

Abstract
We report on four families affected by a clinical presentation of complex hereditary spastic paraplegia (HSP) due to recessive mutations in DDHD2, encoding one of the three mammalian intracellular phospholipases A1 (iPLA1). The core phenotype of this HSP syndrome consists of very early-onset (<2 years) spastic paraplegia, intellectual disability, and a specific pattern of brain abnormalities on cerebral imaging. An essential role for DDHD2 in the human CNS, and perhaps more specifically in synaptic functioning, is supported by a reduced number of active zones at synaptic terminals in Ddhd-knockdown Drosophila models. All identified mutations affect the protein’s DDHD domain, which is vital for its phospholipase activity. In line with the function of DDHD2 in lipid metabolism and its role in the CNS, an abnormal lipid peak indicating accumulation of lipids was detected with cerebral magnetic resonance spectroscopy, which provides an applicable diagnostic biomarker that can distinguish the DDHD2 phenotype from other complex HSP phenotypes. We show that mutations in DDHD2 cause a specific complex HSP subtype (SPG54), thereby linking a member of the PLA1 family to human neurologic disease.
Main Text
Hereditary spastic paraplegia (HSP) is a genetically heterogeneous group of neurodegenerative disorders characterized by a length-dependent, distal axonopathy of fibers of the corticospinal tract, which clinically leads to lower-limb spasticity and weakness. In pure HSP, symptoms are more or less restricted to corticospinal-tract dysfunction, whereas in complex HSP, the disease is more widespread and encompasses additional neurologic and non-neurologic symptoms. Inheritance can follow an X-linked, dominant, or autosomal-recessive pattern. In 1986, the first HSP locus was described,1 and to date, over 50 loci (SPG1–SPG53) have been mapped. In these loci, disease-causing mutations in 29 genes have been identified (Schule et al.2 and Online Mendelian Inheritance in Man). The proteins involved have diverse functions, of which the most prominent are regulation of intracellular trafficking, organelle biogenesis, and shaping of membranes.3 For autosomal-recessive HSP in combination with a thin corpus callosum and white-matter abnormalities, mutations in five genes have been identified: SPG11 (∼20% of autosomal-recessive HSP; MIM 604360),2 ZFYVE26 (SPG15, <3% of autosomal-recessive HSP; MIM 270700),2 SPG21 (in an old-order Amish population; MIM 248900),4 GJC2 (SPG44, one family reported; MIM 613206),5 and AP4B1 (SPG47, two families reported; MIM 607245).6,7
We ascertained two families affected by a complex HSP form consisting of a combination of early-onset spastic paraplegia and intellectual disability (ID) and characterized by a marked thin corpus callosum and subtle periventricular white-matter hyperintensities on MRI and an abnormal lipid peak on cerebral proton magnetic resonance spectroscopy (MRS). This study was approved by the Medical Ethics Committee of the Radboud University Nijmegen Medical Centre, and all participants signed informed consent. Exome sequencing was performed on genomic DNA of the affected boy from family 1 and on both siblings and the father from family 2 (Figure 1). For families 1 and 2, genomic DNA was captured with an Agilent SureSelect Human All Exon 50 Mb Kit (Agilent Technologies, Santa Clara, CA, USA). Captured DNA of family 1 was sequenced on one-fourth of a sequence slide on a SOLiD 4 System and aligned to the reference human genome (hg19) (Life Technologies, Carlsbad, CA, USA). The exome-sequencing experiment and analysis pipeline were previously described by Vissers et al.9 For family 2, captured DNA was sequenced on Illumina HiSeq 2000 according to the manufacturer’s (Illumina, San Diego, CA, USA) protocol. The Fastx toolkit was used for removing adaptor sequences and trimming sequence reads. The Burrows-Wheeler Aligner 0.5.910 and Genome Analysis Toolkit11 were used for aligning reads to the reference human genome (hg19). Single-nucleotide variants and indels were called with SAMtools12 and annotated with ANNOVAR13 and custom scripts. For both families, variant prioritization (including indels) excluded all variants present in fewer than 20% of reads, as well as all nongenic, intronic (other than canonical splice sites) variants and variants leading to synonymous amino acid changes. Next, variants with a frequency > 1% in dbSNP v.134 or the Nijmegen local variant database (∼270 exome-sequencing experiments) were excluded for family 1. For family 2, variants present in dbSNP v.132 or control samples from exome experiments sequenced through the FORGE project (∼400 exome-sequencing experiments) were excluded. Candidate mutations were selected under the assumption of an autosomal-recessive disease model (Tables S1 and S2, available online). There was overlap for candidate mutations in only one gene, DDHD2 (RefSeq accession number NM_015214.2), between both families. For family 1, variant selection resulted in candidate recessive mutations in seven genes; the most striking were two heterozygous mutations in DDHD2 because they both result in a shift of the open reading frame. In family 2, compound heterozygous mutations in only one gene, DDHD2, fit a recessive model after comparison of variants of all three family members (Figure S1). Sanger sequencing showed that the respective mutations segregated with the phenotype in both families (Figures 1A and 1B).
