

Abstract
We examined the burden of large, rare, copy-number variants (CNVs) in 192 individuals with renal hypodysplasia (RHD) and replicated findings in 330 RHD cases from two independent cohorts. CNV distribution was significantly skewed toward larger gene-disrupting events in RHD cases compared to 4,733 ethnicity-matched controls (p = 4.8 × 10−11). This excess was attributable to known and novel (i.e., not present in any database or in the literature) genomic disorders. All together, 55/522 (10.5%) RHD cases harbored 34 distinct known genomic disorders, which were detected in only 0.2% of 13,839 population controls (p = 1.2 × 10−58). Another 32 (6.1%) RHD cases harbored large gene-disrupting CNVs that were absent from or extremely rare in the 13,839 population controls, identifying 38 potential novel or rare genomic disorders for this trait. Deletions at the HNF1B locus and the DiGeorge/velocardiofacial locus were most frequent. However, the majority of disorders were detected in a single individual. Genomic disorders were detected in 22.5% of individuals with multiple malformations and 14.5% of individuals with isolated urinary-tract defects; 14 individuals harbored two or more diagnostic or rare CNVs. Strikingly, the majority of the known CNV disorders detected in the RHD cohort have previous associations with developmental delay or neuropsychiatric diseases. Up to 16.6% of individuals with kidney malformations had a molecular diagnosis attributable to a copy-number disorder, suggesting kidney malformations as a sentinel manifestation of pathogenic genomic imbalances. A search for pathogenic CNVs should be considered in this population for the diagnosis of their specific genomic disorders and for the evaluation of the potential for developmental delay.
Introduction
Congenital malformations of the kidney and urinary tract are present in 3–7 out of 1,000 births,1,2 accounting for 23% of birth defects.3 These malformations account for 40%–50% of pediatric and 7% of adult end-stage renal disease worldwide.4–6 Among these malformations, renal aplasia, agenesis, hypoplasia, and dysplasia (referred to hereafter as renal hypodysplasia [RHD]) represent severe forms of disease with a profound impact on long-term renal survival.6 Currently, the diagnosis is based on prenatal or postnatal imaging studies demonstrating absent or small kidneys with or without additional urinary-tract defects. The etiology of the majority of cases remains unknown. However, multiple lines of evidence suggest a strong genetic contribution to the pathogenesis of these birth defects. For example, many cytogenetic abnormalities and genetic syndromes are associated with RHD, and mutations in genes (e.g., PAX2 [MIM 167409] or HNF1B [MIM 189907]) associated with syndromic forms of disease are detected in up to 10% of individuals with kidney malformations.7–11 Moreover, many familial forms of disease have been reported, and multiple loci have been implicated.12–14 These data suggest that many individuals with RHD have a specific genetic diagnosis that cannot be discerned by clinical evaluation alone.
Recent studies have shown that copy-number variations (CNVs) are a common feature of the human genome.15,16 Rare CNVs, identified by array-based technologies, have been implicated in the pathogenesis of many developmental disorders, such as neuropsychiatric diseases or craniofacial malformations.17–22 It is not known whether CNVs similarly contribute to congenital kidney defects. We performed a large systematic survey of CNV burden in children with congenital renal agenesis and hypodysplasia.
Material and Methods
Cohorts
The discovery cohort (n = 192) and the first replication cohort (n = 196) consisted of white European affected individuals recruited from pediatric centers in Italy, Poland, Macedonia, Croatia, and the Czech Republic (Table S1, available online). All cases were unrelated.
Inclusion criteria included the presence of a primary renal-parenchyma defect—such as renal agenesis, a congenital solitary kidney or renal hypodysplasia (finding of a small or cystic kidney for age)—documented by prenatal or postnatal imaging studies, such as an ultrasound, a computed-tomography scan, or a renal isotopic scan. Additional urinary-tract and extra-urinary-tract defects were also documented. Additional detected urinary-tract defects included vesicoureteral reflux, duplicated ureters, and ureteropelvic-junction obstruction. Extra-urinary-tract manifestations detected in the cohort included cardiac (e.g., atrial or ventricular septal defects), gastrointestinal (e.g., pyloric stenosis or anal atresia), neurologic (e.g., developmental delay or a seizure disorder), genital (e.g., septate uterus), craniofacial (e.g., cleft lip), and skeletal (e.g., brachydactyly) defects. A family history of nephropathy was obtained.
The second replication cohort consisted of 134 multiethnic North American individuals (63% white, 23% African American, and 10% admixed [Table S1]) diagnosed with RHD at the Children’s Hospital of Philadelphia (CHOP). Individuals were identified on the basis of International Statistical Classification of Diseases and Related Health Problems version 9 (ICD-9) codes from a cohort of over 31,638 children and young adults assembled by the Center for Applied Genomics. Chart review and evaluation of electronic medical records were performed for further validation of the ICD-9 codes.
