

Abstract
Neurodegeneration with brain iron accumulation (NBIA) is a group of genetic disorders characterized by abnormal iron deposition in the basal ganglia. We report that de novo mutations in WDR45, a gene located at Xp11.23 and encoding a beta-propeller scaffold protein with a putative role in autophagy, cause a distinctive NBIA phenotype. The clinical features include early-onset global developmental delay and further neurological deterioration (parkinsonism, dystonia, and dementia developing by early adulthood). Brain MRI revealed evidence of iron deposition in the substantia nigra and globus pallidus. Males and females are phenotypically similar, an observation that might be explained by somatic mosaicism in surviving males and germline or somatic mutations in females, as well as skewing of X chromosome inactivation. This clinically recognizable disorder is among the more common forms of NBIA, and we suggest that it be named accordingly as beta-propeller protein-associated neurodegeneration.
Main Text
Neurodegeneration with brain iron accumulation (NBIA) comprises a set of single-gene disorders that manifest as a range of neurological phenotypes and share the feature of high levels of basal-ganglia iron.1 To date, the major genes associated with NBIA include PANK2 (MIM 606157), C19orf12 (MIM 614297), and PLA2G6 (MIM 603604), which encode mitochondria-associated proteins with no clear link to iron homeostasis. Mutations in these genes lead to autosomal-recessive degenerative disorders affecting children and adults.
As specific genetic forms of NBIA were identified, a distinctive phenotype became evident among the remaining idiopathic cohort.2 A subset of study participants had global developmental delay in early childhood and slow motor and cognitive gains until adolescence or early adulthood, when dystonia, parkinsonism, and dementia would manifest and prompt further diagnostic evaluation. Brain MRI after deterioration showed a pattern characteristic of high iron: a markedly hypointense signal on T2-weighted sequences in the substantia nigra and globus pallidus (Figure 1). In addition, cerebral atrophy was observed. A unique feature of this form of NBIA was T1 hyperintensity surrounding a central linear region of signal hypointensity within the substantia nigra and cerebral peduncles (Figure 1), a feature previously suggested to indicate a new NBIA subtype1–3 (also see GeneReviews in Web Resources).
Figure 1.
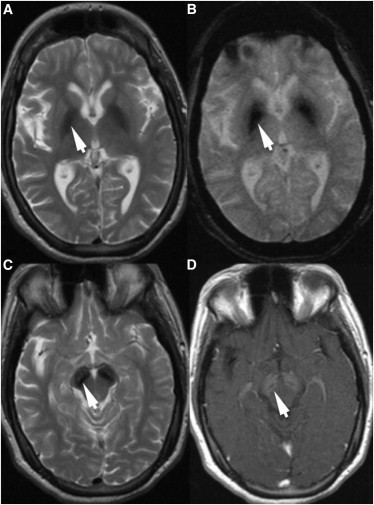
MRI Features Associated with WDR45 Mutations
T2-weighted turbo-spin-echo (TSE) MRI shows (A) a hypointense signal (arrow) in the globus pallidus, which “blooms” on fluid-attenuated inversion-recovery sequence (B, arrow), consistent with iron. A hypointense signal (C, arrow) in the substantia nigra on T2-weighted TSE shows iron, and T1-weighted imaging demonstrates a hyperintense halo (D, arrow) in the substantia nigra.
Among families with affected individuals, none demonstrated a pattern of Mendelian inheritance of this phenotype. Affected individuals were singletons from diverse ethnic and racial groups without known or suspected consanguinity. One was male, and 13 were female. The simplex pattern of disease in families, the similar phenotype in both genders, and the absence of male lethality led us to consider autosomal-recessive inheritance and de novo mutations in an NBIA-associated gene. However, we remained open to the possibility of X-linked inheritance on the basis of the gender bias in our cohort. We therefore undertook exome sequencing to identify their causative mutations. The procedures followed were in accordance with the ethical standards of the institutional review board at Oregon Health & Science University, and proper informed consent was obtained.
Exome sequencing and variant calling were performed as described previously.4 We used the SureSelect Human All Exon 50 Mb kit from Agilent for in-solution enrichment of exonic sequences. Libraries were subsequently sequenced as 100 bp paired-end runs. Read alignment was performed with the Burrows-Wheeler Aligner (v.0.5.8). SAMtools (v.0.1.7) was used for detecting single-nucleotide variants and small insertions and deletions. We produced an average of 10.2 Gb of mappable sequences per sample; the mean was 123× coverage, and >90% of the exome was covered >20×, enabling high-confidence variant detection (Table S1, available online).