Figure 1.

Pedigrees of Families 1–4, Photographs of the Face, and Chromatograms Showing Sanger Confirmation of the DDHD2 Mutations
Arrows indicate the individuals on whom exome sequencing was performed. The following abbreviations are used: M, mutant allele; −, wild-type allele; and C, control.
(A) Family 1 (family identifier: W10-1338) has an affected sister and brother and compound heterozygous frameshift mutations c.1804_1805insT (p.Thr602Ilefs∗18) and c.2057delA (p.Glu686Glyfs∗35).
(B) Family 2 has an affected sister and brother and compound heterozygous frameshift and missense mutations c.1386dupC (p.Ile463Hisfs∗6) and c.1978G>C (p.Asp660His).
(C) Consanguineous family 3 has seven affected individuals and homozygous mutation c.1546C>T (p.Arg516∗). Pedigree numbering is according to the original pedigree by Al-Yahyaee et al.8
(D) Consanguineous family 4 (family identifier: W12-0041) has one affected male individual and homozygous mutation c.859C>T (p.Arg287∗).
(E) The protein structure of DDHD2 includes its four domains (WWE, lipase, SAM, and DDHD), and the position of all identified mutations are indicated.
In family 1, compound heterozygous frameshift mutations c.1804_1805insT and c.2057delA were detected in DDHD2 (Figure 1A). At the protein level, both mutations result in a premature termination codon (PTC), p.Thr602Ilefs∗18 (c.1804_1805insT) and p.Glu686Glyfs∗35 (c.2057delA), within the DDHD domain, which is located at the C-terminal end of the protein (Figure 1E). In family 2, a compound heterozygous frameshift mutation, c.138dupC, and a missense mutation, c.1978G>C, were identified (Figures 1B and 1E). At the protein level, the frameshift mutation introduces a PTC (p.Ile463Hisfs∗6) before the DDHD domain, and the missense mutation results in the substitution of a histidine for an aspartic acid (p.Asp660His) within the RIDYXL motif, which is conserved among the DDHD domains of the three human PLA1s, DDHD1, DDHD2, and SEC23IP (Figure S2A). In addition, the aspartic acid at position 660 is conserved down to Drosophila melanogaster (Figure S2B). The c.1978G>C mutation was found once in the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server database consisting of 13,006 alleles, but not in our local SNP database consisting of 2,302 alleles. The other mutations were not present in either database.
DDHD2 is located in chromosomal region 8p11.23. Previously, in a large consanguineous Omani family (family 3) affected by a similar phenotype, the genetic defect was mapped to a 9 cM interval on chromosome 8p.8 This region (SPG18 [MIM 611225]) encompasses DDHD2 and ERLIN2,8,14,15 but mutation analysis and mRNA gene-expression analysis excluded mutations in ERLIN2 as disease causing, whereas Sanger sequencing of DDHD2 revealed a homozygous c.1546C>T mutation. This mutation introduces a PTC resulting in p.Arg516∗ at the very beginning of the DDHD domain (Figure 1C). A screen of all affected and unaffected individuals showed complete segregation of this mutation with the disease phenotype in this family. The p.Arg516∗ change was absent from 200 alleles of healthy ethnically matched controls and was observed once in the heterozygous state in the NHLBI Exome Variant Server database but was absent from our local SNP database.
In order to further determine the occurrence and phenotypic spectrum of the DDHD2 mutations, we sequenced the protein-coding sequence of DDHD2 in 55 additional complex-HSP-affected individuals in whom SPG11 mutations were excluded and who had a family history suggestive of recessive inheritance. This resulted in the identification of one further homozygous mutation in an affected male individual from a consanguineous Iranian family. This homozygous C-to-T substitution in exon 9 at c.859 introduces a PTC at amino acid position 287 of the protein (family 4, Figure 1D).
Reviewing the phenotypic features of all four families revealed psychomotor delay that develops into intellectual disability and very early-onset, progressive spasticity (Table 1). The first symptoms of spasticity of the affected individuals became apparent before the age of 2 years. Foot contractures developed as a result of progressive and pronounced spasticity. In addition, strabismus and dysarthria were present in 9 out of 12 individuals and were frequently accompanied by dysphagia. Of note, optic-nerve hypoplasia was present in three out of five individuals who had undergone thorough ophthalmologic evaluation. Cerebral imaging gave a consistent pattern of brain abnormalities composed of a marked thin corpus callosum combined with subtle periventricular white-matter hyperintensities (Figure 2 and Figures S3–S6). In five affected individuals from families 1, 2, and 4, cerebral proton MRS was performed, and this revealed an abnormal spectrum with a lipid peak (1.3 ppm) showing the highest intensity around the basal-ganglia and thalamus area (Figure 2C and Figures S3, S4, and S6). This peak is similar to the characteristic, abnormal lipid peak seen in Sjögren-Larssen syndrome16 and is indicative of abnormal brain lipid accumulation. Furthermore, in two male individuals from families 1 and 2, a syrinx was observed on spinal MRI (Figure 2D and Figure S7).