The study was approved by the institutional review boards at Columbia University and the University of Pennsylvania, as well as local ethics review committees in Genoa, Brescia, Parma, Foggia and Milan (Italy), Poznan (Poland), Skopje (Macedonia), Split (Croatia), and Olomouc (Czech Republic).
Controls
The control group consisted of 13,839 anonymized adults and children selected from six cohorts of European (80.4%), Asian (13.4%), and African American ancestry (6.1%) after stringent quality control. These cohorts were genotyped on high-density Illumina platforms as cases or controls for genetic studies of complex traits not related to any developmental phenotypes (Table S2).
Genotyping, CNV Detection, and Burden Analysis
Genomic DNA was purified from peripheral-blood samples collected after informed consent. None of the 522 RHD cases was screened for mutations in known genes, such as PAX2, HNF1B, EYA1. Genome-wide genotyping for CNV analysis was performed with different Illumina platforms (Hap550v1 or higher, Illumina, see Table S3), and genotype calls and quality-control analyses were performed with GenomeStudio v.2010.3 (Illumina) and PLINK software.23
The CNV calls were determined with generalized genotyping methods implemented in the PennCNV program.24 The CNVs were mapped to the human reference genome hg18 and annotated with UCSC RefGene and RefExon (CNVision program25). On the basis of validation studies, we only included CNVs with confidence scores > 30 in the analyses (see Supplemental Material and Methods). CNV frequencies were calculated on the basis of the entire control data set of 13,839 individuals. For the analysis of overlapping events, CNVs were defined as identical if they fulfilled three criteria: (1) same CNV state, (2) ≤30% difference in length, and (3) >70% overlap in span. All CNVs with <70% overlap were not considered identical.
To compare the burden of large, rare CNVs, we utilized a subset of 4,733 controls matched for ethnicity and genotyping platform to cases in the discovery cohort (Illumina Hap-550, 610-Quad or 660W). We selected 4,733 controls from the Glasgow-Malmo Hypertension study, the CHOP CNV study, and the Parkinson Disease in Ashkenazi Jewish populations study in order to exclude individuals of African American, Asian, and Hispanic descent (see Supplemental Material and Methods). Criteria for the inclusion of CNVs for the burden analysis included: (1) confidence score > 30, (2) number of SNPs per CNV > 5, (3) CNV size > 100 kb, (4) CNV frequency < 1% in the total sample set, and (5) no overlap with any known common (frequency > 1%) CNVs. We used Fisher’s exact test (R v.2.12) for testing differences in the distributions of CNV type and CNV size. In addition, we calculated CNV metrics per genome and compared distributions by using nonparametric statistics (the Mann-Whitney U test) and empirical p values. We also examined the population frequency of the largest CNV per genome by using a log-rank test (SPSS IBM v.19). The proportions of cases and controls with the largest CNVs at a given threshold were compared with Fisher’s exact test.
Finally, to address the potential confounding effects of population stratification on CNV-burden analysis, we also performed genetic matching of RHD cases with controls (see in Supplemental Material and Methods and Tables S5 and S6). SNP genotyping data from the discovery cohort of 192 RHD cases have been deposited in the dbGaP repository under accession number phs000565.v1.1.
CNV Annotation and Confirmation
We annotated all rare CNVs across public databases (Gene Reviews, Decipher, Online Mendelian Inheritance in Man, and PubMed) to identify known genomic disorders. To select novel, rare events, we eliminated all CNVs that had identical overlaps in controls or that were encompassed within larger CNVs present at a frequency higher than 0.025% in controls. (In this study, we use “novel” to describe those disorders and variants that, to the best of our knowledge, are not present in any database or in the literature.) Rare or novel CNVs were also annotated against ECARUCA (European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations), a database of rare cytogenetic abnormalities.
Results
CNV-Burden Analysis
The discovery and replication cohorts are described in Tables S1–S3. CNV analysis identified all large, rare CNVs (defined as size > 100 kb and frequency < 1% across the entire population). To avoid the confounding effects of ethnicity or genotyping platform on the CNV-burden analysis, we compared the discovery cohort to a subset of 4,733 controls matched for ethnicity and genotyping platform (Table 1).
Table 1.
