiva, the age at the time of surgery was ascertained. Additionally, the surgical outcome, including any recurrence of the mass, residual pain and neurovascular symptoms, or occurrence of subsequent heterotopic ossification at the site of the excised osteochondroma, was noted.
Results
Ninety-six consecutive individuals with classic features of fibrodysplasia ossificans progressiva were seen and examined in a thirty-month time-period. Of the ninety-six patients, fifty-two (54%) were female and forty-four (46%) were male. The median age of the patients at the time of evaluation was ten years (range, one to fifty-one years).
Ninety-three patients (97%) reported no pain, paresthesias, numbness, or other lower extremity neurological symptoms. Three patients reported transient unilateral or bilateral anteromedial proximal tibial pain associated with a mass. None of the patients who were included in the study reported a flare-up of fibrodysplasia ossificans progressiva or a known history of posttraumatic heterotopic bone formation at the proximal part of the tibia.
On physical examination, eighty-six patients had at least one palpable mass at the proximal part of the tibia. All masses were hard, fixed, and nontender. Twenty (23%) of the eighty-six patients were under the age of six years. In eighty-four (98%) of the eighty-six patients the observed mass or masses were located anteromedially at the level of the pes anserinus insertion (Fig. 1), and in two of the eighty-six patients they were located posteromedially, at the level of the insertion of the popliteus tendon. We did not observe an association between the presence of proximal tibial masses and the gender of the patient. In patients with at least one palpable mass of the proximal part of the tibia, seventy-seven (90%) had a bilateral mass and nine had a unilateral mass.
Fig. 1.

Lateral photograph (A) and anteroposterior radiograph (B) showing a typical anteromedial proximal tibial osteochondroma in a sixteen-year-old patient with fibrodysplasia ossificans progressiva. The lesion appears as a hard, fixed, nontender mass.
Sixty-seven patients (70%) had radiographs that had been previously read by a radiologist and that were reviewed by us. All sets of radiographs included a bilateral view of the proximal part of the tibia. Sixty-two (93%) of the proximal tibial radiographs showed osteochondromas unilaterally or bilaterally, whereas five showed no lesion bilaterally. Radiographs for fifty-six (90%) of these sixty-two patients showed osteochondromas bilaterally, and radiographs for six patients (10%) showed osteochondromas unilaterally. All sixty-two patients who had radiographic evidence of the tumor had at least one osteochondroma at the anteromedial aspect of the proximal part of the tibia, consistent with the location of the osteochondroma on physical examination.
Although available radiographs of other anatomic sites varied between patients, seventeen (25%) of the sixty-seven patients with radiographs had at least one osteochondroma at an anatomic location other than the proximal part of the tibia, including the distal part of the femur, the distal part of the tibia, the proximal part of the fibula, or the first metacarpal. Taking these data into account, a total of sixty-four (96%) of the sixty-seven patients with radiographs had at least one osteochondroma.
The dimensions of 178 osteochondromas (including 104 lesions of the proximal part of the tibia, sixty-eight lesions of the distal part of the femur, four lesions of the proximal part of the fibula, one lesion of the distal part of the tibia, and one lesion of the first metacarpal) were measured and classified as sessile or pedunculated. One hundred and thirty-three osteochondromas (75%) were pedunculated (Fig. 2, A), and forty-five (25%) had a sessile morphology (Fig. 2, B).
Fig. 2.
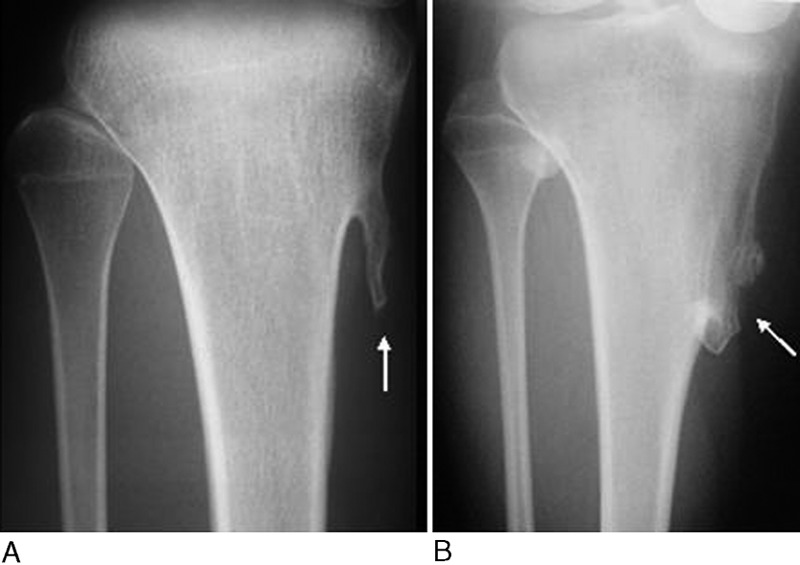
Anteroposterior radiographs of the proximal parts of the right tibiae in two patients with fibrodysplasia ossificans progressiva. In our patients with fibrodysplasia ossificans progressiva, most osteochondromas were pedunculated (A), although some appeared sessile (B). These radiographs show osteochondromas (arrows) composed of cortical and medullary bone that is continuous with the underlying native osseous cortex and medullary canal.
