

Abstract
We have assessed the numbers of potentially deleterious variants in the genomes of apparently healthy humans by using (1) low-coverage whole-genome sequence data from 179 individuals in the 1000 Genomes Pilot Project and (2) current predictions and databases of deleterious variants. Each individual carried 281–515 missense substitutions, 40–85 of which were homozygous, predicted to be highly damaging. They also carried 40–110 variants classified by the Human Gene Mutation Database (HGMD) as disease-causing mutations (DMs), 3–24 variants in the homozygous state, and many polymorphisms putatively associated with disease. Whereas many of these DMs are likely to represent disease-allele-annotation errors, between 0 and 8 DMs (0–1 homozygous) per individual are predicted to be highly damaging, and some of them provide information of medical relevance. These analyses emphasize the need for improved annotation of disease alleles both in mutation databases and in the primary literature; some HGMD mutation data have been recategorized on the basis of the present findings, an iterative process that is both necessary and ongoing. Our estimates of deleterious-allele numbers are likely to be subject to both overcounting and undercounting. However, our current best mean estimates of ∼400 damaging variants and ∼2 bona fide disease mutations per individual are likely to increase rather than decrease as sequencing studies ascertain rare variants more effectively and as additional disease alleles are discovered.
Introduction
Genetic variation contributes to human ill health. Hence, identifying the variants that underlie the disease phenotypes (such variants are referred to here as “disease variants” or “disease alleles”) of affected individuals has been an important goal of medical geneticists for decades. The comprehensive catalogs of both high-penetrance variants underlying Mendelian disorders (Online Mendelian Inheritance in Man and Human Gene Mutation database [HGMD])1 and low-penetrance variants contributing to complex disorders (National Human Genome Research Institute [NHGRI] Catalog of Published Genome-wide Association Studies) attest to the progress made to date. In parallel, researchers have made attempts to predict the functional consequences of DNA variants, particularly missense variants (leading to amino acid substitutions) in protein-coding genes,2,3 with the aim of identifying potentially damaging mutations independently of a known disease association. Apparently healthy individuals can, for a number of reasons, carry many disadvantageous variants without showing any obvious ill effects: (1) they might carry a single disease allele for a severe high-penetrance recessive disorder that requires two alleles to manifest the disease phenotype, (2) the disorder might be late in onset or require additional genetic and/or environmental factors for expression (reduced penetrance), (3) or the clinical phenotype might be mild and classified as lying within the range of normal healthy variation. Indeed, many damaged or entirely inactivated proteins can have no perceptible impact on health and hence can be tolerated. In addition, available catalogs of disease alleles are invariably imperfect: not only are they far from complete, because most variants in the human population are very rare and disease-allele discovery has barely begun in many populations, but, more confusingly, they can also contain entries that have been erroneously included as disease variants. Indeed, as many as 27% of database entries were judged to be potentially unreliable in one recent assessment.4 It is clearly important that such uncertain records be identified in order that genomic sequences can be reliably interpreted in a medical context, and this will be increasingly relevant as we enter a new era of personalized genomics.5
Assessing the magnitude of the “genetic burden” imposed by harmful alleles on the general population has been an aim of medical and population geneticists since the first half of the 20th century. In early theoretical treatments, Muller estimated that “the average individual is probably heterozygous for at least [eight] genes, and possibly for scores, each of which produces a significant but usually slight detrimental effect,”6 whereas Morton et al. calculated from a consideration of consanguineous marriages that “the average person carries heterozygously the equivalent of [three to five] recessive lethals.”7 Subsequently, it has been argued that individuals could carry as many as 100 lethal equivalents,8 and a recent consideration of likely numbers of disease alleles per individual in the general population resulted in a “best guess” of 31.9 Large-scale sequencing studies have suggested that the numbers of variants encoding amino acid substitutions predicted to be damaging to the protein in a sample of 10,150 genes were 426 heterozygotes (range = 340–534) and 92 (range = 77–113) homozygotes per individual;10 numbers over the entire genome might be twice these. Other studies also support the view that many missense variants are likely to be damaging.11–13 Furthermore, the proportion of damaging substitutions appeared to be higher in individuals of European ancestry than in those of African ancestry.10 Although these studies focused on coding variation, noncoding positions in the human genome can be equally evolutionarily constrained; variation at such sites might provide the majority of functional variation in each individual9 but is currently not as easily studied.14,15 In an alternative approach, a survey of 437 genes known to underlie recessive Mendelian diseases revealed 2.8 (range = 0–7) severe mutations per individual.4 It is difficult to estimate the numbers per complete genome, but they would clearly be higher. Similarly, another survey of 417 recessive Mendelian variants discovered ∼0.4 recessive lethals per individual and also that ∼0.3% of the study population was homozygous or compound heterozygous for a severe condition,16 and a study of the Hutterite population showed ∼1.1 recessive disease mutations per individual.17 Personal genome sequences have not only provided estimates of the number of disease variants (e.g., 19) carried by each subject18 but have also given us a glimpse of the likely complexity of the functional interpretation of such data.19 Taken together, theoretical and experimental studies suggest that individuals typically carry hundreds of mildly disadvantageous variants and several severe disease alleles. Such findings emphasize the difficulties of interpreting variant function and hence point to the need for additional information.
Advances in sequencing technology now allow genetic variation to be discovered efficiently throughout most of the genome in population samples.20 In a pilot study for the 1000 Genomes Project, we reported the numbers of disease alleles, defined by reference to the disease-causing mutations (DMs) listed in HGMD,1 in population samples of African, European, and East Asian origin. These numbers were surprisingly high, for example, 57–80 disease alleles per individual (interquartile range) in a sample of 179 participants. Moreover, further examination of these numbers showed that 191 disease alleles were present in the homozygous state in at least one individual and hence were not present simply because their effects were masked by a normal allele. Although little phenotypic information other than sex, ethnicity, place of origin, and relationship to other participants is available for the 1000 Genome Project donors, the project’s ethical process requires that sample donors be nonvulnerable adults who are competent to consent to participation in the project. It seems unlikely that they will have suffered from overt genetic disease at the time of sampling. Instead, it seems important to seek some other explanation for the high numbers of DMs. For example, the penetrance of the disease alleles might be lower than previously realized. Most past studies have identified a genotype given a disease phenotype; this is very different from identifying a phenotype given a (disease-associated) genotype. Alternatively, these alleles might not actually be pathogenic at all—they might have been erroneously reported and then inappropriately entered into the mutation database. Finally, they might represent sequencing errors in the 1000 Genomes Project data.21 These considerations prompted us both to re-examine the disease-allele findings of the 1000 Genomes Pilot Project and to extend the analyses to other categories of potentially harmful variants.