Comparison of Global CNV Counts between 192 RHD Cases and 4,733 Controls Matched for Ethnicity and Genotyping Platform
| Global CNV Metrics | RHD Cases (n = 192) | Controls (n = 4,733) | p Value (Exact Test) |
|---|---|---|---|
| Total number of rare CNVs | ncnv = 351 | ncnv = 7,970 | - |
| Size Distribution of All CNVs | |||
| 100–250 kb | 168 (47.9%) | 4,908 (61.6%) | 4.8 × 10−11 |
| 250–500 kb | 107 (30.5%) | 2,234 (28.0%) | |
| 500–1,000 kb | 52 (14.8%) | 673 (8.4%) | |
| >1,000 kb | 24 (6.8%) | 155 (1.9%) | |
| Size Distribution of All Deletions | |||
| 100–250 kb | 77 (56.2%) | 3,251 (66.9%) | 1.2 × 10−11 |
| 250–500 kb | 28 (20.4%) | 1,238 (25.5%) | |
| 500–1,000 kb | 16 (11.7%) | 317 (6.5%) | |
| >1,000 kb | 16 (11.7%) | 54 (1.1%) | |
| Size Distribution of Gene-Disrupting Deletions | |||
| 100–250 kb | 41 (47.1%) | 2,430 (65.6%) | 7.9 × 10−13 |
| 250–500 kb | 20 (23.0%) | 990 (26.7%) | |
| 500–1,000 kb | 11 (12.6%) | 240 (6.5%) | |
| >1,000 kb | 15 (17.2%) | 47 (1.3%) | |
| Size Distribution of All Duplications | |||
| 100–250 kb | 91 (42.5%) | 1,657 (53.3%) | 0.011 |
| 250–500 kb | 79 (36.9%) | 996 (32.0%) | |
| 500–1,000 kb | 36 (16.8%) | 356 (11.4%) | |
| >1,000 kb | 8 (3.7%) | 101 (3.2%) | |
| Size Distribution of Gene-Disrupting Duplications | |||
| 100–250 kb | 65 (43.9%) | 1,255 (51.2%) | 0.010 |
| 250–500 kb | 44 (29.7%) | 802 (32.7%) | |
| 500–1,000 kb | 32 (21.6%) | 295 (12.0%) | |
| >1,000 kb | 7 (4.7%) | 97 (4.0%) | |
The following abbreviations are used: CNV, copy-number variant; and RHD, renal hypodysplasia.
The frequency of rare CNVs was only nominally higher in cases than in controls (77% versus 70%, p = 0.036). However, RHD cases were significantly enriched with larger events (p = 4.8 × 10−11 [Table 1 and Figure 1A]). The enrichment of large CNVs among cases was most evident for gene-disrupting events (p = 1.8 × 10−5) and particularly for large deletions (p = 7.9 × 10−13 [Table 1]). For example, 29.2% of gene-disrupting deletions were larger than 500 kb in cases, whereas only 7.3% were larger than 500 kb in controls (Table 1).
Figure 1.
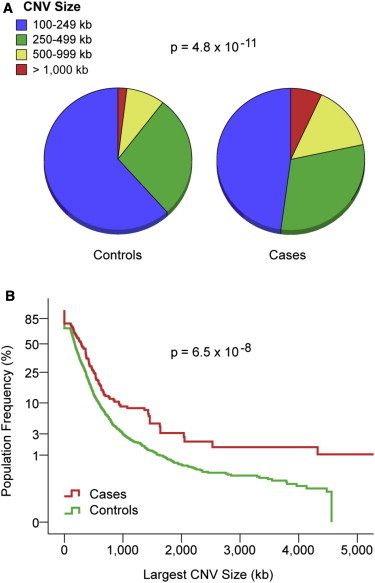
CNV Burden Comparison between Cases and Controls
(A) Distribution of large (>100 kb), rare (<1%) CNVs by size in 192 RHD cases and 4,733 controls matched for ethnicity and genotyping platform.
(B) Comparison of the largest CNV per genome shows enrichment of larger events among RHD cases.
The y axis describes the proportion of individuals with CNV size above each size threshold (x axis). Note that the y axis in (B) is on an exponential scale. The p values for differences in the distribution are indicated.
To verify that the excess of large CNVs was not attributable to a few cases with an unusually high CNV load, we further calculated CNV burden per genome (Table 2). Consistent with the analysis of global CNV distribution, the CNV rate per genome was not different between cases and controls (1.83 versus 1.68, p = 0.21), but cases demonstrated a significantly greater average CNV size (366.1 kb versus 197.1 kb, p = 1.5 × 10−6), average total CNV span (868 kb versus 476 kb, p = 2.1 × 10−5), and average largest CNV size per genome (518.5 kb versus 260.4 kb, p = 2.71 × 10−6). Comparison of the largest CNV per genome showed clear differences above the 250 kb threshold: 54.7% of cases harbored a CNV greater than 250 kb, whereas only 37.5% of controls did (odds ratio = 2.01, p = 2.2 × 10−6 [Table 2 and Figure 1B]), suggesting that as much as 17.2% of RHD cases in this cohort is attributable to CNVs larger than 250 kb.
Table 2.