Only one patient had a history of surgical excision of an osteochondroma, and the case of that patient was reported previously2. At the age of twelve years, prior to the diagnosis of fibrodysplasia ossificans progressiva, the patient complained of left anteromedial proximal tibial pain associated with a palpable mass. Plain radiographs revealed an osteochondroma. An excisional biopsy of the lesion was performed, and histopathological evaluation of the mass confirmed the diagnosis. Subsequently, the patient experienced robust heterotopic ossification at the surgical site, consistent with the natural history of fibrodysplasia ossificans progressiva, and was later definitively diagnosed with the condition.
Discussion
The present study demonstrates the previously undocumented common occurrence of proximal tibial osteochondromas in individuals who have fibrodysplasia ossificans progressiva. This study specifically establishes proximal tibial osteochondromas as a common phenotypic feature of fibrodysplasia ossificans progressiva and expands the clinical consequences of endochondral bone formation associated with the recurrent activating mutation in ACVR1 to include not only congenital malformations and heterotopic skeletogenesis but also benign osteochondral neoplasms or orthotopic lesions of skeletal modeling.
Ninety-six percent of the patients with routine radiographs had at least one skeletal osteochondroma. Other studies have demonstrated isolated osteochondromas in patients with fibrodysplasia ossificans progressiva, but not as a generalized feature of the condition. In a case series of thirty-four patients that was performed to define the natural history of the disease, Connor and Evans reported that eleven patients had fibrodysplasia ossificans progressiva with osteochondromas, most commonly of the proximal part of the tibia9. O’Reilly and Renton also reported the case of a patient with fibrodysplasia ossificans progressiva who had a proximal tibial osteochondroma25. Two cases2,24 have been reported in which osteochondromas were excised from the proximal part of the tibia and the coronoid process of the mandible prior to the diagnosis of fibrodysplasia ossificans progressiva.
The present study had limitations. Because of the rarity of fibrodysplasia ossificans progressiva and the wide dispersion of patients throughout the world, it was difficult to evaluate all 500 known patients in a reasonable period of time. We therefore limited our study to an arbitrary thirty-month time-period during which we were able to examine approximately 20% of all known individuals with fibrodysplasia ossificans progressiva worldwide. Our study included patients from five continents: North America, South America, Europe, Australia, and Asia. We have no reason to believe that the temporal limitations of patient accrual would have biased our results.
Another limitation of the present study involved the method of assessment. Because invasive procedures can stimulate heterotopic bone formation in patients with fibrodysplasia ossificans progressiva8,10, pathologic evidence was not available for diagnosis; thus, we used physical examination and radiographic studies to identify the osteochondromas. For the thirty-three patients (34%) for whom radiographs were not available, we used physical examination alone. Physical examination may not be a sufficient means of diagnosing osteochondromas because a proximal tibial mass in a patient with fibrodysplasia ossificans progressiva could also represent heterotopic ossification or other benign or malignant lesions. However, a few factors make these other diagnoses unlikely. The lack of a history of a fibrodysplasia ossificans progressiva flare-up or heterotopic ossification involving the proximal part of the tibia in all patients makes flare-ups an unlikely cause of the lesions. Also, the proximal tibial masses that were examined in the present study had consistent size and physical characteristics and had a predominantly anteromedial location, making other diagnoses less likely. Furthermore, in the group of patients who were evaluated on the basis of both a physical examination and a review of radiographs, there were no cases in which the findings of our examination and the radiographic diagnosis were discordant. Of note, data derived after the exclusion of patients who lacked radiographic evidence did not differ from data derived from all patients. Thus, our results do not rely on the group of patients who were diagnosed on the basis of physical examination alone.