For this follow-up study, we adopted two different approaches. We evaluated the predicted properties of missense variants in the data set irrespective of whether they are currently annotated (in the HGMD) as disease alleles. In addition, we expanded the sample of disease alleles (defined as those present in the HGMD) to include other categories. HGMD entries are currently classified in two distinct ways: first by function (DMs, disease-associated polymorphisms [DPs], DPs with additional supporting functional evidence [DFPs], or in vitro or in vivo functional polymorphisms [FPs]) and second by variant type (base substitutions [further subdivided into missense, nonsense, regulatory, and essential splice variants], microindels, microdeletions, microinsertions, and some additional categories not employed here). We therefore identified the overlap of each of these categories (by combining microindels, microdeletions, and microinsertions into one category, henceforward referred to as “indels”) with the 1000 Genomes Pilot data and concentrated on the Low-Coverage Pilot because the Exon Pilot identified fewer overlaps and because these have already been reanalyzed in some detail.22 We then applied a number of different approaches to evaluate the likely functional impact of these overlapping variants and hence the apparent “genetic burden” experienced by the human population.
Material and Methods
Data Sources
1000 Genomes Low-Coverage Pilot data20 were downloaded from the 1000 Genomes Project website. This data set is based on the NCBI human reference genome build 36, which is used here as well. HGMD data were obtained from HGMD Professional release 2009.4. Overlaps were identified as variants sharing the same genomic coordinate and, for base substitutions, the same nucleotide. The number of overlapping DMs was corrected from 57820 to 577 (Table 1). Indel overlaps were those present in both the Low-Coverage Pilot data and the “small insertion,” “small deletion,” and “small indel” classes of HGMD data. We initially allowed ± 10 bp in genomic coordinate and subsequently manually examined the indels to assess whether or not the variant structures were the same. The ancestral states used were those annotated in the 1000 Genomes genotype (.vcf) files; the few sites lacking ancestral annotation were excluded from analyses requiring this information.
Table 1.
Summary of HGMD Data
| Variant Types |
DM |
FP |
DFP |
DP |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HGMD Total | Overlap | Homozygous Overlap | HGMD Total | Overlap | Homozygous Overlap | HGMD Total | Overlap | Homozygous Overlap | HGMD Total | Overlap | Homozygous Overlap | |
| Missense and nonsense SNPs | 50,361 | 577 | 191 | 1,210 | 313 | 157 | 273 | 208 | 168 | 1,068 | 767 | 633 |
| Regulatory SNPs | 626 | 44 | 19 | 341 | 245 | 208 | 378 | 340 | 317 | 338 | 294 | 271 |
| Essential splice sites | 5,830 | 7 | 0 | 40 | 3 | 1 | 2 | 2 | 2 | 7 | 3 | 1 |
| Other splice sites | 2,229 | 95 | 36 | 62 | 37 | 31 | 39 | 39 | 36 | 157 | 141 | 124 |
| Small indels | 1,319 | 0 | 0 | 14 | 0 | 0 | 4 | 0 | 0 | 4 | 0 | 0 |
| Small deletions | 14,411 | 5 | 3 | 70 | 7 | 6 | 4 | 1 | 1 | 21 | 4 | 3 |
| Small insertions | 5,934 | 1 | 0 | 29 | 0 | 0 | 5 | 0 | 0 | 7 | 1 | 1 |
“HGMD Total” represents variants found in the HGMD data set. “Overlap” represents variants found in both the HGMD and the 1000 Genomes Low-Coverage Pilot data sets. “Homozygous overlap” represents those variants found in both data sets and observed in the homozygous state in one or more individuals. The following abbreviations are used: DM, disease-causing mutation (pathological mutation reported to be disease causing in the report entered into HGMD); FP, in vitro or in vivo functional polymorphism (polymorphism reported to affect the structure, function, or expression of the gene [or gene product] but with no disease association reported as yet); DFP, disease-associated polymorphism with additional supporting functional evidence (polymorphism reported to be in significant association [p < 0.05] with disease and that has evidence of being of direct functional importance [e.g., as a consequence of altered expression, mRNA studies, etc.]); and DP, disease-associated polymorphism (polymorphism reported to be in significant association [p < 0.05] with a disease or phenotype and that is assumed to be functional [e.g., as a consequence of location, evolutionary conservation, replication studies, etc], although there might not yet be any direct evidence [e.g., from an expression study] of a functional effect).
HGMD DM Genotype Validation
Two approaches were used for validation of the HGMD DM genotype calls. Twenty of the pilot samples were sequenced to high coverage by Complete Genomics with independent technology.23 In these overlapping samples, 318 of the 577 DM disease alleles were called by the 1000 Genomes Project. Among these, the disease allele was also called by Complete Genomics at 316 sites, although in a different individual in one case. These 316 DM site calls were regarded as validated, and two were not validated, giving a site validation rate of 99.4%. Genotype concordance was 97.9% (6,224/6,360, Table S1, available online) and 100% among the DM sites that were variable and invariable, respectively, in these 20 individuals. In addition, five sites, including all HGMD DMs in the filtered list found as homozygotes and not already validated, were tested by capillary sequencing. All were confirmed as being homozygous for the disease allele (Figure S1).
Statistical Analyses
Derived allele-frequency (DAF) spectra were compared with a Mann-Whitney U test, and the overrepresentation or underrepresentation of the lowest frequency bin (0%–10%) was assessed with a Chi-square test. The dependence of DAF on consensus deleteriousness (Condel) scores24 was analyzed with Spearman’s and Kendall’s rank correlations.
Manual Curation of Variants
We sought further data on variants of interest in PubMed and Google by using the dbSNP reference SNP ID number, gene name, or disorder name as a search term and by identifying publications that cited the reference listed in the HGMD PubMed Unique Identifier field. Disease association was accepted when association with disease in the original reference or an additional source was statistically significant or when functional data, such as cell line or model-organism studies, identified a relevant biological effect. All of the overlapping indels and essential splice sites, as well as an initial 20 missense DMs chosen randomly from the list of 577 and all 71 filtered DMs listed in Table 2 and Table S2, were assessed in this way.
Table 2.