Comparison of CNV Burden per Genome between 192 RHD Cases and 4,733 Controls Matched for Ethnicity and Genotyping Platform
| Cases (n = 192) | Controls (n = 4,733) | Asymptotic p Valuea | Empiric p Valueb | OR (95% CI) | p Value (Fisher exact) | |
|---|---|---|---|---|---|---|
| Metric | ||||||
| Average CNV rate | 1.83 | 1.68 | 0.13 | 0.21 | - | - |
| Average CNV size (median) in kb | 366.1 (218.7) | 197.1 (161.1) | 1.5 × 10−6 | <1 × 10−6 | - | - |
| Average largest CNV size (median) in kb | 518.5 (289.6) | 260.4 (178.5) | 2.7 × 10−6 | 3.0 × 10−6 | - | - |
| Average total CNV span (median) in kb | 868.1 (417.0) | 476.1 (234.2) | 2.1 × 10−5 | 1.9 × 10−5 | - | - |
| Distribution of the Largest CNV per Genome | ||||||
| Individuals with largest CNV size > 1,000 kb | n = 17 (18.9%) | n = 142 (3.0%) | - | - | 3.14 (1.74–5.35) | 1.4 × 10−4 |
| Individuals with largest CNV size > 500 kb | n = 49 (29.5%) | n = 629 (13.3%) | - | - | 2.24 (1.56–3.15) | 8.8 × 10−6 |
| Individuals with largest CNV size > 250 kb | n = 105 (54.7%) | n = 1,774 (37.5%) | - | - | 2.01 (1.49–2.72) | 2.2 × 10−6 |
The following abbreviations are used: OR, odds ratio; CI, confidence interval; and CNV, copy-number variant.
Nonparametric (Mann-Whitney U) test for quantitative variables, Poisson-rate ratio test for rates, and Fisher’s exact test for proportions.
Based on 1,000,000 permutations.
The burden analysis was also repeated after we genetically matched the discovery cohort with a different set of controls. This analysis confirmed a highly significant excess of large CNVs among cases, demonstrating that differences in CNV load are not due to population stratification (see Supplementary Material and Methods, Tables S4–S6, and Figures S1 and S2).
Moreover, we repeated the analysis by using only the pediatric controls from the CHOP study. The results from this analysis are nearly identical to the original findings on the larger controls data set and the genetically matched cohort that included adults, thereby ruling out bias due to the inclusion of adult controls (see Table S7 and Figure S3).
Identification of Known Copy-Number Disorders in 10.5% of RHD Cases
The consistent overrepresentation of large CNVs among cases indicated the presence of genomic disorders. We therefore annotated all large, rare CNVs that disrupted coding segments in the discovery cohort and replicated findings in two cohorts recruited from European (n = 196) and North American (n = 134) medical centers.
All together, 55/522 RHD individuals (10.5%) in the combined discovery and two replication cohorts harbored a known genomic disorder for a total of 34 distinct, known syndromes (Table 3 and Table S8). We identified CNVs diagnostic of 17 known genomic disorders in 25 (13%) cases in the discovery cohort, 21 known genomic disorders in 18 (9.2%) individuals in the European replication cohort, and 14 known genomic disorders in 12 (9%) cases in the North American cohort (Table 3). The same disorders were present in only 30 (0.2%) of 13,839 population controls (Fisher exact p value = 1.2 × 10−58 versus RHD cases). These data independently confirm that genomic disorders represent a very common etiology for RHD (Table 3).
Table 3.
Thirty-Four Known Genomic Disorders Identified in 522 RHD Cases
| Chromosomal Region | CNV Type | Start (Mb) | End (Mb) | Size (Mb) | Syndrome | Discovery (n = 192) | Replication 1 (n = 196) | Replication 2 (n = 134) | Combined (n = 522) | Controls (n = 13,839) | p Value | Prior Association with RHD/Neuropsychiatric Traits? |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1p36 | dup | 2.91 | 3.65 | 0.74 | 1p36 dup | 0 | 0 | 2 | 2 | 0 | 1.32 × 10−3 | N/Y |
| 1p22 | dup | 89.50 | 89.97 | 0.47 | 1p22.2-p31.1 dupa | 0 | 1 | 1 | 2 | 0 | 1.32 × 10−3 | N/Y |
| 1q21 | del | 144.11 | 144.63 | 0.52 | 1q21 TAR delb | 1 | 0 | 0 | 1 | 1 | 0.071 | Y/Y |