To identify the disease-causing mutations, we analyzed exome sequence in 14 unrelated persons with a homogeneous clinical and radiological phenotype; this allowed us to use a stringent in silico filtering strategy to address the major challenge of distinguishing disease-causing mutations from benign sequence variants. To prioritize candidate genes, we step-wise filtered the identified DNA variants on the basis of the following assumptions: (1) Because the disease is rare and severe, we expected the mutations to alter the protein sequence and to have a very low frequency in control populations. We excluded variants with a minor allele frequency > 0.3% in public SNP databases (HapMap and 1000 Genomes) and in 1,429 exomes. (2) Although we favored de novo mutations exerting their effects in a dominant fashion, we also considered an autosomal-recessive mode of inheritance. (3) Given the distinct phenotype of the individuals studied, we postulated that most would share defects in the same gene. For the joint analysis of dominant variants, we excluded highly variable genes with >70 different rare nonsynonymous variants in 1,414 exomes. Searching for compound heterozygous or homozygous mutations yielded approximately ten mutant genes, none of which were known to be associated with NBIA, per individual (Table S2). The only gene with compound heterozygous or homozygous mutations in more than two individuals was HR (Table 1), considered an unlikely candidate given its role in hair growth (MIM 203655).
Table 1.
Identification of the Associated Gene by Joint Analysis of 14 Exomes
|
Minimal Cases |
||||||
|---|---|---|---|---|---|---|
| ≥2 | ≥3 | ≥4 | ≥5 | ≥6 | ≥13 | |
| Dominant-model candidate genes (≥1 rare nonsynonymous variant in the same gene) | >500 | 224 | 32 | 2 (MYLK, WDR45) | 1 (WDR45) | 1 (WDR45) |
| Recessive-model candidate genes (≥2 rare nonsynonymous variants in the same gene) | 45 | 1 (HR) | 0 | 0 | 0 | 0 |
In more than 2,000 X chromosomes, only one missense variant, c.763G>T (p.Ala255Ser) (with benign or neutral effects as predicted by in silico analysis by PolyPhen-2), was identified in WDR45. Loss-of-function alleles were not detected, nor were any found in the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server listing a total of >8,700 alleles. Nonsynonymous variants include missense, nonsense, stop-loss, and splice-site mutations, as well as insertions and deletions. “Rare” indicates a frequency < 0.3% in 1,429 control exomes in the in-house database, HapMap, and the 1000 Genomes project. Genes in bold are those that carry the causal mutations.
We next searched for single variants affecting the same gene shared by an increasing number of cases. This filtering left a single gene with variants identified in more than five individuals: X chromosomal WDR45 (RefSeq accession number NM_007075.3), which encodes a WD40 repeat protein. In fact, 13 of 14 individuals carried a mutated allele, each with a different variant; 11 of these variants were predicted to cause loss of function, and two were predicted to change an evolutionarily conserved amino acid residue (Figure 2). One missense mutation, c.38G>C, was predicted to change the arginine at position 13 to proline and to skip the first coding exon, leading to a protein starting with methionine at position 25 (GENSCAN). Mutations in WDR45 were independently confirmed by Sanger sequencing.
Figure 2.
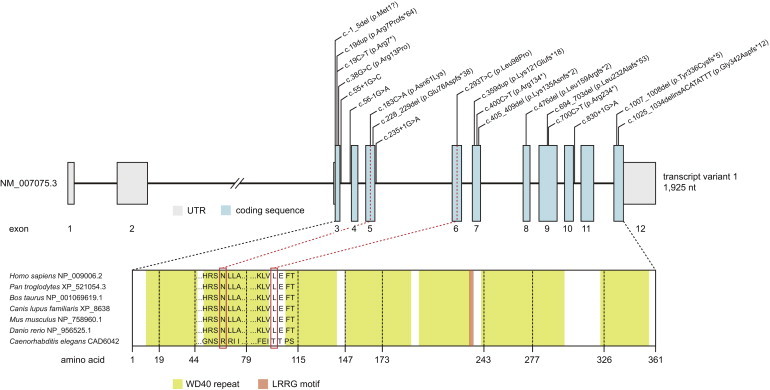
WDR45 Structure and Conservation of Mutated Residues
Exon-intron structure of WDR45 with localization and conservation of amino acid residues harboring substitutions that we predict as the cause of BPAN.