Another limitation of our study was the limited scope of the anatomic sites that were evaluated. Although osteochondromas have been identified at a variety of anatomic sites both in our study and in previous reports, the proximal part of the tibia is the only anatomic site that we systematically evaluated in all patients. The proximal part of the tibia is one of the most common sites of osteochondroma formation in patients with solitary osteochondromas, multiple hereditary exostoses, and fibrodysplasia ossificans progressiva7,26,27. Furthermore, the site is easy to examine because of its superficial nature. Because soft tissues can mask the physical findings at other anatomic sites and skeletal surveys were not available for all patients, we focused our evaluation on proximal tibial osteochondromas. While our results show a strong association, they likely represent an underestimation of the general association of osteochondromas with fibrodysplasia ossificans progressiva, given the prevalence of these lesions that we found radiographically at other locations.
Our results have important clinical implications. Because of the rarity of fibrodysplasia ossificans progressiva, physicians often fail to recognize the disorder when patients present with congenital malformation of the great toes or preosseous soft-tissue masses during childhood. Consequently, surgical correction of the malformed toes and biopsy of soft-tissue lesions both lead to robust heterotopic ossification at the site of surgery, resulting in poor patient outcomes2,8,24,25. The frequent association of proximal tibial osteochondromas with fibrodysplasia ossificans progressiva adds a simple, diagnostically useful dimension to the disease, raising the physician’s level of suspicion and allowing an earlier recognition of the diagnosis that should help to preclude inappropriate and dangerous invasive procedures.
From the perspective of clinical management, one must approach osteochondromas in patients with fibrodysplasia ossificans progressiva differently from those in patients with other hereditary disorders. Because of the consequences of invasive procedures, osteochondromas should not be excised or biopsied in these patients2,24. Our data indicate that these patients rarely have pain, persistent irritation, or neurological compromise secondary to osteochondromas. Furthermore, none of the patients in our study had a known history of malignant degeneration of an osteochondroma, and we are not aware of any reports of such occurrences in the medical literature28,29. Therefore, as for individuals with multiple hereditary exostoses, we do not recommend routine imaging of the osteochondromas for the purpose of monitoring for malignant transformation. Only patients who have an increase in lesion size or a change in symptoms should undergo magnetic resonance imaging to evaluate the thickness of the cartilage cap, a marker of malignancy30.
The present study expands the current understanding of the pathology of fibrodysplasia ossificans progressiva as a disorder of both orthotopic and heterotopic endochondral ossification. Histopathologic analysis of lesional tissue obtained from patients prior to the definitive diagnosis of fibrodysplasia ossificans progressiva uniformly shows normal endochondral osteogenesis at heterotopic sites2. Considering the number of patients under the age of six years who had osteochondromas in our study, it is likely that these benign orthotopic lesions represent developmental malformations that occur as a consequence of the same molecular dysregulation in BMP signaling responsible for other features of the disease3,19.
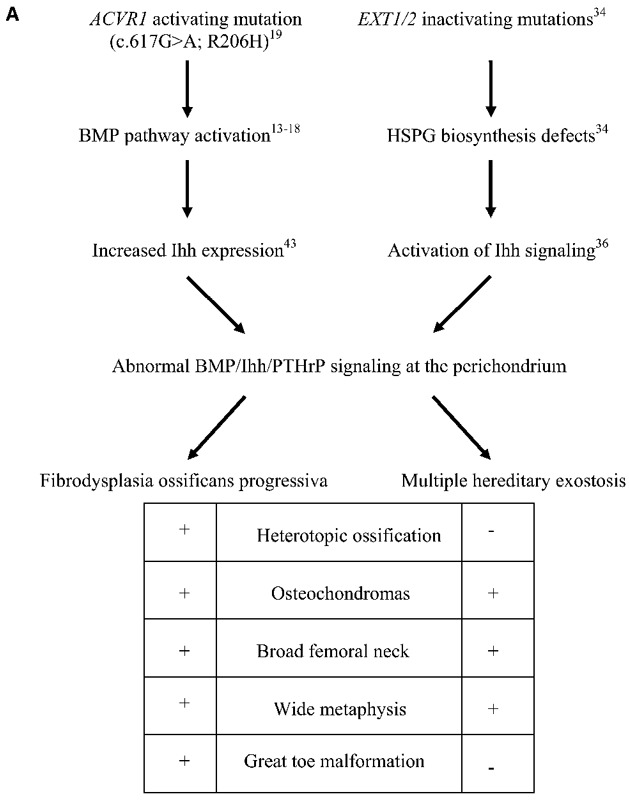
Fibrodysplasia ossificans progressiva is on a short list of hereditary disorders in which osteochondromas occurs. The most common disorder is multiple hereditary exostoses (OMIM #13700), an autosomal dominant condition characterized clinically by numerous osteochondromas occurring predominantly at the metaphyses and less commonly at the diaphyses of long bones. Deformities of the hands, forearms, and legs occur frequently secondary to these lesions. Patients with multiple hereditary exostoses additionally exhibit defective remodeling of the metaphyses of long bones, leading to shortening31,32. It is difficult to assess height and body proportions when there are fixed deformities of the spine and limbs as in patients with fibrodysplasia ossificans progressiva. However, the results of the present study strongly suggest the possibility of a subtle skeletal dysplasia in individuals who have fibrodysplasia ossificans progressiva. A detailed study to address this question is presently underway. In addition to the development of multiple osteochondromas, fibrodysplasia ossificans progressiva shares other skeletal similarities with multiple hereditary exostoses, including short broad femoral necks and metaphyseal widening, suggesting that the two conditions may share overlapping pathophysiologic mechanisms24,25,32,33.