| 1q21 | del | 144.80 | 145.86 | 1.06 | 1q21 distal delb | 1 | 3 | 0 | 4 | 4 | 1.07 × 10−4 | Y/N |
| 1q43-q44 | del | 240.61 | 245.67 | 5.06 | 1q43-q44 del | 1 | 0 | 0 | 1 | 0 | 0.036 | Y/Y |
| 2q37 | dup | 240.99 | 242.44 | 1.45 | 2q37 dupc | 0 | 1 | 0 | 1 | 0 | 0.036 | Y/Y |
| 3p26 | dup | 1.35 | 2.18 | 0.83 | 3pter-p25 del | 2 | 0 | 0 | 2 | 8 | 0.049 | N/Y |
| 4p16 | del | 0.06 | 17.29 | 17.23 | Wolf-Hirschhornd | 0 | 1 | 1 | 2 | 0 | 1.32 × 10−3 | Y/Y |
| 5p15 | dup | 0.11 | 10.96 | 10.85 | 5p distal dupd | 0 | 0 | 1 | 1 | 0 | 0.036 | Y/Y |
| 5q14-q23 | del | 91.46 | 114.55 | 23.09 | 5q interstitial del | 0 | 0 | 1 | 1 | 0 | 0.036 | N/Y |
| 6q13-q14 | dup | 70.29 | 70.76 | 0.47 | 6q13-q14 del | 1 | 0 | 0 | 1 | 0 | 0.036 | Y/Y |
| 7p22 | dup | 6.82 | 7.27 | 0.45 | 7p interstitial dup | 0 | 0 | 1 | 1 | 0 | 0.036 | Y/Y |
| 7p21 | dup | 16.80 | 17.71 | 0.91 | 7p interstitial dup | 0 | 0 | 1 | 1 | 1 | 0.071 | Y/Y |
| 7p15 | del | 23.68 | 27.43 | 3.75 | 7p15.1-p21.1 del | 0 | 1 | 0 | 1 | 0 | 0.036 | Y/N |
| 7q34-q36 | del | 141.53 | 158.81 | 17.28 | 7q36 del | 1 | 0 | 1 | 2 | 0 | 1.32 × 10−3 | Y/Y |
| 8p23 | dup | 8.13 | 11.94 | 3.81 | 8p23.1 dup | 1 | 0 | 0 | 1 | 1 | 0.071 | Y/Y |
| 9p22e | del | 14.81 | 14.97 | 0.17 | 9p22.3 del | 0 | 1 | 0 | 1 | 0 | 0.036 | N/N |
| 16p13 | dup | 0.04 | 15.09 | 15.04 | 16p subtelomeric dupf | 0 | 1 | 0 | 1 | 0 | 0.036 | Y/Y |
| 16p13 | dup | 15.03 | 15.80 | 0.77 | 16p13.11 dup | 1 | 0 | 0 | 1 | 5 | 0.199 | N/Y |
| 16p11 | del | 29.55 | 31.86 | 2.31 | 16p11.2 distal del | 0 | 2 | 0 | 2 | 0 | 1.32 × 10−3 | Y/Y |
| 16p11 | dup | 29.50 | 30.05 | 0.55 | 16p11.2 distal dup | 0 | 0 | 1 | 1 | 3 | 0.138 | N/Y |
| 17p11-p12 | dup | 16.41 | 20.23 | 3.82 | Potocki-Lupski syndrome | 1 | 0 | 1 | 2 | 0 | 1.32 × 10−3 | Y/Y |
| 17q11-q12 | del | 31.89 | 33.35 | 1.46 | renal cysts and diabetes (HNF1B)g | 5 | 5 | 1 | 11 | 0 | 1.32 × 10−16 | Y/Y |
| 17q11-q12 | dup | 31.89 | 33.25 | 1.36 | 17q12 dup (HNF1B) | 1 | 0 | 0 | 1 | 1 | 0.071 | Y/Y |
| 17q21 | del | 40.94 | 41.41 | 0.47 | 17q21.31 del | 1 | 0 | 0 | 1 | 2 | 0.105 | Y/Y |
| 20p11-p13 | dup | 0.11 | 24.77 | 24.66 | 20p partial trisomya | 0 | 1 | 0 | 1 | 0 | 0.036 | Y/Y |
| 21q22 | del | 40.51 | 46.91 | 6.40 | 21q partial monosomy | 0 | 0 | 1 | 1 | 0 | 0.036 | N/Y |
| 22q11 | dup | 15.29 | 18.61 | 3.32 | 22q11.2 dup (VCFS region)c | 0 | 1 | 0 | 1 | 0 | 0.036 | Y/Y |
| 22q11 | del | 17.27 | 19.79 | 2.52 | DiGeorge/VCFS del | 3 | 1 | 0 | 4 | 0 | 1.73 × 10−6 | Y/Y |
| 22q13 | del | 42.94 | 49.52 | 6.58 | Phelan-McDermid syndromef,g | 0 | 1 | 1 | 2 | 0 | 1.32 × 10−3 | Y/Y |
| X | gain | XXY | XXY | - | Klinefelter syndrome | 1 | 0 | 0 | 1 | 0 | 0.044 | Y/Y |
| Xp22 | del | 6.46 | 8.10 | 1.64 | Xp22.31 del | 2 | 0 | 0 | 2 | 0 | 1.92 × 10−3 | Y/Y |
| Xp22 | dup | 8.19 | 8.67 | 0.48 | Kallman syndrome region (KAL1) | 2 | 1 | 0 | 3 | 4 | 1.5 × 10−3 | Y/Y |
| Xq27 | dup | 139.36 | 139.91 | 0.55 | mental retardation with panhypopituitarism syndrome | 1 | 0 | 0 | 1 | 0 | 0.044 | N/Y |
| Total number of known pathogenic CNVs | 26 | 21 | 14 | 61 | 30 | 9.9 × 10−66 | - | |||||
| Total number of individuals with at least one pathogenic CNV | 25 (13%) | 18 (9.2%) | 12 (9%) | 55 (10.5%) | 30 (0.21%) | 1.22 × 10−58 | - | |||||
CNV start and end positions are based on UCSC genome build hg18. The symbol for the causal gene at each locus is indicated when known. Fisher’s exact p values for comparison of CNV frequency between combined cohorts (n = 522) and controls (n = 13,839) are indicated. The last row compares the total number of individuals carrying at least one of the CNVs listed in this table (Fisher’s exact test). The following abbreviations are used: CNV, copy-number variation; RHD, renal hypodysplasia; dup, duplication; del, deletion; N, no; Y, yes; and VCFS, velocardiofacial syndrome.