We then broadened our search to include other study participants with idiopathic NBIA and a suggestive phenotype. Mutations were found in seven additional individuals, including two males. Segregation analysis in the families from which parental DNA was available revealed only reference alleles of WDR45 in all samples, indicating that the mutations in the probands were de novo. In total, 19 mutations were found in 20 subjects, and none were present in parental DNA samples, arguing for de novo mutations as the de facto mechanism underlying the disease (Table 2). Two affected individuals harbored an identical mutation (c.1007_1008del [p.Tyr336Cysfs∗5]). Clinical testing for WDR45 mutations is now available.
Table 2.
Genetic Characteristics of Study Subjects and Family Members
| ID | Sex | Ancestry |
WDR45 Mutations Identified |
Mutation Status of Family Members |
|||
|---|---|---|---|---|---|---|---|
| cDNA (RefSeq NM_007075.3) | Protein (RefSeq NP_009006) | Mother | Father | Sibling (n) | |||
| 60251 | F | German, Irish, English, Austrian | c.1007_1008del | p.Tyr336Cysfs∗5 | WT | WT | WT (1) |
| 63700 | F | German, Sioux, Cherokee | c.38G>C | p.Arg13Proa | WT | WT | WT (2) |
| 63701 | F | German, French, Irish | c.-1_5del | p.Met1? | WT | N/A | N/A |
| 63702 | F | African American | c.293T>C | p.Leu98Pro | WT | N/A | WT (1) |
| 63703 | F | African American | c.476del | p.Leu159Argfs∗2 | N/A | N/A | N/A |
| 63704 | F | Hispanic, Puerto Rican | c.19C>T | p.Arg7∗ | N/A | N/A | N/A |
| 63705 | F | Romanian, French | c.56-1G>A | splicing defect | N/A | N/A | N/A |
| 63706 | F | German, Irish, English | c.700C>T | p.Arg234∗ | WT | N/A | WT (4) |
| 63707 | F | unknown | c.400C>T | p.Arg134∗ | WT | WT | WT (3) |
| 63708 | M | German, Irish, Scottish | c.228_229del | p.Glu76Aspfs∗38 | WT | WT | N/A |
| 63709 | F | Italian, northern European, Native American | c.405_409del | p.Lys135Asnfs∗2 | N/A | N/A | N/A |
| 63711 | F | Dutch | c.359dup | p.Lys121Glufs∗18 | WT | WT | WT (4) |
| 63712 | F | Scottish, Irish | c.830+1G>A | splicing defect | WT | WT | N/A |
| 49841 | M | German | c.19dup | p.Arg7Profs∗64 | N/A | N/A | N/A |
| 411-201 | F | Italian | c.235+1G>A | splicing defect | WT | WT | N/A |
| HH56 | F | unknown | c.1007_1008del | p.Tyr336Cysfs∗5 | WT | WT | N/A |
| HH84 | F | unknown | c.694_703del | p.Leu232Alafs∗53 | WT | WT | N/A |
| NBIA10 | F | Pakistani | c.183C>A | p.Asn61Lys | WT | WT | N/A |
| 463 | M | English | c.1025_1034delinsACATATTT | p.Gly342Aspfs∗12 | N/A | N/A | N/A |
| 152 | F | unknown | c.55+1G>C | splicing defect | WT | WT | WT (1) |
Numbers in parentheses indicate the amount of tested siblings of the proband. The following abbreviations are used: F, female; M, male; WT, wild-type; and N/A, not available.
This alteration is predicted to cause skipping of exon 3 and usage of an alternate start methionine at position 25.
WD40 repeat proteins compose a large family of molecules with repeating units containing a conserved core of 40+ amino acids that terminate with tryptophan-aspartic-acid (WD) residues. WD40 proteins regulate the assembly of multiprotein complexes to serve diverse functions including signal transduction, transcriptional regulation, autophagy, and cell-cycle control.5 The structure of these proteins is key to their role in orchestrating protein-protein interactions. WD40 repeat proteins assume a highly symmetrical, seven-bladed, beta-propeller platform structure for coordinating protein-protein interactions. WDR45 contains a conserved motif for interaction with phospholipids, and the protein binds or associates with known autophagy proteins ATG2A and ATG2B.5,6
WD40 repeat proteins are defective in other neurologic disorders.7 Lissencephaly-1 (LIS1 [MIM 607432]) was the first WD repeat gene identified as the cause of a human disease. Also, type A Cockayne syndrome (MIM 216400) is associated with mutations in a gene encoding a WD40-repeat-protein family member. Another X-linked gene, TBL1X (MIM 300196), encodes the protein transducing-β-like, and its mutations lead to sensorineural deafness. Finally, achalasia-addisonianism-alacrima syndrome (MIM 231550) is caused by mutations in a gene encoding a WD40 repeat protein, aladin. Defective aladin leads to abnormal development of the autonomic nervous system and late-onset neurodegeneration. Interestingly, all of these disorders are characterized by defects in brain development or in the maintenance of normal brain function.