Table 1.
Phenotype of Individuals with DDHD2 Mutations
|
Family 1 |
Family 2 |
Family 3a |
Family 4 |
Total | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| II-1 | II-2 | II-1 | II-2 | IV-3 | IV-4 | IV-10 | IV-11 | IV-12 | IV-13 | V-30 | II-1 | ||
| Descent | Dutch Filipino | Canadian | Oman | Iran | |||||||||
| Consanguinity | − | − | + | + | − | ||||||||
| Mutation (cDNA) | c.1804_1805insT; c.2057delA | c.1386dupC; c.1978G>C | c.1546C>T | c.859C>T | − | ||||||||
| Alteration (protein) | p.Thr602Ilefs∗18; p.Glu686Glyfs∗35 | p.Ile463Hisfs∗6; p.Asp660His | p.Arg516∗ | p.Arg287∗ | − | ||||||||
| Gender | F | M | F | M | − | M | F | F | F | M | F | M | − |
| Age at investigation (years) | 5 | 3 | 10 | 7 | − | 10 | 21 | 15 | 11 | 10 | 8 | 30 | − |
| Clinical Features | |||||||||||||
| ID and/or DD | + | + | + | + | + | + | + | + | + | + | + | + | 12/12 |
| Hypomimia | − | − | + | + | − | − | − | − | − | − | − | + | 3/12 |
| Strabismus | − | − | + | + | + | + | + | + | + | + | N/A | + | 9/12 |
| Optic-nerve hypoplasia | + | + | − | − | N/A | N/A | N/A | N/A | N/A | N/A | N/A | + | 3/5 |
| Dysartria | − | − | + | + | + | + | + | + | + | + | N/A | + | 9/12 |
| Dysphagia | − | − | − | + | − | − | + | + | + | + | + | − | 6/12 |
| Constipation | + | + | + | + | + | − | + | + | − | − | − | − | 7/12 |
| Urinary incontinence | + | + | + | + | − | − | − | − | − | − | − | − | 4/12 |
| Fecal incontinence | + | + | + | − | − | − | − | − | − | − | − | − | 3/12 |
| Upper limbs | |||||||||||||
| Spasticity | − | − | moderate | moderate | mild | mild | − | mild | − | − | − | − | 5/12 |
| Distal weakness | − | − | + | + | − | − | − | − | − | − | − | + | 3/12 |
| Rigidity | − | − | − | + | − | − | − | − | − | − | − | + | 2/12 |
| Lower limbs | |||||||||||||
| Spastic paraplegia | + | + | + | + | + | + | + | + | + | + | + | + | 12/12 |
| Hyperreflexia | + | + | + | + | + | + | + | + | + | + | + | + | 12/12 |
| Distal weakness | − | − | + | + | + | + | + | + | + | + | N/A | + | 9/12 |
| Pes cavus | − | − | − | − | − | − | + | + | − | − | − | − | 2/12 |
| Foot contractures | + | + | + | + | + | + | + | + | + | + | + | + | 12/12 |
| Radiological Findings | |||||||||||||
| Thin corpus callosum | + | + | + | + | + | + | N/A | + | + | + | + | + | 11/11 |
| PWMH | + | + | + | + | + | + | N/A | + | + | + | + | + | 11/11 |
| Lipid peakb | + | + | + | + | N/A | N/A | N/A | N/A | N/A | N/A | N/A | + | 5/5 |
| Syrinx | − | + | N/A | + | N/A | N/A | N/A | N/A | N/A | N/A | N/A | − | 2/4 |
| Mitochondrial Function | normal | N/A | normal | normal | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | 0/3 |
RefSeq accession number NM_015214.2 was used in naming mutations. The following abbreviations are used: +, presence of clinical features; –, absence of clinical features; F, female; M, male; ID, intellectual disability; DD, developmental delay; PWMH, periventricular white-matter hyperintensities; and N/A, not available.
Family 3 has previously been described by Al-Yahyaee et al.8
Lipid peak at 1.3 ppm (as measured by proton MRS) and the highest signal intensity in the basal-ganglia and thalamus area.
Figure 2.