Six individuals, corresponding to letters a–d, f, and g, were each diagnosed with two of these syndromes (e.g. “a” indicates that one individual had a 1p22.2-p31.1 deletion and 20p partial trisomy). Additional information and references are reported in Table S8.
Thirteen disorders were detected across different cohorts or affected individuals of different nationalities, implicating independent events. Deletions at the HNF1B locus in chromosomal region 17q11-12 and at the locus for DiGeorge syndrome (DGS [MIM 188400]) and velocardiofacial syndrome (VCFS [MIM 192430]) (hereafter called the DGS/VCFS locus) in chromosomal region 22q11 were the most frequent findings (11 and 4 cases, respectively). However, the majority of disorders were detected in a single individual, indicating significant genetic heterogeneity of the trait. We detected four inherited and five de novo events among the eight cases with parental DNA available in the discovery cohort. Six cases, distributed across all three cohorts, carried two known genomic imbalances. Twenty (59%) diagnostic CNVs were flanked by segmental duplications, implicating nonallelic homologous recombination as the underlying mechanism. Finally, genomic disorders were detected in individuals with isolated RHD (n = 31), as well as in those with multiorgan manifestations (n = 24).
Identification of Novel or Rare Copy-Number Disorders in up to 6.1% of RHD
After exclusion of individuals with diagnostic CNVs, there was still evidence of excess CNV burden among the RHD cases (Table S9). We therefore searched for additional novel or rare genomic disorders by identifying CNVs that were larger than 100 kb, disrupted coding segments, and were absent from or extremely rare in the 13,839 controls (CNV frequency ≤ 1:4,000).
All together, we identified 38 independent events fulfilling these criteria in 32/522 cases (6.1% of the RHD cases, Table S10), defining candidate novel genomic disorders. Similar to the situation with known disorders, the majority of imbalances were encountered in a single individual, with or without multiorgan manifestations, and among the 12 cases with parental DNA available, six CNVs occurred de novo.
If we use highly conservative criteria—selecting only CNVs that occurred de novo, were recurrent, or were larger than 1 Mb—this analysis identified 15 rare or novel genomic disorders (five recurrent duplications, three de novo deletions, two de novo duplications, and four CNVs > 1,000 kb, Table 4) in 20 RHD cases. This indicates a lower bound of 3.8% for novel or rare genomic disorders in the combined cohort.
Table 4.
Fifteen Novel or Rare Genomic Disorders Identified in 522 RHD Cases on the Basis of the Strictest Criteria
| Chromosomal Region | CNV Type | Start (Mb) | End (Mb) | Size (Mb) | Inheritance | Discovery (n = 192) | Replication 1 (n = 196) | Replication 2 (n = 134) | Combined (n = 522) | Controls (n = 13,839) | p Value |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1q32 | del | 162.68 | 163.19 | 0.51 | de novo | 1 | 0 | 0 | 1 | 0 | 0.036 |
| 2p25 | dup | 0.02 | 3.65 | 3.63 | N/A | 1 | 0 | 1 | 2 | 0 | 1.32 × 10−3 |
| 2p11 | dup | 88.16 | 89.24 | 1.08 | N/A | 0 | 0 | 1 | 1 | 0 | 0.036 |
| 3q13-q22 | del | 118.15 | 133.11 | 14.96 | de novo | 1 | 0 | 0 | 1 | 0 | 0.036 |
| 3q29 | dup | 199.17 | 199.32 | 0.15 | N/A | 1 | 1 | 0 | 2 | 3 | 0.012 |
| 4p13 | dup | 44.12 | 44.75 | 0.63 | de novo | 1 | 0 | 0 | 1 | 0 | 0.036 |
| 5q34 | dup | 159.53 | 160.58 | 1.05 | N/A | 0 | 2 | 0 | 2 | 0 | 1.32 × 10−3 |
| 7q21 | del | 79.33 | 80.91 | 1.58 | N/A | 0 | 1 | 0 | 1 | 0 | 0.036 |
| 10p11 | dup | 42.10 | 42.71 | 0.61 | N/A | 2 | 0 | 0 | 2 | 0 | 1.32 × 10−3 |
| 11p11 | dup | 49.58 | 50.52 | 0.94 | N/A | 0 | 2 | 0 | 2 | 1 | 3.86 × 10−3 |
| 12q24 | dup | 124.67 | 132.29 | 7.52 | de novo | 1 | 0 | 0 | 1 | 0 | 0.036 |
| 13q11 | del | 22.44 | 23.80 | 1.36 | de novo | 1 | 0 | 0 | 1 | 3 | 0.138 |
| 13q12 | dup | 36.28 | 37.51 | 1.23 | inherited | 1 | 0 | 0 | 1 | 0 | 0.036 |
| 16q22 | del | 73.39 | 73.90 | 0.51 | de novo | 1 | 0 | 0 | 1 | 0 | 0.036 |
| 17q25 | dup | 71.00 | 78.63 | 7.63 | N/A | 0 | 0 | 1 | 1 | 0 | 0.036 |
CNV start and end positions are based on UCSC genome build hg18. These rare CNVs were selected on the basis of a frequency < 0.025% in controls and occurrence in ≥ 2 RHD cases or on the basis of de novo status or a size > 1 Mb. A complete list of novel, rare CNVs and additional information are reported in Table S10. The following abbreviations are used: CNV, copy-number variation; del, deletion; dup, duplication; and N/A, not available.