Although WDR45 is located on the X chromosome and undergoes inactivation, the clinical features of this form of NBIA do not follow a pattern typical of an X-linked disorder. The phenotype of affected males is indistinguishable from that of females; indeed, there is a striking uniformity to the clinical features and the natural history of the disease in all 20 study participants. All three males harbor a mutation that is predicted to render the protein nonfunctional, as is also the case for many of the females. How might this be explained? Sex-chromosome aneuploidy could account for the observations; however, normal karyotypes were reported on the two males from whom they were available. Although males could harbor a separate activating mutation in one of several WDR45 pseudogenes (of which there are at least three) or a tandem duplication of WDR45 (with a subsequent mutation in only one gene copy), the more likely explanation is a postzygotic mutation leading to somatic mosaicism in males. This mechanism could explain the phenotypic similarities between genders and would be consonant with the finding of exclusively de novo mutations, which could occur in germline or somatic tissues. Males with germline mutations are probably nonviable; males and females with somatic mutations manifest a phenotype that is most likely determined by the stage of development at the time of mutation and by the tissue distribution of mutated cells. Females might harbor germline or somatic mutations. A further corollary is that more and less severe phenotypes are likely to be found in males on the basis of whether somatic mutations occur earlier or later in embryogenesis, respectively. This pattern has been observed in Rett syndrome (MIM 312750), another X-linked dominant disorder.8 Females could also present with milder phenotypes if they harbor a later somatic mutation. In support of this mechanism, sequencing of DNA from the blood of the second male identified two WDR45 exon 12 amplicons (one wild-type and one deleted with discrepant peak amplitudes; see Figure 3), which would be seen if there were two different populations of DNA molecules, as in the case of somatic mosaicism. Genetic analysis of multiple tissues might be necessary for identifying WDR45 mutations in mildly affected individuals.
Figure 3.
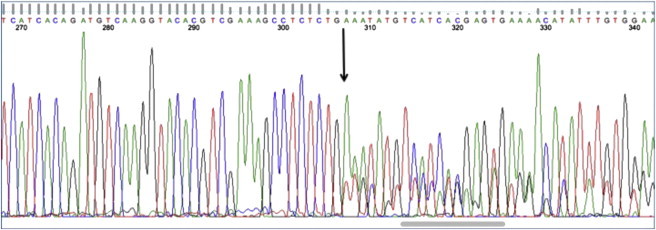
Evidence for Somatic Mutation
A sequencing electropherogram from male subject 463 shows two DNA traces of differing peak amplitudes to the right of the arrow.
To also help explain the phenotypic similarity between males and females, we found evidence to suggest skewing of X chromosome inactivation. Using established methods9 to analyze peripheral blood from 12 women with mutations in WDR45, we rated the X-inactivation pattern as random in two subjects (50:50 to 75:25), skewed in six subjects (75:25 to 90:10), and extremely skewed in four subjects (>90:10). Although these results suggest that a skewed methylation pattern might contribute to disease pathogenesis, more detailed studies will be needed for determining whether the mutated allele is preferentially inactivated and whether patterns in peripheral blood reflect those in the brain in this disorder.
Despite clear evidence of a causal role for mutations in WDR45 in this phenotype with prominent parkinsonism, Xp11.23 has not been reported as a PARK locus. Predictably, both linkage and association studies would lack signal because of the combination of de novo mutations and low reproductive fitness of those affected. Given the phenotype, we propose WDR45 as a PARK gene. As with other forms of NBIA, the phenotypic spectrum associated with mutations in WDR45 is likely to broaden and to include other parkinsonism syndromes as more individuals are screened.
In conclusion, we have identified mutations in WDR45 as a cause of a distinctive X-linked dominant form of NBIA. De novo mutations in this gene underlie a phenotype of childhood developmental disability with adolescent or adult onset of dystonia, parkinsonism, and dementia. In keeping with the naming convention of other forms of NBIA (PKAN, PLAN, MPAN), we propose the term “beta-propeller protein-associated neurodegeneration” (BPAN) for the disease associated with mutations in WDR45. We recommend that the previously referenced term “SENDA” (static encephalopathy of childhood with neurodegeneration in adulthood) no longer be used1–3 (see also GeneReviews in Web Resources). Our understanding of the pathogenesis of BPAN will increase with further investigation of typical, as well as atypical, affected persons who challenge the usual pattern of X-linked disease.