A comparison of the exostoses of fibrodysplasia ossificans progressiva with those of multiple hereditary exostoses reveals several important differences (Table I). In multiple hereditary exostoses, most exostoses are sessile. Those that are pedunculated have a narrow stalk with a bulbous tip, typically pointing away from the joint26,33. In the present study, we found that 75% of the exostoses in patients with fibrodysplasia ossificans progressiva were pedunculated and pointed away from the joint but lacked a bulbous tip. In patients with multiple hereditary exostoses, focal pain, persistent irritation, and neurovascular compromise are common reasons for excision of exostoses31,32. Our findings indicate that such symptoms are uncommon in patients with fibrodysplasia ossificans progressiva, perhaps because of the differences in osteochondroma morphology.
TABLE I.
Comparison of Characteristics of Osteochondromas in Multiple Hereditary Exostoses and Fibrodysplasia Ossificans Progressiva
| Multiple Hereditary Exostoses | Fibrodysplasia Ossificans Progressiva | |
| Tumor morphology | Sessile > pedunculated | Pedunculated > sessile |
| Symmetry | Uncommon | Common |
| Metaphyseal widening | Yes | Yes |
| Associated bursitis | Common | Rare |
| Associated pain | Common | Rare |
| Associated neuropathy | Common | Rare |
| Response to excision | Normal healing | Exuberant heterotopic ossification |
| Malignant degeneration | Relatively common | Not reported |
| Affected gene | EXT1/EXT2 | ACVR1 |
| Type of mutation | Inactivating | Activating |
Transformation of osteochondromas to chondrosarcomas is a relatively common occurrence in patients with multiple hereditary exostoses, especially for lesions in the axial skeleton. This malignant transformation has not been reported to date in patients who have fibrodysplasia ossificans progressiva, nor have we seen it in our experience with >500 patients who were followed for as long as eighteen years28,29. At the present time, we can only speculate why there have been no reports of malignant degeneration of osteochondromas in individuals who have fibrodysplasia ossificans progressiva. Possible reasons include the rarity of this disease and the resultant limited sample pool, failure of diagnosis, decreased lifespan of individuals with reduced time for malignant degeneration to occur, and unique features of the pathophysiology of these lesions that do not predispose them to malignant degeneration.
Multiple hereditary exostoses is a disorder caused by mutations in the EXT1 or EXT2 genes, which encode tumor suppressors and glycosyltransferases involved in the biosynthesis of heparan sulfate, essential components of heparan sulfate proteoglycans34. Cell surface heparan sulfate proteoglycans are abundant molecules that bind to and modulate the activity of a vast array of growth factors and cell surface receptors, which direct cellular processes of proliferation, differentiation, adhesion, and intracellular signaling35.
The canonical molecular model of exostosis formation in multiple hereditary exostoses links the causative mutations in the EXT genes to the Indian hedgehog (Ihh)/parathyroid hormone-related peptide (PTHrP) pathway36. EXT genes are required for the synthesis of cell surface heparan sulfate proteoglycans, which play a seminal role in the local distribution of secreted morphogens, such as Ihh and BMPs, to receptive cells, thus establishing morphogen gradients37. EXT1 deficiency in mice leads to defective heparan sulfate proteoglycan synthesis, defective Ihh transport, and increased levels of intracellular Ihh in perichondrial cells. The abnormal modulation of the tightly regulated Ihh/PTHrP negative feedback loop has been proposed as a molecular model of osteochondroma formation in patients with multiple hereditary exostoses36,38-41. More recently, Stickens et al. reported that mice deficient in EXT2 had defective heparan sulfate proteoglycan synthesis and had development of exostoses in a pattern and distribution identical to that seen in mice deficient in EXT141, an important finding that supports a recent hypothesis on the role of BMP signaling in exostoses20,42.