Rare Intergenic and Single-Gene CNVs
We also searched for rare intergenic CNVs and CNVs disrupting a single gene in the discovery cohort. We identified 27 intergenic CNVs and 13 single-gene-disrupting CNVs that were absent in all 13,839 controls and in a recent study that identified CNVs at a resolution reaching 1 kb (Tables S12 and S13).26 These CNVs identify candidate genes for RHD. For example, a gene-disrupting deletion and an intergenic deletion identify EFEMP1 (RefSeq accession number NM_001039348; MIM 601548) as a potential causal gene for RHD (Tables S12 and S13).
Annotation of Genes within CNVs
We examined phenotypes resulting from inactivation of the murine orthologs of the genes located within the 72 known and candidate pathogenic CNVs. We identified 53 positional candidates whose inactivation results in kidney developmental defects in mice, and there is at least one gene implicated in kidney developmental defects in 32% of these CNV intervals (Table S8 and S10). For example, disruption of murine orthologs of KIF26B (MIM 614026)25 and PBX1 (MIM 176310)26 leads to renal agenesis or hypoplasia, suggesting that these are most likely the culprit genes within the de novo deletion in chromosomal regions 1q43-q44 and 1q32, respectively. Many of these genes are associated with both renal and neurodevelopmental defects, suggesting pleiotropism. For example, inactivation of Fgfrl1 produces kidney and brain morphological defects that recapitulate many of the clinical features of Wolf-Hirschhorn syndrome (MIM 194190).27,28 The identification of credible candidate genes within the majority of these loci further supports the pathogenicity of the imbalances.
Clinical Correlations
There were no differences in gender, ethnicity, or family history of nephropathy between individuals with or without a genomic disorder. Deletions and duplications were also similarly distributed between these two groups. However, genomic disorders were detected more frequently among cases with malformations outside the urinary tract (32/142 [22.5%]) than among those with isolated urinary-tract defects (55/380 [14.5%], Fisher’s exact p = 0.03 for comparison between the groups). Fourteen individuals (2.7% of the RHD cohort), distributed across all three cohorts, harbored two or more diagnostic or rare CNVs; nine (64%) of these individuals manifested multiorgan defects, consistent with their high CNV load (Table S11). Among the cases with ten inherited CNVs, four individuals had familial disease (the CNVs segregated with disease in three of these individuals), and one had parents with an unknown renal phenotype and an unavailable affected sibling; therefore, the segregation pattern is not discernible (Tables S8 and S10). Finally, consistent with the identification of many positional candidates involved in neurological defects, 90% of the known imbalances listed in Table 3 are associated with an increased risk of neuropsychiatric disease, such as autism, schizophrenia, intellectual disability, or seizures (e.g., 1q21 deletion [MIM 612474],21 2q37 deletion [MIM 600430],27 or Potocki-Lupski syndrome [MIM 610883]).28
Discussion
Nephrogenesis requires a complex sequence of mutually inductive signals between two intermediate mesenchymal progenitors: the metanephric mesenchyme and the ureteric bud.29 Consistent with the complex signaling cascade involved in this process, we identified very diverse genetic lesions resulting in kidney developmental defects. Our findings were robust to many alternative analyses and were consistent across all three RHD cohorts, excluding an analytic bias. The significant etiological heterogeneity of congenital kidney malformations was not detectable by clinical evaluation, and the fact that most of the structural variants were below the resolution of standard cytogenetic analysis indicates that high-resolution genomic methods are required for identifying the specific etiology of disease in the RHD population.