Filtered Disease Variants either Causing Dominant Disease or Causing Recessive Disease and Observed in the Homozygous State
| RefSeq Accession Number | Disease (MIM Number) | Inheritance | Gene | Chr | Position | HGVS cDNA Mutation | Protein Alteration | Total Homozygotes | Total Heterozygotes | Comments |
|---|---|---|---|---|---|---|---|---|---|---|
| NM_000051.3 | ataxia telangiectasia (MIM 607585) | AR | ATM | 11 | 107,665,560 | c.4258C>T | p.Leu1420Phe | 1 | 3 | low-penetrance breast cancer susceptibility allele |
| NM_206933.2 | Usher syndrome type IIA (MIM 276901) | AR | USH2A | 1 | 214,490,898 | c.2137G>C | p.Gly713Arg | 3 | 22 | literature is ambiguous; probable complex pathogenicity; neutral in YRI? |
| NM_015102.3 | nephronophthisis 4 (MIM 606966) | AR | NPHP4 | 1 | 5,862,830 | c.2542C>T | p.Arg848Trp | 1 | 2 | growth retardation; adult-onset renal disease |
| NM_000529.2 | Cushing syndrome (MIM 607397) | AR | MC2R | 18 | 13,874,685 | c.833T>G | p.Phe278Cys | 1 | 10 | hormonal disorder; variable sex-specific symptoms; variant is functionally defective in vitro |
| NM_000443.3 | low-phospholipid-associated cholelithiasis (MIM 171060) | AR | ABCB4 | 7 | 86,887,281 | c.2363G>A | p.Arg788Gln | 2 | 9 | adult onset |
| NM_000256.3 | cardiomyopathy, hypertrophic (MIM 115197) | AD | MYBPC3 | 11 | 47,320,810 | c.1519G>A | p.Gly507Arg | 0 | 2 | late onset; incomplete penetrance |
| NM_139281.2 | glaucoma, primary open angle (MIM 609887) | AD | WDR36 | 5 | 110,473,878 | c.1586G>A | p.Arg529Gln | 0 | 1 | adult onset; variant is functionally defective in vitro |
| NM_000249.3 | colorectal cancer, nonpolyposis (MIM 609310) | AD | MLH1 | 3 | 37,064,024 | c.1742C>T | p.Pro581Leu | 0 | 1 | adult onset; variant is functionally defective in vitro |
| NM_144997.5 | renal cell carcinoma (MIM 144700) | AD | FLCN | 17 | 17,066,604 | c.715C>T | p.Arg239Cys | 0 | 1 | late onset |
| NM_000185.3 | heparin cofactor 2 deficiency (MIM 612356) | AD | SERPIND1 | 22 | 19,464,223 | c.623G>A | p.Arg208His | 0 | 2 | deficiency state; no overt disease; risk factor for thrombophilia |
| NM_005577.2 | Lp(a) deficiency (MIM 152200) | AD | LPA | 6 | 160,926,067 | c.4289+1G>A | essential splice site | 0 | 5 | risk factor in heart disease; late onset |
See Table S2 for full details of these variants. The following abbreviations are used: chr, chromosome; AR, autosomal recessive; AD, autosomal dominant; and YRI, Yoruba in Ibadan, Nigeria.
Functional Prediction of Missense-Variant Deleterious Properties
We used Ensembl (release 65)-computed modified Condel scores,24 which combine PolyPhen-22 and SIFT3 scores. We established Condel score ≥ 0.99 as a cutoff for disease variants on the basis of the discrimination between Condel scores for HGMD DMs that do not overlap with the 1000 Genomes Project Pilot data set on the one hand and 1000 Genomes Project variants that have a frequency > 10% and at least one homozygote and that do not overlap with HGMD DMs on the other (Figure 1). The accuracy of the Condel predictions has been estimated to be about 88%–90%.24
Figure 1.
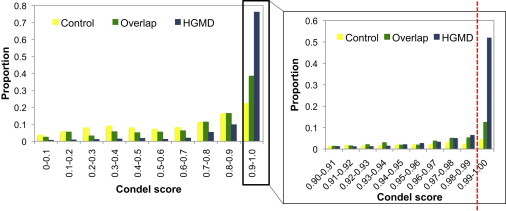
Condel-Score Distribution for HGMD-Only DMs, Variants Only in the 1000 Genomes Low-Coverage Pilot, and Overlap Variants
Shown are Condel scores for HGMD-only DMs (blue), variants only in the 1000 Genomes Low-Coverage Pilot data (yellow), and overlap variants (green). Condel scores range from 0 (the amino acid change is predicted to not damage the protein) to 1 (the amino acid change is predicted to damage the protein). The top decile of Condel scores is further subdivided in the panel on the right-hand side.
Results
Characterization of Missense Variants in Low-Coverage Pilot Sequences
The most readily recognized deleterious variants are those that disrupt a protein-coding gene either by leading to loss of function (e.g., nonsense or frameshift variants) or by altering an amino acid. Because the former category has been the subject of an independent follow-up study,25 we have focused here mainly on missense variants, considering the eight validated loss-of-function variants overlapping with HGMD only in the final filtered data set. We counted the number of derived heterozygous and homozygous missense variants carried by each individual in the 1000 Genomes Low-Coverage Pilot data (Table 1 and Table S3). This analysis differs slightly from the earlier study,20 where nonreference allele numbers were reported. Derived missense-allele numbers per individual ranged from ∼13,000 (range = 12,253–13,868) for the YRI (Yoruba in Ibadan, Nigeria) samples to ∼12,000 (range = 11,715–12,439 for CEU [Utah residents with ancestry from northern and western Europe from the CEPH collection] and 11,197–12,352 for CHB [Han Chinese in Beijing, China] + JPT [Japanese in Tokyo, Japan]) for the European and Asian samples, as expected from the known higher level of genetic variation in African populations.26
We next classified these variants according to the extent of their predicted damaging effect on the protein.24 Around 3% fell into the most damaging class with a Condel score ≥ 0.99, and some differences were evident between populations: there were, per individual, 377 [324–468] for YRI, 321 [281–375] for CEU, and 435 [342–515] for CHB + JPT). Homozygous variant numbers in this most damaging class were considerably lower: 53 (40–70), 62 (53–75), and 70 (51–85), respectively, for the same three populations. These numbers, corresponding to a few hundred damaging missense variants per individual, are in line with previous estimates,10 although they are somewhat lower here, probably because a more stringent prediction of damaging effect was employed. We could not confirm the previously reported higher proportion of damaging variants in individuals of European ancestry (2.7% in CEU vs. 2.9% in YRI), although we did find a greater proportion in East Asians (3.7%). However, the biological interpretation of this discrepancy is not straightforward because the differences between populations in our data are confounded by differences in both read depth and error rate between the populations.20
Characterization of HGMD Variants in Low-Coverage Pilot Sequences
We identified a total of 2,630 HGMD entries that were present in the 1000 Genomes Low-Coverage Pilot data (Table 1 and Table S4). These were very unevenly distributed between the different categories. Forty-two percent of the combined polymorphism classes (DP + DFP + FP) were represented, whereas only 0.9% of the DMs were present (Figure 2A). A large difference might be expected because those entries classified as polymorphisms have small (perhaps negligible) effects on disease risk and received the designation “polymorphism” largely because of their estimated ≥1% frequency, whereas DMs are detrimental and generally rare. Similarly, there were also large differences between the different variant types; for example, 55% of the regulatory SNPs were present, whereas only 0.08% of the combined “indel” classes were (Figure 2B). These differences partly reflect the incomplete discovery of indels in the 1000 Genomes data, but there are also differences within the SNP class. SNP types other than regulatory SNPs are less frequently represented: 2.6% of missense and nonsense SNPs, 0.2% of essential splice SNPs, and 10% of nonessential splice SNPs. Frequency differences between the SNP types are likely to reflect their differential functional impacts.