In fibrodysplasia ossificans progressiva, the molecular mechanism of osteochondroma formation is predicted to be different from that of multiple hereditary exostoses, albeit involving the same feedback loops in the growth plate and perichondrium. Recently, the genetic basis of fibrodysplasia ossificans progressiva was discovered to be a recurrent heterozygous missense mutation in the glycine-serine activation domain of the activin A receptor type-I gene (ACVR1), which encodes a bone morphogenetic protein type-I receptor that is expressed in a variety of tissues, including chondrocytes, skeletal muscle, and perichondrium19,43. Protein modeling predicts that the mutation in fibrodysplasia ossificans progressiva leads to enhanced activation of the receptor, resulting in abnormal intracellular signaling19. Interestingly, this is the second mutation in a bone morphogenetic type-I protein receptor putatively associated with the formation of benign neoplasms. Inactivating mutations of bone morphogenetic protein receptor IA (BMPRIA) are the cause of juvenile polyposis syndrome44.
Recent studies involving a genetically-engineered chicken limb bud model with clinical, genetic, and molecular features remarkably similar to fibrodysplasia ossificans progressiva have shown robust activation of Ihh expression as a result of constitutively active ACVR1. Zhang et al. used this model to show in vivo that ACVR1 is a BMP receptor that is robustly expressed within the perichondrium and periosteum throughout the developing limb43. Furthermore, transfection of constitutively active ACVR1, similar to the fibrodysplasia ossificans progressiva mutation, resulted in dramatic upregulation of Ihh at the perichondrium and a delay in the temporal and anatomic pattern of chondrocyte differentiation. Taken together, these findings allow us to expand our understanding of the Ihh/PTHrP feedback loop to include important regulatory contributions from the BMP signaling pathway. Thus, constitutively active ACVR1 robustly stimulates Ihh expression in perichondrial cells in a chicken model of fibrodysplasia ossificans progressiva and, as a result, distorts the Ihh/PTHrP negative feedback loop, leading to abnormal endochondral ossification at periarticular and perichondrial locations43.
It is thus reasonable to suggest that osteochondroma formation in both fibrodysplasia ossificans progressiva and multiple hereditary exostoses is mediated by disruption of the BMP/Ihh/PTHrP negative feedback loop at the perichondrium (Fig. 3). In fibrodysplasia ossificans progressiva, this feedback loop is likely affected directly through the increased activity of mutated ACVR1, whereas in multiple hereditary exostoses the feedback loop is affected indirectly through reduction in heparan sulfate proteoglycans, leading to a functional increase of BMP and Ihh morphogens that regulate BMP/Ihh/PTHrP signaling. This schema integrates knowledge of the tumor suppressor genes EXT1 and EXT2 with emerging knowledge of mutant ACVR1 to elucidate the pathophysiology of osteochondromas. Additional studies will be necessary to examine this schema in detail in a knock-in model of fibrodysplasia ossificans progressiva.
Fig. 3.

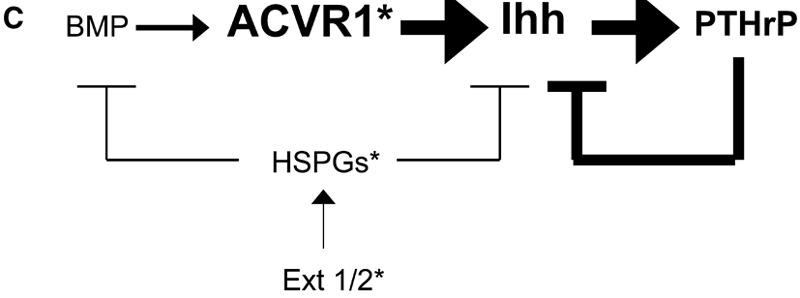
Hypothetical model of the molecular pathogenesis of osteochondromas in multiple hereditary exostoses and fibrodysplasia ossificans progressiva. A, Multiple hereditary exostoses is a condition caused by mutations within the EXT family of genes, leading to deficient biosynthesis of heparan sulfate proteoglycans (HSPGs), hyperactivity of Indian hedgehog (Ihh), and a proposed resulting dysregulation of the BMP/Ihh/PTHrP feedback loop in the growth plate and perichondrium. Fibrodysplasia ossificans progressiva is caused by a mutation in the gene encoding ACVR1, a bone morphogenetic protein type-I receptor, leading to abnormal BMP signaling and a proposed dysregulation of the BMP/Ihh/PTHrP feedback loop in the growth plate and perichondrium. The model proposes that the BMP/Ihh/PTHrP regulatory loop is the common signaling pathway in the pathogenesis of osteochondromas in the two genetic diseases. B, Normal regulation of the BMP/Ihh/PTHrP pathway. Arrows signify stimulation, and lines ending in bars signify inhibition. C, Abnormal signaling pathways in multiple hereditary exostoses and fibrodysplasia ossificans progressiva. For multiple hereditary exostoses, abnormal EXT1/2 (EXT1/2*) leads to deficient synthesis of heparan sulfate proteoglycans (HSPGs*), resulting in increased Ihh and BMP activity via diminished inhibition (unbolded lines and bars). For fibrodysplasia ossificans progressiva, constitutively active ACVR1 (ACVR1*) is postulated to cause hyperstimulation of Ihh (left bolded arrow) and, subsequently, of PTHrP (right bolded arrow). In both multiple hereditary exostoses and chicken models having fibrodysplasia ossificans progressiva-like features, hyperactive Ihh results in abnormalities in the regulation of chondrocyte differentiation and leads to osteochondroma formation.