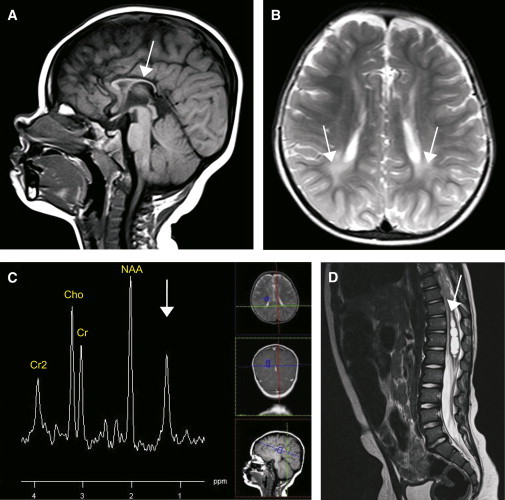
CNS Imaging of Individual II-2 of Family 1
(A) Midsagittal T1-weighted MRI of the brain shows a marked thin corpus callosum (arrow).
(B) Transverse T2-weighted MRI of the brain shows subtle white-matter hyperintensities (arrows).
(C) Proton MRS obtained at a magnetic field of 1.5 tesla. Voxel was fixed just cranial of the basal-ganglia and thalamus area, of which proton MRS at long echo time (144 ms) was obtained. It shows the prominent pathologic lipid peak at 1.3 ppm (arrow), apart from the common spectral peaks of choline (Cho), creatine (Cr), and N-acetylaspartate (NAA).
(D) Sagittal T2-weighted MRI of the spine shows a spinal syrinx (arrow). Similar brain abnormalities and proton MRS were found in the other families (Figures S3–S7).
Four of the six mutations identified are predicted to introduce a premature termination codon into the DDHD2 mRNA open reading frame. Three of these are positioned more than 55 nt before the last exon-exon boundary and hence might give rise to nonsense-mediated RNA decay (NMD). To test this prediction, we measured DDHD2 mRNA levels in Epstein-Barr-virus-transformed lymphoblastoid cell lines of the affected individuals from family 1 and 4 and in a cultured fibroblast cell line from individual IV-10 in family 3 by real-time quantitative PCR analysis. We observed that in affected individuals of family 1, expression levels of DDHD2 mRNA were 84% of those of the eight control individuals (p = 0.04; Figure S8). Because NMD usually results in 15%–58% expression as compared to controls,17,18 this suggests NMD for only one of the two alleles, which is in line with the fact that one of the mutations, c.2057delA, is located in the last coding exon and hence does not target the mRNA for NMD. This mutation, however, is predicted to alter the protein sequence of the last 14 amino acids, of which seven are conserved down to the Drosophila melanogaster, of the DDHD2 domain, (Figure S2B). For family 4, 46% expression of DDHD2 mRNA levels was observed (p < 0.0001), which suggests that both alleles are subject to NMD. For individual IV-10 of family 3, we found that DDHD2 mRNA expression was eight times lower than in two healthy control samples. As expected for mutations that result in NMD, DDHD2 mRNA levels in families 1 and 4 were restored to normal when cells of affected individuals were treated with protein-translation inhibitor cycloheximide (Figure S8). Further mRNA studies comparing DDHD2 expression levels among different human tissues showed markedly high expression in the adult CNS, which is in line with the observed phenotypic features (Figure S9).
DDHD2 (DDHD-domain-containing 2) belongs to the mammalian intracellular phospholipase A1 (iPLA1) family. This protein family consists of three paralogs—DDHD1, DDHD2, and SEC23IP—that share a conserved lipase motif (GxSxG) and a DDHD domain. They exhibit enzymatic activity and hydrolyze an acyl group of phospholipids at the sn-1 position. All three members have been implicated in organelle biogenesis and membrane trafficking.19–22 From the N terminus to the C terminus, DDHD2 contains a WWE domain, a GxSxG lipase motif, a sterile alpha motif (SAM), and a DDHD domain (Figure 1E). The DDHD domain is a multifunctional domain that, together with the SAM domain, is essential for its phospholipase activity.19 DDHD2 preferentially hydrolyzes phosphatidic acid but also exhibits activity toward several other phospholipids, such as phosphatidylethanolamine. The protein localizes to the cis-Golgi and also to the endoplasmic-reticulum (ER)-Golgi intermediate compartment.19,21,23 Furthermore, RNA-interference experiments in cellular systems have indicated functions for DDHD2 in transport from the Golgi to the plasma membrane.19 In view of this, it has been hypothesized that DDHD2 determines (local) membrane curvature and facilitates membrane and vesicle fusion by the modification of membranes through phospholipid hydrolysis.23
Given the function of DDHD2 in phospholipid metabolism and the observed lipid accumulation by brain proton MRS, we investigated fibroblasts of affected individuals for abnormalities in lipid metabolism. Oil-red-O staining of fibroblasts from individuals II-1 of family 1, II-2 of family 2, and II-1 of family 4 showed no difference in appearance and number of lipid droplets between affected individuals (n = 3) and controls (n = 3) (Figure S10). We also investigated the effect of the mutations on cellular organelle morphology because DDHD2 has been described as an intracellular-transport protein involved in organelle biogenesis.19–22 Electron microscopy of fibroblasts from individuals II-1 of family 1, IV-10 of family 3, and II-1 of family 4 showed a normal appearance of the ER, golgi, mitochondria, and nucleus in fibroblasts of affected individuals (n = 3) and no difference in organelle distribution between fibroblasts of affected and control individuals (n = 4) (Figure S11). In all fibroblast cell lines, dense lysosomes and small empty vacuoles could be found in various amounts; however, in a minority of cells from individual II-1 of family 1, large empty vacuoles were localized at the cellular membrane and were surrounded by a single membrane and glycogen (Figure S11I). These large vacuoles were not observed in fibroblast cells of any of the other affected individuals or in the control individuals and are thus of uncertain significance. These results do not support gross abnormalities in lipid metabolism or organelle morphology in cultured fibroblast cells of affected individuals.