Figure 2.
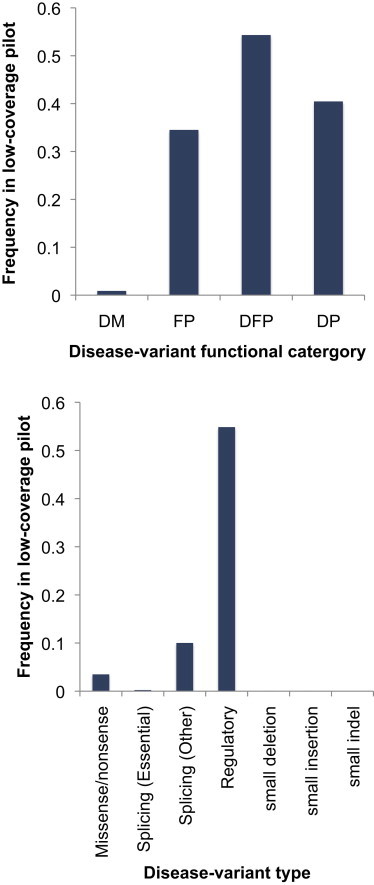
Representation of HGMD Variant Classes in the 1000 Genomes Low-Coverage Pilot
(A) HGMD variants subdivided by functional category. The following abbreviations are used: DM, disease-causing mutation; FPs, in vitro or in vivo functional polymorphism; DFP, disease-associated polymorphism with additional supporting functional evidence; and DP, disease-associated polymorphism.
(B) HGMD variants subdivided by variant type. The first four types are all SNPs.
In addition to the simple presence or absence of HGMD variants in the Low-Coverage Pilot data, their frequency in the population is likely to be informative. This can usefully be summarized as a derived allele-frequency spectrum and assessed by comparison with a variant class assumed to be approximately neutral, such as synonymous SNPs, which avoids interpretation biases as a result of the frequency-dependent ascertainment bias inherent in low-coverage sequencing. As reported by the Pilot Project,20 the spectrum for nonsynonymous SNPs is skewed more toward lower frequencies than is that for synonymous SNPs in all populations, reflecting their tendency to be slightly deleterious (Figure 3). Of the HGMD overlap SNPs, all of the polymorphism classes were skewed toward higher frequencies, probably as a consequence of their ascertainment as “polymorphisms;” however, the DMs were skewed toward lower frequencies, consistent with their presumed deleterious nature. Most of these differences were highly significant (Figure 3; Table S3). This skew was most extreme in the CEU samples, perhaps as a result of the better ascertainment of low-frequency variants in this population and/or a European publication bias influencing entry into HGMD.
Figure 3.
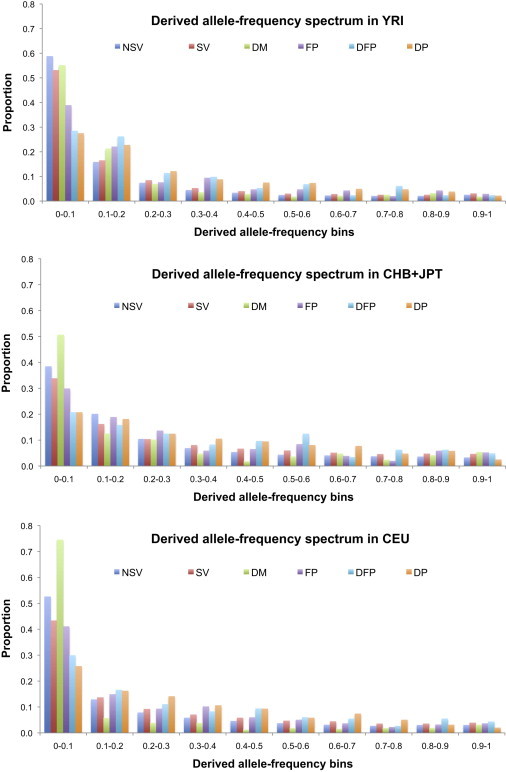
Derived Allele-Frequency Spectra of Nonsynonymous and Synonymous Variants and HGMD SNPs in Three 1000 Genomes Low-Coverage Pilot Samples
The following abbreviations are used: NSV, nonsynonymous variant; SV, synonymous variant; DM, disease-causing mutation; FP, in vitro or in vivo functional polymorphism; DFP, disease-associated polymorphism with additional supporting functional evidence; and DP, disease-associated polymorphism.
As a consequence of these differences in frequency, the numbers of homozygous variants per individual differ greatly between classes. They are generally high for the polymorphism classes (Table 1), but the DM missense variants still show an average of 15 (9–24), 10 (5–17), and 11 (3–22) for the YRI, CEU, and CHB + JPT samples, respectively. In addition, small numbers of disease-causing essential splice-site SNPs and indels were noted (Table 1).
Overall, the proportion of DP, DFP, and FP entries found in the Low-Coverage Pilot data, as well as their allele-frequency spectra, can be understood in terms of the consequences of their relatively mild phenotypic effects and ascertainment. However, although the low proportion and skewed allele-frequency spectra of DMs are consistent with, and indicative of, their expected deleterious properties, these variants are still surprisingly numerous and frequent, both as heterozygotes and homozygotes. Validation rates of these specific variants were high, both for sites (>99%) and for individual genotypes (∼98%), so these numbers are unlikely to reflect genotyping errors (Table S1). We therefore investigated their disease implications in more detail.