In summary, we have shown that proximal tibial osteochondromas are a common phenotypic feature of fibrodysplasia ossificans progressiva, a finding that has important clinical, pathological, and basic-science implications. The present study expands the spectrum of endochondral disorders caused by recurrent activating mutations in ACVR1 to include not only congenital skeletal malformations and progressive heterotopic skeletogenesis but also benign osteochondral neoplasms or orthotopic lesions of skeletal remodeling. This finding provides insight into the genetic cause of osteochondromas in fibrodysplasia ossificans progressiva and possibly in more common conditions in which such neoplasms occur. Finally, this work illustrates that the molecular basis of a disease can be revealed not only by discovering the genetic cause of the disease but also by discovering unanticipated clinical features of a condition whose genetic cause is already known.
Footnotes
Disclosure: In support of their research for or preparation of this work, one or more of the authors received, in any one year, outside funding or grants in excess of $10,000 from the International Fibrodysplasia Ossificans Progressiva Association, the Ian Cali Endowment, the Whitney Weldon Endowment, the Isaac & Rose Nassau Professorship of Orthopaedic Molecular Medicine, and the National Institutes of Health (R01-AR41916). Neither they nor a member of their immediate families received payments or other benefits or a commitment or agreement to provide such benefits from a commercial entity. No commercial entity paid or directed, or agreed to pay or direct, any benefits to any research fund, foundation, division, center, clinical practice, or other charitable or nonprofit organization with which the authors, or a member of their immediate families, are affiliated or associated.
References
- 1.Kaplan FS, McCluskey W, Hahn G, Tabas JA, Muenke M, Zasloff MA. Genetic transmission of fibrodysplasia ossificans progressiva. Report of a family. J Bone Joint Surg Am. 1993;75:1214-20 [DOI] [PubMed] [Google Scholar]
- 2.Kaplan FS, Tabas JA, Gannon FH, Finkel G, Hahn GV, Zasloff MA. The histopathology of fibrodysplasia ossificans progressive. An endochondral process. J Bone Joint Surg Am. 1993;75:220-30 [DOI] [PubMed] [Google Scholar]
- 3.Schroeder HW, Jr, Zasloff M. The hand and foot malformations in fibrodysplasia ossificans progressiva. Johns Hopkins Med J. 1980;147:73-8 [PubMed] [Google Scholar]
- 4.Cohen RB, Hahn GV, Tabas JA, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. A study of forty-four patients. J Bone Joint Surg Am. 1993;75:215-9 [DOI] [PubMed] [Google Scholar]
- 5.Rogers JG, Geho WB. Fibrodysplasia ossificans progressiva. A survey of forty-two cases. J Bone Joint Surg Am. 1979;61:909-14 [PubMed] [Google Scholar]
- 6.Kaplan FS, Glaser DL, Shore EM, Deirmengian GK, Gupta R, Delai P, Morhart R, Smith R, Le Merrer M, Rogers JG, Connor M, Kitterman JA. The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:183-8 [Google Scholar]
- 7.Smith R, Athanasou NA, Vipond SE. Fibrodysplasia (myositis) ossificans progressiva: clinicopathological features and natural history. QJM. 1996;89:445-56 [DOI] [PubMed] [Google Scholar]
- 8.Kitterman JA, Kantanie S, Rocke DM, Kaplan FS. Iatrogenic harm caused by diagnostic errors in fibrodysplasia ossificans progressiva. Pediatrics. 2005;116:e654-61 [DOI] [PubMed] [Google Scholar]
- 9.Connor JM, Evans DA. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J Bone Joint Surg Br. 1982;64:76-83 [DOI] [PubMed] [Google Scholar]