Acknowledgments
The authors gratefully acknowledge the study participants and families, as well as the support of our advocacy partners: the NBIA Disorders Association, Hoffnungsbaum e.V., and Associazione Italiana Sindromi Neurodegenerative da Accumulo di Ferro. This work was funded by the NBIA Disorders Association and was made possible with support from the Oregon Clinical and Translational Research Institute (UL1 RR024140 NCRR), a component of the National Institutes of Health (NIH) and NIH Roadmap for Medical Research. P.H., N.N., T.M., H.P., and S.J.H. participate in the TIRCON consortium (European Commission Seventh Framework Program, FP7/2007-2013, HEALTH-F2-2011, grant agreement 277984). We thank Evelyn Botz and Carola Fischer for technical support. T.M. and H.P. were supported by the German Federal Ministry of Education and Research (Systems Biology of Metabotypes grant SysMBo 0315494A) and the German Network for Mitochondrial Disorders (mitoNET 01GM0867). T.M. and T.M.S. were supported by the European Commission Seventh Framework Program (N. 261123), Genetic European Variation in Disease Consortium, and German Ministry for Education and Research (01GR0804-4). M.C.K. receives support from the American Academy of Neurology and American Philosophical Society. We thank the Cell line and DNA bank of pediatric movement disorders of the Telethon Genetic Biobank Network (GTB07001). M.A.K.’s research is supported by Action Medical Research. We thank the UK Parkinson’s Disease Consortium, University College London Institute of Neurology, University of Sheffield, University of Dundee, Medical Research Council, Parkinson’s Disease Foundation, Dystonia Medical Research Foundation, and Brain Research Trust.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GeneReviews, Gregory, A., and Hayflick, S.J. (2002). Pantothenate Kinase-Associated Neurodegeneration, http://www.ncbi.nlm.nih.gov/books/NBK1490/
GENSCAN, http://genes.mit.edu/GENSCAN.html
GeneTests, www.genetests.org
MutationTaster, http://www.mutationtaster.org/
NHLBI Exome Sequencing Project Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
References
- 1.Gregory A., Hayflick S.J. Genetics of neurodegeneration with brain iron accumulation. Curr. Neurol. Neurosci. Rep. 2011;11:254–261. doi: 10.1007/s11910-011-0181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gregory A., Polster B.J., Hayflick S.J. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J. Med. Genet. 2009;46:73–80. doi: 10.1136/jmg.2008.061929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kruer M.C., Boddaert N., Schneider S.A., Houlden H., Bhatia K.P., Gregory A., Anderson J.C., Rooney W.D., Hogarth P., Hayflick S.J. Neuroimaging features of neurodegeneration with brain iron accumulation. AJNR Am. J. Neuroradiol. 2012;33:407–414. doi: 10.3174/ajnr.A2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mayr J.A., Haack T.B., Graf E., Zimmermann F.A., Wieland T., Haberberger B., Superti-Furga A., Kirschner J., Steinmann B., Baumgartner M.R. Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am. J. Hum. Genet. 2012;90:314–320. doi: 10.1016/j.ajhg.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behrends C., Sowa M.E., Gygi S.P., Harper J.W. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Proikas-Cezanne T., Waddell S., Gaugel A., Frickey T., Lupas A., Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene. 2004;23:9314–9325. doi: 10.1038/sj.onc.1208331. [DOI] [PubMed] [Google Scholar]
- 7.Li D., Roberts R. WD-repeat proteins: Structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 2001;58:2085–2097. doi: 10.1007/PL00000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moog U., Smeets E.E., van Roozendaal K.E., Schoenmakers S., Herbergs J., Schoonbrood-Lenssen A.M., Schrander-Stumpel C.T. Neurodevelopmental disorders in males related to the gene causing Rett syndrome in females (MECP2) Eur. J. Paediatr. Neurol. 2003;7:5–12. doi: 10.1016/s1090-3798(02)00134-4. [DOI] [PubMed] [Google Scholar]
- 9.Allen R.C., Zoghbi H.Y., Moseley A.B., Rosenblatt H.M., Belmont J.W. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am. J. Hum. Genet. 1992;51:1229–1239. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.