To further demonstrate an essential function for DDHD2 in the CNS, we targeted the Drosophila DDHD2 ortholog, CG8552, by using the UAS-Gal4 system24 and inducible RNA interference (RNAi)25 with the pan-neuronal UAS-dicer2; elav-Gal4 driver. Of note, CG8552 (from here on referred to as Ddhd) is equally related to DDHD2 and its paralog SEC23IP, encoding one of the other human intracellular iPLAs1. Three different Ddhd RNAi lines (vdrcGD35956, vdrcGD35957, and vdrcKK108121), representing two nonoverlapping RNAi target sequences, were utilized and compared to two control fly lines that represent the same genetic background as the mutant lines (vdrcGD60000 for vdrcGD35956 and vdrcGD35957; vdrcKK60100 for vdrcKK108121). The effect of neuron-specific Ddhd knockdown on synaptic and subsynaptic organization was studied at the neuromuscular junction (NMJ). The Drosophila larval NMJ is a well-established synaptic model system that shares major features with central excitatory synapses in the mammalian brain26 and has successfully been used for characterizing a number of Drosophila models of neurological diseases, including HSP and ID disorders.27–30 Staining of these synapses with an antibody against the scaffolding protein disc large 1 (dlg1) highlights the overall NMJ morphology. Quantitative measurement of the amount of synaptic branches and branching points revealed normal overall architecture of synaptic terminals in all three knockdown conditions. Synaptic terminals of one of the knockdown lines (vdrcKK108121) were smaller, as reflected in both area (91%, p = 0.012) and length (88%, p = 0.001) (Figure S12). We also determined the amount of chemical synapses, so-called active zones, within synaptic terminals, which represent the presynaptic sites of neurotransmitter release. Active zones were visualized with an antibody against the active-zone component bruchpilot (brp). Knockdown of Ddhd resulted in a mild but highly significant decrease in active-zone number per synaptic terminal as compared to the appropriate genetic-background controls. This phenotype was consistent in all three RNAi lines (vdrcGD35956, 90%, p = 0.029; vrdcGD35957, 80%, p < 0.0001; and vdrcKK108121, 88%, p = 0.002 [Figure 3]). No obvious motor abnormalities were observed in the three knockdown lines of 2- and 10-day-old flies. Because brp and active zones are crucial for synaptic transmission and plasticity,31 these observations support an essential role for human DDHD2 in synaptic organization and transmission.
Figure 3.
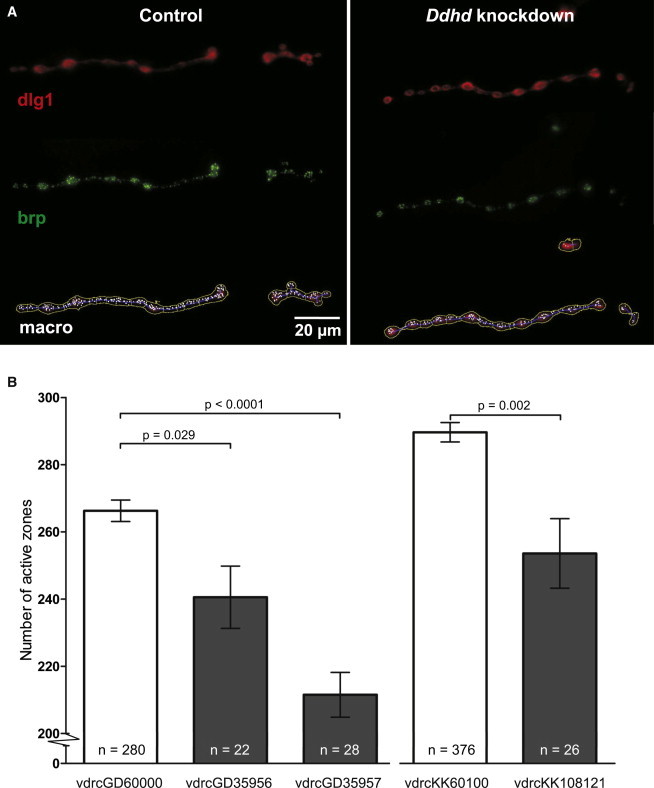
Synapse Morphology and Organization at the Drosophila NMJ of Control and Ddhd-Knockdown Flies
Three different RNAi lines, vdrcGD35956, vdrcGD35957, and vdrcKK108121, from the Vienna Drosophila Research Center, were used and compared to their genetic background lines, vdrcGD60000 (for vdrcGD35956 and vdrcGD35957) and vdrcKK60100 (for vdrcKK108121). RNAi was induced with the pan-neuronal UAS-dicer2; elav-Gal4 driver. Drosophila muscle 4 type 1b NMJs were analyzed as previously described.30
(A) Anti-dlg1 (upper panel) and anti-brp immunolabeling (middle panel) at the NMJ of control (vdrcGD60000) and Ddhd-knockdown (vdrcGD35956) larvae, as well as output of computer-assisted analysis with an in house-developed macro (bottom panel). Each white dot represents one active zone.