Refinement of HGMD DMs Present in Low-Coverage Pilot Sequences
We next adopted two approaches to further assess the overlapping DMs. First, we undertook manual recuration of a sample to determine whether there was any additional published evidence for or against their pathogenicity. Manual recuration was applied to all the disease-causing “indels” and essential splice-site SNPs, as well as to a proportion of the missense variants. Among the five indels in this class, one (and 0/14 in the polymorphism classes), a single-base deletion (c.50delT in PRF1 [MIM 170280; RefSeq accession number NM_001083116.1], associated with the severe disease familial haemophagocytic lymphohistiocytosis) found in the heterozygous state in a single YRI sample, was judged likely to be causative (entry CD993068 in the supplemental data sheet). Of the seven essential splice-site SNPs, all were considered likely to be causative, although they were associated with mild phenotypes in four cases and with a moderate phenotype, deafness, in a fifth case. In all seven, the disease allele was observed only in the heterozygous state in one to ten individuals. Among the missense DMs, we found known pathological variants, such as HBB (MIM 603903; RefSeq NM_000518.4) c.20A>T (p.Glu7Val), which leads to increased resistance to malaria in heterozygotes but to sickle cell disease in homozygotes27 (there were 12 heterozygotes and 1 homozygote in YRI, but 0 in CEU or CHB + JPT). In addition, we found variants exemplified by USH2A (MIM 276901; RefSeq NM_206933.2) c.2138G>C (p.Gly713Arg), reported as being causal for Usher syndrome type 2,28 a recessive disorder characterized by combined deafness and blindness and found in the homozygous state in three YRI samples, despite the fact that it represents a phenotype unlikely to be found among the sample donors. In the case of this mutation, a subsequent report concluded that this variant was unlikely to be causal for the disease given that it was found in 2/200 controls;29 this complex variant is considered further in the Discussion. Thus, manual curation revealed the presence of three variant classes among the DMs: (1) plausible severe disease-causing variants, (2) variants convincingly causative for pathological conditions yet compatible with adult life, and (3) probably incorrect disease-status assignments. However, it also became apparent that little or no relevant additional information was available for the majority of variants examined, and therefore, alternative approaches to reassessment are required.
In a second approach, we used the damage prediction, as measured by the Condel score implemented in Ensembl, to stratify all 577 missense variants. Using a score of ≥ 0.99 as a filter, we found that 55 DMs were predicted to be damaging. Compared with the initial 577 variants, these 55 were present at a significantly lower frequency in the population (p = 0.007, Mann-Whitney U test). A smaller proportion was seen as homozygotes, and the number per individual was also much smaller and ranged from 0 to 7, of which 0 or 1 were homozygotes (Table 1). Thus, this filter served to greatly reduce the number of probable disease alleles per individual.
The 55 damaging missense DMs, the eight validated nonsense DMs, and the splice-site and single-base-deletion DMs discussed above (the filtered shortlist of disease alleles), along with the outcome of their recuration, are summarized in Table 2, Table S2, and the supplemental data sheet. Compared with the HGMD as a whole, this filtered set did not include any significantly overrepresented disease categories, perhaps because the small numbers of alleles provided low power for identifying such effects (Figure S2). Of the 71 entries, recuration caused three to be reclassified as FPs, 22 to be considered probably not pathogenic, and two to be considered truncating variants, leaving 45 judged to be pathogenic. Most of these cause recessive disorders and were found only in heterozygotes; this class of variant is expected to be present in the general population. Two groups, however, were unexpected. Six DMs cause dominant disorders, and five DMs cause recessive disorders but were found in the homozygous state in one or more individuals. These 11 variants are discussed further below.
Discussion
The 1000 Genomes Project Pilot data, as the first available population-scale whole-genome sequence data set, has provided an opportunity for researchers to assess the issues accompanying the interpretation of apparently harmful genetic variants in the human population. These data must, however, be interpreted in the light of some significant limitations. False disease-allele genotype calls, however, are very low. False-discovery rates in the project as a whole were controlled to be <5%, so the variants reported are mostly reliable. Further, the erroneous variant calls are unlikely to correspond to real variants logged in mutation databases. In support of this, validation experiments confirmed that genotyping error contributes very little (<2%) to the results presented in this study; the particularly low error rate most likely reflects the above-average quality of the read mapping to gene regions. Similarly, somatic mutations in the cell lines sequenced by the project are also unlikely to correspond to HGMD entries. False-negative rates in the Low-Coverage Pilot data were, however, substantial for rare variants and represent a limitation of this data set. Discovery in the Low-Coverage Pilot was near complete for variants of frequency ≥ 5% but was only about 30% for variants of 1% frequency. Thus, disease variants, which are generally rare in the population,22 tend to be underrepresented. Consequently, the numbers in the current study should be regarded as conservative lower bounds to the actual numbers in the general population.
One clear conclusion from this and other studies4 is that there is an urgent need to improve disease-allele annotation. Of the 577 HGMD DMs present in the Low-Coverage Pilot data set, >90% were not predicted to be severely damaging to the protein. Such predictions have error rates > 10%,24 but the fact that higher scores are strongly associated with lower allele frequencies (Table S5 and Figure S3) indicates that they are, on average, subject to stronger negative selection and are thus enriched with deleterious alleles. Protein damage does not, of course, invariably give rise to disease; this is most strikingly illustrated by the finding that each individual carries ∼100 severe loss-of-function variants, ∼20 of which are in the homozygous state, without manifesting any evidence of overt genetic disease.25 Conversely, the well-known HBB sickle cell allele is clearly disease causing but has a Condel score of 0.956 and hence lies outside our severely damaging category. Like many loss-of-function variants, some of the homozygous DMs are deleterious on a purely biochemical level and hence manifest as a presumed deficiency state (e.g., c.280C>T [p.Arg94∗] in MOK [formerly RAGE1 (MIM 605762; RefSeq NM_014226.1)]; Table S2) without causing any overt disease in the individual concerned. Some homozygotes are population specific, leading to the possibility that these alleles might well be deleterious in one population, but not in another (e.g., c.598A>G [p.Met200Val] in DMC1 [MIM 602721; RefSeq NM_007068.2]).30,31 In addition, several of the homozygotes occur in association with phenotypes that require a specific environmental trigger (e.g., STOX1 variants and preeclampsia predisposition).32,33 Such considerations point to the complexities of the annotation process required. Nevertheless, it seems probable that many of the 191 DMs identified here in the homozygous state are likely to represent disease-allele-annotation errors. Because HGMD inclusion is largely made on the basis of publication in the peer-reviewed literature, we can infer that the editorial criteria for accepting a given variant as disease causing in the original literature need to be made more stringent. We suggest, for example, that missense variants found in one or a few disease cases and in zero out of a few hundred controls should no longer automatically be accepted as disease causing in the absence of additional functional evidence, particularly because the availability of data from the 1000 Genomes Project and other studies now makes determining allele frequencies in many populations relatively easy. This should serve to reduce the proportion of putatively pathological missense variants that turn out to be spurious, albeit at the price of falsely excluding some truly pathogenic variants.