- 10.Lanchoney TF, Cohen RB, Rocke DM, Zasloff MA, Kaplan FS. Permanent heterotopic ossification at the injection site after diphtheria-tetanus-pertussis immunizations in children who have fibrodysplasia ossificans progressiva. J Pediatr. 1995;126:762-4 [DOI] [PubMed] [Google Scholar]
- 11.Scarlett RF, Rocke DM, Kantanie S, Patel JB, Shore EM, Kaplan FS. Influenza-like viral illnesses and flare-ups of fibrodysplasia ossificans progressiva. Clin Orthop Relat Res. 2004;423:275-9 [DOI] [PubMed] [Google Scholar]
- 12.Kaplan FS, Glaser DL. Thoracic insufficiency syndrome in patients with fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:213-6 [Google Scholar]
- 13.de la Pena LS, Billings PC, Fiori JL, Ahn J, Kaplan FS, Shore EM. Fibrodysplasia ossificans progressiva (FOP), a disorder of ectopic osteogenesis, misregulates cell surface expression and trafficking of BMPRIA. J Bone Miner Res. 2005;20:1168-76 [DOI] [PubMed] [Google Scholar]
- 14.Fiori JL, Billings PC, de la Pena LS, Kaplan FS, Shore EM. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP). J Bone Miner Res. 2006;21:902-9 [DOI] [PubMed] [Google Scholar]
- 15.Kaplan FS, Fiori JL, de la Pena LS, Ahn J, Billings PC, Shore EM. Dysregulation of the BMP-4 signaling pathway in fibrodysplasia ossificans progressiva. Ann N Y Acad Sci. 2006;1068:54-65 [DOI] [PubMed] [Google Scholar]
- 16.Shafritz AB, Shore EM, Gannon FH, Zasloff MA, Taub R, Muenke M, Kaplan FS. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. N Engl J Med. 1996;335:555-61 [DOI] [PubMed] [Google Scholar]
- 17.Ahn J, Serrano de la Pena L, Shore EM, Kaplan FS. Paresis of a bone morphogenetic protein-antagonist response in a genetic disorder of heterotopic skeletogenesis. J Bone Joint Surg Am. 2003;85:667-74 [DOI] [PubMed] [Google Scholar]
- 18.Glaser DL, Economides AN, Wang L, Liu X, Kimble RD, Fandl JP, Wilson JM, Stahl N, Kaplan FS, Shore EM. In vivo somatic cell gene transfer of an engineered Noggin mutein prevents BMP4-induced heterotopic ossification. J Bone Joint Surg Am. 2003;85:2332-42 [DOI] [PubMed] [Google Scholar]
- 19.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Mohart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525-7 [DOI] [PubMed] [Google Scholar]
- 20.Nakase T, Myoui A, Shimada K, Kuriyama K, Joyama S, Miyaji T, Tomita T, Yoshikawa H. Involvement of BMP-2 signaling in a cartilage cap in osteochondroma. J Orthop Res. 2001;19:1085-8 [DOI] [PubMed] [Google Scholar]
- 21.Porter DE, Simpson AH. The neoplastic pathogenesis of solitary and multiple osteochondromas. J Pathol. 1999;118:119-25 [DOI] [PubMed] [Google Scholar]
- 22.Murphey MD, Choi JJ, Kransdorf MJ, Flemming DJ, Gannon FH. Imaging of osteochondroma: variants and complications with radiologic-pathologic correlation. Radiographics. 2000;20:1407-34 [DOI] [PubMed] [Google Scholar]
- 23.Chichareon V, Arpornmaeklong P, Donsakul N. Fibrodysplasia ossificans progressiva and associated osteochondroma of the coronoid process in a child. Plast Reconstr Surg. 1999;103:1238-43 [DOI] [PubMed] [Google Scholar]
- 24.Cremin B, Connor JM, Beighton P. The radiological spectrum of fibrodysplasia ossificans progressiva. Clin Radiol. 1982;33:499-508 [DOI] [PubMed] [Google Scholar]
- 25.O’Reilly M, Renton P. Metaphyseal abnormalities in fibrodysplasia ossificans progressiva. Br J Radiol. 1993;66:112-6 [DOI] [PubMed] [Google Scholar]
- 26.Shapiro F, Simon S, Glimcher MJ. Hereditary multiple exostoses. Anthropometric, roentgenographic, and clinical aspects. J Bone Joint Surg Am. 1979;61:815-24 [PubMed] [Google Scholar]