(B) Quantification of active zones shows a significant reduction in all three RNAi lines compared to their genetic-background controls. The p values are from two-sided t tests. Error bars indicate the SEM. The following abbreviation is used: n, number of quantified synaptic terminals.
We also compared the clinical characteristics of the emerging phenotype of recessive mutations in DDHD2 to complex HSP phenotypes that give rise to autosomal-recessive spastic paraplegia with a thin corpus callosum and white-matter abnormalities, i.e., SPG11, SPG15, SPG21, SPG44, and SPG47. This shows a clear clinical overlap between the phenotype of recessive DDHD2 mutations and those of SPG11 (mutations in SPG11), SPG15 (mutations in ZFYVE26), and SPG47 (mutations in AP4B1). However, individuals with both SPG11 and SPG15 show, in general, a later age of onset of spasticity, namely in the second decade of life. Although individuals with SPG47 do show onset in the first decade of life, they present with more severe ID than do the affected individuals in our families. The overlap with individuals with SPG21 and SPG44 is limited to the brain pattern of a thin corpus callosum and white-matter abnormalities.5 The proteins encoded by SPG11, ZFYVE26, and AP4B1—spatascin, spastizin, and AP4B1 respectively—are all proposed to be involved in intracellular trafficking.3,32,33 The overlap in function between these three proteins and DDHD2 might relate to the phenotypic overlap. Notably, abnormal lipid accumulation on MRS has not been reported in any of these complex autosomal-recessive HSPs,34–36and this finding therefore represents a valuable parameter for distinguishing the phenotype of recessive DDHD2 mutations from those of the other complex HSPs.
In conclusion, we identified recessive mutations in DDHD2, encoding an iPLA1, and have defined a complex form of HSP, designated SPG54. The core phenotype of the mutations in DDHD2 consists of very early onset of spasticity, ID, and a specific pattern of structural and metabolic brain abnormalities; the latter represents a useful distinguishing biomarker in clinical evaluation. An essential role for DDHD2 in lipid metabolism in the CNS is underlined by lipid accumulation that we observed in the brains of affected individuals, by markedly high expression of DDHD2 mRNA in the human CNS, and by the reduced number of active zones at synaptic terminals of the Drosophila Ddhd-knockdown nervous system.
Acknowledgments
We are grateful to the studied individuals and their families for their support and cooperation. We thank Prof. N. Knoers for her contribution to the collection of the follow-up cohort and A. Heister for homozygosity analysis in family 4. This work was completed with collaboration from the FORGE Canada Consortium—supported by the Government of Canada through Genome Canada, the Canadian Institutes of Health Research, and the Ontario Genomics Institute (OGI-049)—as well as from the Netherlands Organization for Health Research and Development (VIDI grants 917-86-319 to B.B.A.d.V. and 917-96-346 to A.S.), the GENCODYS project (EU-7th-2010-241995 to H.v.B., A.S., A.T.V.v.S., and B.B.A.d.V.), and the Dutch Brain Foundation (2010(1)-30 to A.P.M.d.B. and 2009(1)-22 to B.B.A.d.V.). The laboratories of L.A. and B.R.A. are funded by the Dubai Harvard Foundation for Medical Research and the United Arab Emirates University.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
FASTX-Toolkit, http://hannonlab.cshl.edu/fastx_toolkit/
NHLBI Exome Variant Server Exome Sequencing Project, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Kenwrick S., Ionasescu V., Ionasescu G., Searby C., King A., Dubowitz M., Davies K.E. Linkage studies of X-linked recessive spastic paraplegia using DNA probes. Hum. Genet. 1986;73:264–266. doi: 10.1007/BF00401241. [DOI] [PubMed] [Google Scholar]
- 2.Schüle R., Schöls L. Genetics of hereditary spastic paraplegias. Semin. Neurol. 2011;31:484–493. doi: 10.1055/s-0031-1299787. [DOI] [PubMed] [Google Scholar]