Nevertheless, simply tightening the criteria for inclusion in mutation databases would not address the relevant biological issue of reduced penetrance. One of the best characterized examples of incomplete penetrance is provided by the c.845G>A (p.Cys282Tyr) mutation in the hemochromatosis-associated gene HFE (MIM 613609; RefSeq NM_000410.3). p.Cys282Tyr homozygotes are found at a frequency of approximately 1 in 200 people of northern European origin.34,35 Available data suggest that 38%–50% of p.Cys282Tyr homozygotes develop iron overload and that 10%–25% develop some form of hemochromatosis-associated morbidity. However, there appears to be a sex-dependent effect: large studies that have specifically assessed liver disease in newly diagnosed p.Cys282Tyr homozygotes have shown that disease manifests in 24%–43% of males and 1%–14% of females.36 The extent of clinically relevant reduced penetrance in mutation databases remains poorly understood but is an issue that urgently needs further consideration.
Our conclusions about the high frequency of spurious reported disease alleles do not, of course, apply to the vast majority of HGMD DM entries, which were not found in the 1000 Genomes samples. A simple best estimate of the number of questionable entries in HGMD would lie between 191 (overlapping homozygous DMs) and 577 (total overlapping DMs), i.e., 0.4%–0.9% of the total number of entries, and >99% of these would be bona fide by this criterion. Nevertheless, investigators sequencing individuals (in a clinical context) or personal-genomics participants in order to understand their genetic disease status are likely to encounter many erroneous disease alleles. What steps can be taken for facilitating interpretation in such a situation? One approach that might be considered would be to regard all variants found in the 1000 Genomes Project samples as being noncausative for disease. This, however, would be overly conservative because disease variants are to be expected in such a population, and indeed, a number of compelling dysfunctional variants are observed. An alternative approach would be to flag variants according to their frequency in the 1000 Genomes Project samples, their presence in the homozygous state, and/or their predicted damage as part of the HGMD entry so that investigators can make an informed judgment about their medical relevance. In the longer term, more comprehensively annotated databases of pathological or disease-associated variants will be required; in the meantime, however, we believe that the type of iterative approach to variant validation advocated here is the most efficient and hence most appropriate way to go about constructing a comprehensive lexicon of DMs, loss-of-function alleles, and damaging but nonpathogenic amino acid substitutions; this will be essential if personalized genomics is to enter the realm of routine clinical practice.
After applying all the criteria for identifying true disease alleles available to us, we were left with a list of 45 filtered candidates (Table 2 and Table S2). Of these, 34 were present only in carriers and do not require further discussion here. Among the remaining 11, the 6 linked to dominant disorders can be understood because they either have late onset (e.g., c.1519G>A [p. Gly507Arg] in MYBPC3 [MIM 115197; RefSeq NM_000256.3]) or no overt disease phenotype (e.g., c.623G>A [p.Arg208His] in SERPIND1 [MIM 612356; RefSeq NM_000185.3]). Similarly, the presence of homozygotes for four of the five recessive disorders can be explained by late onset and/or reduced penetrance. Accounting for c.2138G>C (p.Gly713Arg) in USH2A (MIM 276901; RefSeq NM_206933.2) is the most difficult; this variant is strongly predicted to be damaging to the protein by multiple approaches, including in silico modeling and in vitro laboratory analysis.37 Despite this, the variant is evidently nonpathogenic in some populations, such as the YRI, yet is regarded as pathogenic in other populations.4 One explanation for this apparent contradiction could be that in the YRI population, the USH2A locus is subject to copy-number variation38 that could either reflect the nonessential nature of the locus in YRI or provide functional complementation of the mutant gene. Usher syndrome type IIA is a rare disease found primarily in populations of European descent39,40 and might not be found in the YRI population at all, lending support to this postulate. However, this possibility needs to be investigated in a future study. Such ambiguities could perhaps be clarified by genotype-based recall for further phenotypic analyses or further CNV evaluation of study participants; these approaches are not applicable to the fully anonymized 1000 Genomes Project participants but could be informative in other studies.
Our findings are also relevant to discussions about “incidental findings,” which are, in this context, the unintentional discovery of information about the future disease prospects of the participants.41 Particularly relevant are the dominant diseases, as well as the recessive diseases for which homozygotes were found. In all, 19/179 (11%) individuals were affected (Table 2 and Table S2). In most of these, the primary explanation for the absence of disease at the time of recruitment is likely to be the age of onset, although penetrance is often variable as well, and some phenotypes, such as loose anagen hair syndrome (caused by c.1009G>A [p.Glu337Lys] in KRT75 [MIM 600628; RefSeq NM_004693.2]; Table S2), might not even be regarded as “diseases.” Nevertheless, several participants might later develop a genetic disease. In this situation, the participants might benefit from advice about risks to avoid or monitoring as they age. 1000 Genomes Project participants cannot be recontacted, but our findings suggest that incidental findings relevant to health and well-being might be detected in ∼11% of subjects sequenced; this number is likely to be an underestimate because of our incomplete discovery of rare variants.