- 27.Bottner F, Rodl R, Kordish I, Winklemann W, Gosheger G, Lindner N. Surgical treatment of symptomatic osteochondroma. A three- to eight-year follow-up study. J Bone Joint Surg Br. 2003;85:1161-5 [DOI] [PubMed] [Google Scholar]
- 28.Jaffe HL. Hereditary multiple exostoses. Arch Path. 1943;36:335-57 [Google Scholar]
- 29.Voutsinas S, Wynne-Davies R. The infrequency of malignant disease in diaphyseal aclasis and neurofibromatosis. J Med Genet. 1983;20:345-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hudson TM, Springfield DS, Spanier SS, Enneking WF, Hamlin DJ. Benign exostoses and exostotic chondrosarcomas: evaluation of cartilage thickness by CT. Radiology. 1984;152:595-9 [DOI] [PubMed] [Google Scholar]
- 31.Peterson HA. Multiple hereditary osteochondromata. Clin Orthop Relat Res. 1989;239:222-30 [PubMed] [Google Scholar]
- 32.Greenfield GB. Radiology of bone diseases. Philadelphia: Lippincott; 1990. Hereditary multiple exostoses (diaphyseal aclasis); p 672-8.
- 33.Vanhoenacker FM, Van Hul W, Wuyts W, Willems PJ, De Schepper AM. Hereditary multiple exostoses: from genetics to clinical syndrome and complications. Eur J Radiol. 2001;40:208-17 [DOI] [PubMed] [Google Scholar]
- 34.McCormick C, Leduc Y, Martindale D, Mattison K, Esford LE, Dyer AP, Tufaro F. The putative tumour suppressor EXT1 alters the expression of cell-surface heparan sulfate. Nat Genet. 1998;19:158-61 [DOI] [PubMed] [Google Scholar]
- 35.Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729-77 [DOI] [PubMed] [Google Scholar]
- 36.Koziel L, Kunath M, Kelly OG, Vortkamp A. Ext1-dependent heparan sulfate regulates the range of Ihh signaling during endochondral ossification. Dev Cell. 2004;6:801-13 [DOI] [PubMed] [Google Scholar]
- 37.Jiao X, Billings PC, O’Connell MP, Kaplan FS, Shore EM, Glaser DL. Heparan sulfate proteoglycans (HSPGS) modulate BMP2 osteogenic bioactivity in C2C12 cells. J Biol Chem. 2006;282:1080-6 [DOI] [PubMed] [Google Scholar]
- 38.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332-6 [DOI] [PubMed] [Google Scholar]
- 39.Stickens D, Brown D, Evans GA. EXT genes are differentially expressed in bone and cartilage during mouse embryogenesis. Dev Dyn. 2000;218:452-64 [DOI] [PubMed] [Google Scholar]
- 40.Hilton MJ, Tu X, Cook J, Hu H, Long F. Ihh controls cartilage development by antagonizing Gli3, but requires additional effectors to regulate osteoblast and vascular development. Development. 2005;132:4339-51 [DOI] [PubMed] [Google Scholar]
- 41.Stickens D, Zak BM, Rougier N, Esko JD, Werb Z. Mice deficient in Ext2 lack heparan sulfate and develop exostoses. Development. 2005;132:5055-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bornemann DJ, Duncan JE, Staatz W, Selleck S, Warrior R. Abrogation of heparan sulphate synthesis in Drosophila disrupts the Wingless, Hedgehog and Decapentaplegic signaling pathways. Development. 2004;131:1927-38 [DOI] [PubMed] [Google Scholar]
- 43.Zhang D, Schwarz EM, Rosier RN, Zuscik MJ, Puzas JE, O’Keefe RJ. ALK2 functions as a BMP type I receptor and induces Indian hedgehog in chondrocytes during skeletal development. J Bone Miner Res 2003;18:1593-1604 [DOI] [PubMed] [Google Scholar]
- 44.Howe JR, Blair JL, Sayed MG, Anderson ME, Mitros MA, Peterson GM, Velculescu VE, Traverso G, Vogelstein B. Germline mutations of the gene encoding bone morphogenetic protein receptor IA in juvenile polyposis. Nat Genet. 2001;28:105-7 [DOI] [PubMed] [Google Scholar]