- 3.Blackstone C., O’Kane C.J., Reid E. Hereditary spastic paraplegias: Membrane traffic and the motor pathway. Nat. Rev. Neurosci. 2011;12:31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Simpson M.A., Cross H., Proukakis C., Pryde A., Hershberger R., Chatonnet A., Patton M.A., Crosby A.H. Maspardin is mutated in mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am. J. Hum. Genet. 2003;73:1147–1156. doi: 10.1086/379522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orthmann-Murphy J.L., Salsano E., Abrams C.K., Bizzi A., Uziel G., Freidin M.M., Lamantea E., Zeviani M., Scherer S.S., Pareyson D. Hereditary spastic paraplegia is a novel phenotype for GJA12/GJC2 mutations. Brain. 2009;132:426–438. doi: 10.1093/brain/awn328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Abou Jamra R., Philippe O., Raas-Rothschild A., Eck S.H., Graf E., Buchert R., Borck G., Ekici A., Brockschmidt F.F., Nöthen M.M. Adaptor protein complex 4 deficiency causes severe autosomal-recessive intellectual disability, progressive spastic paraplegia, shy character, and short stature. Am. J. Hum. Genet. 2011;88:788–795. doi: 10.1016/j.ajhg.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer P., Leshinsky-Silver E., Blumkin L., Schlipf N., Schröder C., Schicks J., Lev D., Riess O., Lerman-Sagie T., Schöls L. Mutation in the AP4B1 gene cause hereditary spastic paraplegia type 47 (SPG47) Neurogenetics. 2012;13:73–76. doi: 10.1007/s10048-012-0314-0. [DOI] [PubMed] [Google Scholar]
- 8.Al-Yahyaee S., Al-Gazali L.I., De Jonghe P., Al-Barwany H., Al-Kindi M., De Vriendt E., Chand P., Koul R., Jacob P.C., Gururaj A. A novel locus for hereditary spastic paraplegia with thin corpus callosum and epilepsy. Neurology. 2006;66:1230–1234. doi: 10.1212/01.wnl.0000208501.52849.dd. [DOI] [PubMed] [Google Scholar]
- 9.Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 10.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K., Li M., Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alazami A.M., Adly N., Al Dhalaan H., Alkuraya F.S. A nullimorphic ERLIN2 mutation defines a complicated hereditary spastic paraplegia locus (SPG18) Neurogenetics. 2011;12:333–336. doi: 10.1007/s10048-011-0291-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yıldırım Y., Orhan E.K., Iseri S.A., Serdaroglu-Oflazer P., Kara B., Solakoğlu S., Tolun A. A frameshift mutation of ERLIN2 in recessive intellectual disability, motor dysfunction and multiple joint contractures. Hum. Mol. Genet. 2011;20:1886–1892. doi: 10.1093/hmg/ddr070. [DOI] [PubMed] [Google Scholar]
- 16.Willemsen M.A., IJlst L., Steijlen P.M., Rotteveel J.J., de Jong J.G., van Domburg P.H., Mayatepek E., Gabreëls F.J., Wanders R.J. Clinical, biochemical and molecular genetic characteristics of 19 patients with the Sjögren-Larsson syndrome. Brain. 2001;124:1426–1437. doi: 10.1093/brain/124.7.1426. [DOI] [PubMed] [Google Scholar]
- 17.Coene K.L., Roepman R., Doherty D., Afroze B., Kroes H.Y., Letteboer S.J., Ngu L.H., Budny B., van Wijk E., Gorden N.T. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am. J. Hum. Genet. 2009;85:465–481. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wortmann S.B., Vaz F.M., Gardeitchik T., Vissers L.E., Renkema G.H., Schuurs-Hoeijmakers J.H., Kulik W., Lammens M., Christin C., Kluijtmans L.A. Mutations in the phospholipid remodeling gene SERAC1 impair mitochondrial function and intracellular cholesterol trafficking and cause dystonia and deafness. Nat. Genet. 2012;44:797–802. doi: 10.1038/ng.2325. [DOI] [PubMed] [Google Scholar]
- 19.Inoue H., Baba T., Sato S., Ohtsuki R., Takemori A., Watanabe T., Tagaya M., Tani K. Roles of SAM and DDHD domains in mammalian intracellular phospholipase A1 KIAA0725p. Biochim. Biophys. Acta. 2012;1823:930–939. doi: 10.1016/j.bbamcr.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 20.Nakajima K., Sonoda H., Mizoguchi T., Aoki J., Arai H., Nagahama M., Tagaya M., Tani K. A novel phospholipase A1 with sequence homology to a mammalian Sec23p-interacting protein, p125. J. Biol. Chem. 2002;277:11329–11335. doi: 10.1074/jbc.M111092200. [DOI] [PubMed] [Google Scholar]