It is now abundantly clear that the number of functional genes in the human genome varies between individuals, perhaps by up to 10%, because of large-scale duplications and deletions,42,43 small loss-of-function mutations,25,44 damaging missense substitutions,11,12and other forms of genetic variation. It is also clear that much of this variation has, at most, minor consequences for health. Nevertheless, it is noteworthy that DMs, loss-of-function alleles, and damaging amino acid substitutions (Table S5) as classes are rarer in the population than variants that are approximately neutral, indicating that they are disadvantageous and subject to purifying selection on an evolutionary timescale. They thus contribute to the “genetic burden” in the general population. From the 1000 Genomes Pilot data, we now see that an average individual typically carries ∼60 missense variants that severely damage protein structure and ∼100 loss-of-function variants.25 These numbers are probably biased upward in the sense that not all damaged or inactivated protein-coding genes are harmful to health. However, they are also certainly biased downward not only because the use of a Condel-score cutoff of 0.99 is arbitrary, and many variants with lower scores are likely to be deleterious to health, but also because the contribution of indels and regulatory variants to these categories is likely to have been seriously underestimated. Similarly, the observation of 2.0 (range = 0–7) filtered DMs per individual is affected by a likely residue of noncausative variants that are not removed by our filtering scheme but is also far from complete because many disease-causing mutations still remain to be discovered and entered into HGMD.9 Therefore, giving a definitive estimate of the number of either deleterious or disease-causing variants per individual is impossible at this stage. We nevertheless speculate that as our understanding improves, the effect of removing spurious variants in combination with discovering many more truly harmful ones will lead to a net increase in these numbers. On the basis of our current findings, we predict that the average individual might eventually be found to carry >400 damaging variants and >2 disease-causing ones (∼5, if we assume that most disease mutations would be singletons in this data set and apply a simple correction for incomplete singleton ascertainment), estimates remarkably consistent with several of the early theoretical studies.6,7 Now, however, in addition to knowing the numbers, we also know the identities of at least some of these damaging and disease-causing variants.
Acknowledgments
We thank Ville Mustonen for discussion and suggestions about estimating selective coefficients, A. Kondrashov and two other reviewers for comments, Sarah Ratzel for help with nomenclatural complexities, and the donors of samples used by the 1000 Genomes Project, whose generosity made this study possible. Work at the Wellcome Trust Sanger Institute was supported by the Wellcome Trust (WT098051), and BIOBASE GmbH provides funding for the Human Gene Mutation Database.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
Complete Genomics 69 Genomes Data, http://www.completegenomics.com/public-data/69-Genomes/
Complete Genomics public data in vcf format from the 1000 Genomes Project, ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/technical/working/20111010_completeGenomics_vcf/
Database of Genomic Variants (for USH2A), http://projects.tcag.ca/cgi-bin/variation/xview?source=hg19&view=variation&id=Variation_74898
Google, http://www.google.co.uk/
HGMD, http://www.hgmd.org
HGMD Professional, http://www.biobase-international.com/product/hgmd
NHGRI Catalog of Published Genome-wide Association Studies, http://www.genome.gov/gwastudies/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Stenson P.D., Mort M., Ball E.V., Howells K., Phillips A.D., Thomas N.S., Cooper D.N. The Human Gene Mutation Database: 2008 update. Genome Med. 2009;1:13. doi: 10.1186/gm13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bell C.J., Dinwiddie D.L., Miller N.A., Hateley S.L., Ganusova E.E., Mudge J., Langley R.J., Zhang L., Lee C.C., Schilkey F.D. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Transl. Med. 2011;3:ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yngvadottir B., MacArthur D.G., Jin H., Tyler-Smith C. The promise and reality of personal genomics. Genome Biol. 2009;10:237. doi: 10.1186/gb-2009-10-9-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muller H.J. Our load of mutations. Am. J. Hum. Genet. 1950;2:111–176. [PMC free article] [PubMed] [Google Scholar]
- 7.Morton N.E., Crow J.F., Muller H.J. An estimate of the mutational damage in man from data on consanguineous marriages. Proc. Natl. Acad. Sci. USA. 1956;42:855–863. doi: 10.1073/pnas.42.11.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kondrashov A.S. Contamination of the genome by very slightly deleterious mutations: Why have we not died 100 times over? J. Theor. Biol. 1995;175:583–594. doi: 10.1006/jtbi.1995.0167. [DOI] [PubMed] [Google Scholar]
- 9.Cooper D.N., Chen J.M., Ball E.V., Howells K., Mort M., Phillips A.D., Chuzhanova N., Krawczak M., Kehrer-Sawatzki H., Stenson P.D. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum. Mutat. 2010;31:631–655. doi: 10.1002/humu.21260. [DOI] [PubMed] [Google Scholar]
- 10.Lohmueller K.E., Indap A.R., Schmidt S., Boyko A.R., Hernandez R.D., Hubisz M.J., Sninsky J.J., White T.J., Sunyaev S.R., Nielsen R. Proportionally more deleterious genetic variation in European than in African populations. Nature. 2008;451:994–997. doi: 10.1038/nature06611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyko A.R., Williamson S.H., Indap A.R., Degenhardt J.D., Hernandez R.D., Lohmueller K.E., Adams M.D., Schmidt S., Sninsky J.J., Sunyaev S.R. Assessing the evolutionary impact of amino acid mutations in the human genome. PLoS Genet. 2008;4:e1000083. doi: 10.1371/journal.pgen.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kryukov G.V., Pennacchio L.A., Sunyaev S.R. Most rare missense alleles are deleterious in humans: Implications for complex disease and association studies. Am. J. Hum. Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goode D.L., Cooper G.M., Schmutz J., Dickson M., Gonzales E., Tsai M., Karra K., Davydov E., Batzoglou S., Myers R.M., Sidow A. Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Res. 2010;20:301–310. doi: 10.1101/gr.102210.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ponting C.P., Hardison R.C. What fraction of the human genome is functional? Genome Res. 2011;21:1769–1776. doi: 10.1101/gr.116814.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pheasant M., Mattick J.S. Raising the estimate of functional human sequences. Genome Res. 2007;17:1245–1253. doi: 10.1101/gr.6406307. [DOI] [PubMed] [Google Scholar]
- 16.Lazarin G.A., Haque I.S., Nazareth S., Iori K., Patterson A.S., Jacobson J.L., Marshall J.R., Seltzer W.K., Patrizio P., Evans E.A., Srinivasan B.S. An empirical estimate of carrier frequencies for 400+ causal Mendelian variants: results from an ethnically diverse clinical sample of 23,453 individuals. Genet. Med. 2012 doi: 10.1038/gim.2012.114. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chong J.X., Ouwenga R., Anderson R.L., Waggoner D.J., Ober C. A population-based study of autosomal-recessive disease-causing mutations in a founder population. Am. J. Hum. Genet. 2012;91:608–620. doi: 10.1016/j.ajhg.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asan X., Xu Y., Jiang H., Tyler-Smith C., Xue Y., Jiang T., Wang J., Wu M., Liu X., Tian G. Comprehensive comparison of three commercial human whole-exome capture platforms. Genome Biol. 2011;12:R95. doi: 10.1186/gb-2011-12-9-r95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashley E.A., Butte A.J., Wheeler M.T., Chen R., Klein T.E., Dewey F.E., Dudley J.T., Ormond K.E., Pavlovic A., Morgan A.A. Clinical assessment incorporating a personal genome. Lancet. 2010;375:1525–1535. doi: 10.1016/S0140-6736(10)60452-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nothnagel M., Herrmann A., Wolf A., Schreiber S., Platzer M., Siebert R., Krawczak M., Hampe J. Technology-specific error signatures in the 1000 Genomes Project data. Hum. Genet. 2011;130:505–516. doi: 10.1007/s00439-011-0971-3. [DOI] [PubMed] [Google Scholar]
- 22.Marth G.T., Yu F., Indap A.R., Garimella K., Gravel S., Leong W.F., Tyler-Smith C., Bainbridge M., Blackwell T., Zheng-Bradley X., 1000 Genomes Project The functional spectrum of low-frequency coding variation. Genome Biol. 2011;12:R84. doi: 10.1186/gb-2011-12-9-r84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drmanac R., Sparks A.B., Callow M.J., Halpern A.L., Burns N.L., Kermani B.G., Carnevali P., Nazarenko I., Nilsen G.B., Yeung G. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science. 2010;327:78–81. doi: 10.1126/science.1181498. [DOI] [PubMed] [Google Scholar]
- 24.González-Pérez A., López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am. J. Hum. Genet. 2011;88:440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jobling M.A., Hurles M.E., Tyler-Smith C. Garland Science; Abingdon, UK: 2004. Human Evolutionary Genetics: Origins, Peoples and Disease. [Google Scholar]
- 27.Ballas S.K. Defining the phenotypes of sickle cell disease. Hemoglobin. 2011;35:511–519. doi: 10.3109/03630269.2011.610477. [DOI] [PubMed] [Google Scholar]
- 28.Dreyer B., Tranebjaerg L., Rosenberg T., Weston M.D., Kimberling W.J., Nilssen O. Identification of novel USH2A mutations: Implications for the structure of USH2A protein. Eur. J. Hum. Genet. 2000;8:500–506. doi: 10.1038/sj.ejhg.5200491. [DOI] [PubMed] [Google Scholar]
- 29.Nájera C., Beneyto M., Blanca J., Aller E., Fontcuberta A., Millán J.M., Ayuso C. Mutations in myosin VIIA (MYO7A) and usherin (USH2A) in Spanish patients with Usher syndrome types I and II, respectively. Hum. Mutat. 2002;20:76–77. doi: 10.1002/humu.9042. [DOI] [PubMed] [Google Scholar]
- 30.Mandon-Pépin B., Touraine P., Kuttenn F., Derbois C., Rouxel A., Matsuda F., Nicolas A., Cotinot C., Fellous M. Genetic investigation of four meiotic genes in women with premature ovarian failure. Eur. J. Endocrinol. 2008;158:107–115. doi: 10.1530/EJE-07-0400. [DOI] [PubMed] [Google Scholar]
- 31.Hikiba J., Hirota K., Kagawa W., Ikawa S., Kinebuchi T., Sakane I., Takizawa Y., Yokoyama S., Mandon-Pépin B., Nicolas A. Structural and functional analyses of the DMC1-M200V polymorphism found in the human population. Nucleic Acids Res. 2008;36:4181–4190. doi: 10.1093/nar/gkn362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Dijk M., Mulders J., Poutsma A., Könst A.A., Lachmeijer A.M., Dekker G.A., Blankenstein M.A., Oudejans C.B. Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat. Genet. 2005;37:514–519. doi: 10.1038/ng1541. [DOI] [PubMed] [Google Scholar]
- 33.van Dijk M., van Bezu J., van Abel D., Dunk C., Blankenstein M.A., Oudejans C.B., Lye S.J. The STOX1 genotype associated with pre-eclampsia leads to a reduction of trophoblast invasion by alpha-T-catenin upregulation. Hum. Mol. Genet. 2010;19:2658–2667. doi: 10.1093/hmg/ddq152. [DOI] [PubMed] [Google Scholar]
- 34.Weiss G. Genetic mechanisms and modifying factors in hereditary hemochromatosis. Nat Rev Gastroenterol Hepatol. 2010;7:50–58. doi: 10.1038/nrgastro.2009.201. [DOI] [PubMed] [Google Scholar]
- 35.Rochette J., Le Gac G., Lassoued K., Férec C., Robson K.J. Factors influencing disease phenotype and penetrance in HFE haemochromatosis. Hum. Genet. 2010;128:233–248. doi: 10.1007/s00439-010-0852-1. [DOI] [PubMed] [Google Scholar]
- 36.Rossi E., Olynyk J.K., Jeffrey G.P. Clinical penetrance of C282Y homozygous HFE hemochromatosis. Expert Rev. Hematol. 2008;1:205–216. doi: 10.1586/17474086.1.2.205. [DOI] [PubMed] [Google Scholar]
- 37.Bhattacharya G., Kalluri R., Orten D.J., Kimberling W.J., Cosgrove D. A domain-specific usherin/collagen IV interaction may be required for stable integration into the basement membrane superstructure. J. Cell Sci. 2004;117:233–242. doi: 10.1242/jcs.00850. [DOI] [PubMed] [Google Scholar]
- 38.Matsuzaki H., Wang P.H., Hu J., Rava R., Fu G.K. High resolution discovery and confirmation of copy number variants in 90 Yoruba Nigerians. Genome Biol. 2009;10:R125. doi: 10.1186/gb-2009-10-11-r125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dreyer B., Tranebjaerg L., Brox V., Rosenberg T., Möller C., Beneyto M., Weston M.D., Kimberling W.J., Cremers C.W., Liu X.Z., Nilssen O. A common ancestral origin of the frequent and widespread 2299delG USH2A mutation. Am. J. Hum. Genet. 2001;69:228–234. doi: 10.1086/321269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenberg T., Haim M., Hauch A.M., Parving A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin. Genet. 1997;51:314–321. doi: 10.1111/j.1399-0004.1997.tb02480.x. [DOI] [PubMed] [Google Scholar]
- 41.Kohane I.S., Hsing M., Kong S.W. Taxonomizing, sizing, and overcoming the incidentalome. Genet. Med. 2012;14:399–404. doi: 10.1038/gim.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., Wellcome Trust Case Control Consortium Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yngvadottir B., Xue Y., Searle S., Hunt S., Delgado M., Morrison J., Whittaker P., Deloukas P., Tyler-Smith C. A genome-wide survey of the prevalence and evolutionary forces acting on human nonsense SNPs. Am. J. Hum. Genet. 2009;84:224–234. doi: 10.1016/j.ajhg.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.