- 21.Sato S., Inoue H., Kogure T., Tagaya M., Tani K. Golgi-localized KIAA0725p regulates membrane trafficking from the Golgi apparatus to the plasma membrane in mammalian cells. FEBS Lett. 2010;584:4389–4395. doi: 10.1016/j.febslet.2010.09.047. [DOI] [PubMed] [Google Scholar]
- 22.Tani K., Mizoguchi T., Iwamatsu A., Hatsuzawa K., Tagaya M. p125 is a novel mammalian Sec23p-interacting protein with structural similarity to phospholipid-modifying proteins. J. Biol. Chem. 1999;274:20505–20512. doi: 10.1074/jbc.274.29.20505. [DOI] [PubMed] [Google Scholar]
- 23.Morikawa R.K., Aoki J., Kano F., Murata M., Yamamoto A., Tsujimoto M., Arai H. Intracellular phospholipase A1gamma (iPLA1gamma) is a novel factor involved in coat protein complex I- and Rab6-independent retrograde transport between the endoplasmic reticulum and the Golgi complex. J. Biol. Chem. 2009;284:26620–26630. doi: 10.1074/jbc.M109.038869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 25.Dietzl G., Chen D., Schnorrer F., Su K.C., Barinova Y., Fellner M., Gasser B., Kinsey K., Oppel S., Scheiblauer S. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- 26.Koh Y.H., Gramates L.S., Budnik V. Drosophila larval neuromuscular junction: molecular components and mechanisms underlying synaptic plasticity. Microsc. Res. Tech. 2000;49:14–25. doi: 10.1002/(SICI)1097-0029(20000401)49:1<14::AID-JEMT3>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 27.Bayat V., Jaiswal M., Bellen H.J. The BMP signaling pathway at the Drosophila neuromuscular junction and its links to neurodegenerative diseases. Curr. Opin. Neurobiol. 2011;21:182–188. doi: 10.1016/j.conb.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu Z., Huang Y., Zhang Y., Chen D., Zhang Y.Q. Drosophila Acyl-CoA synthetase long-chain family member 4 regulates axonal transport of synaptic vesicles and is required for synaptic development and transmission. J. Neurosci. 2011;31:2052–2063. doi: 10.1523/JNEUROSCI.3278-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schenck A., Bardoni B., Langmann C., Harden N., Mandel J.L., Giangrande A. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron. 2003;38:887–898. doi: 10.1016/s0896-6273(03)00354-4. [DOI] [PubMed] [Google Scholar]
- 30.Zweier C., de Jong E.K., Zweier M., Orrico A., Ousager L.B., Collins A.L., Bijlsma E.K., Oortveld M.A., Ekici A.B., Reis A. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am. J. Hum. Genet. 2009;85:655–666. doi: 10.1016/j.ajhg.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wichmann C., Sigrist S.J. The active zone T-bar—a plasticity module? J. Neurogenet. 2010;24:133–145. doi: 10.3109/01677063.2010.489626. [DOI] [PubMed] [Google Scholar]
- 32.Burgos P.V., Mardones G.A., Rojas A.L., daSilva L.L., Prabhu Y., Hurley J.H., Bonifacino J.S. Sorting of the Alzheimer’s disease amyloid precursor protein mediated by the AP-4 complex. Dev. Cell. 2010;18:425–436. doi: 10.1016/j.devcel.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Murmu R.P., Martin E., Rastetter A., Esteves T., Muriel M.P., El Hachimi K.H., Denora P.S., Dauphin A., Fernandez J.C., Duyckaerts C. Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol. Cell. Neurosci. 2011;47:191–202. doi: 10.1016/j.mcn.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 34.Dreha-Kulaczewski S., Dechent P., Helms G., Frahm J., Gärtner J., Brockmann K. Cerebral metabolic and structural alterations in hereditary spastic paraplegia with thin corpus callosum assessed by MRS and DTI. Neuroradiology. 2006;48:893–898. doi: 10.1007/s00234-006-0148-2. [DOI] [PubMed] [Google Scholar]
- 35.Hobson G.M., Garbern J.Y. Pelizaeus-Merzbacher disease, Pelizaeus-Merzbacher-like disease 1, and related hypomyelinating disorders. Semin. Neurol. 2012;32:62–67. doi: 10.1055/s-0032-1306388. [DOI] [PubMed] [Google Scholar]
- 36.Stromillo M.L., Malandrini A., Dotti M.T., Battaglini M., Borgogni F., Tessa A., Storti E., Denora P.S., Santorelli F.M., Gaudiano C. Structural and metabolic damage in brains of patients with SPG11-related spastic paraplegia as detected by quantitative MRI. J. Neurol. 2011;258:2240–2247. doi: 10.1007/s00415-011-6106-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.