All together, we detected 72 distinct known or novel genomic disorders in 16.6% of RHD cases (10.5% with known disorders and 6.1% with rare or novel disorders), indicating a large proportion of rare pathogenic imbalances in this population. This number is consistent with the CNV-burden analysis, which suggested that 17.2% of RHD cases are attributable to CNVs larger than 250 kb (Table 2). These data identify candidate genes or loci that impart a large effect on RHD and most likely disrupt critical nodes in the renal developmental program. We detected a single pathogenic imbalance in most individuals (only 2.7% of cases had two or more large CNVs), suggesting a model of rare mutations with large effect. Among the 21 individuals with available parents, 11 (52%) had de novo CNVs, whereas 10 (48%) had inherited structural variants, suggesting incomplete penetrance in this second group. Strikingly, 90% of the known disorders detected in our study have been shown to predispose to developmental delay or neuropsychiatric disease, suggesting shared pathways between renal and neural developmental programs.
Rearrangements in chromosomal region 17q12 were the most common genomic disorders detected in the cohort and accounted for 2.3% of cases.30,31 HNF1B mutations, resulting in renal cysts and diabetes, are the cause of RHD at this locus. This finding is consistent with prior studies showing that HNF1B mutations are a common cause of RHD and that RHD is the most consistent and earliest manifestation of this syndrome, whereas additional phenotypes, such as diabetes or hyperuricemia, develop at a later age.8,9,11,32 Neuropsychiatric disease is also an increasingly recognized complication of rearrangements in chromosomal region 17q12.33,34 DGS/VCFS was the next most frequent disorder, consistent with the known occurrence of urologic defects in nearly 40% of individuals with this syndrome.30,31 Disruption of different genes within the DGS/VCFS locus is thought to account for the spectrum of developmental, metabolic, and immunologic defects in this syndrome, but the specific genetic lesion(s) responsible for the kidney malformations have not been clarified. Our study identified deletions in the distal 370 kb region of the DiGeorge locus (the LCRC-LCRD region) in three cases with isolated RHD, suggesting that the gene responsible for the urinary-tract defects is located in this segment. The other known syndromes occurred mostly as singleton cases. Of clinical importance, about half of the individuals with these copy-number disorders presented with isolated RHD, suggesting that kidney defects might be an early or sensitive manifestation of pathogenic genomic imbalances.
In addition to known syndromes, we also identified large, rare, gene-disrupting CNVs in another 32 individuals (6.1% of the cohort [Table 4 and Table S10]). We found evidence of 15 recurrent, de novo, or large events in 20 individuals, indicating a lower bound of 3.8% for novel or rare genomic disorders. These novel or rare CNVs share many common characteristics with the diagnostic CNVs discovered in this cohort: they have a similar proportion of deletions, duplications, and de novo events but were slightly smaller and less frequently flanked by segmental duplications, suggesting that many arise from mechanisms other than nonallelic homologous recombination. Finally, we identified many unique intergenic and single-gene-disrupting CNVs. These findings offer a list of candidate genes and genomic disorders that can be confirmed in independent human cohorts or via the creation of animal models. For example, we found two rare events involving EFEMP1, a member of the fibulin family of extracellular-matrix glycoproteins. Although a single amino acid substitution (p.Arg345Trp) has been associated with Malattia Leventinese and Doyne honeycomb retinal dystrophy in humans,35 targeted disruption in mice does not produce a retinal phenotype but rather a widespread aging phenotype with early kidney atrophy.36 Thus, loss-of-function mutations in humans could result in early arrest of kidney growth and atrophy, causing reduced kidney size (that can be diagnosed as renal hypoplasia) in childhood.
Our findings are comparable to a recent study showing that diverse pathogenic CNVs account for 14.2% of disease in a large series of children with developmental delay and/or intellectual disability and variable organ malformations.20 However, only 13 of the 34 known genomic disorders detected in the present study overlap with those identified by Cooper et al.,20 indicating both shared and distinct genetic lesions between these two traits. Our findings suggest that similar to neural development, nephrogenesis is very sensitive to variation in gene dosage, and the presence of kidney malformations should alert clinicians to the possibility of pathogenic genomic imbalances. Because kidney malformations can be detected prenatally or at birth, a CNV screen might identify the potential for complications such as developmental delay, autism, or cognitive defects before they become clinically evident. In addition to informing family discussions, identification of RHD-affected individuals with genomic imbalances can better define the burden and trajectory of disease in this subgroup. Finally, this study offers a list of candidate genes and loci that can help dissect the complex signaling pathway required for nephrogenesis.
Acknowledgments
We thank all patients and family members for their participation. This study was supported by 1R01DK080099 (A.G.G.) and Italian Telethon Foundation grant GGP08050 (G.M.G.). S.S.C. is supported by American Heart Association grant 0930151N and the American Society of Nephrology (ASN) Carl W. Gottschalk Research Scholar Grant. K.K. is supported by National Institute of Diabetes and Digestive and Kidney Diseases K23-DK090207. G.M.G. is supported by the Fondazione Malattie Renali nel Bambino. S.N.N. is supported by the ASN and Doris Duke Charitable Foundation. A.M.K., A.L.B., and The Polish Registry of Congenital Malformations are supported by the Polish Ministry of Health. We thank the HYPERGENES Consortium (HEALTH-F4-2007-201550; INTEROMICS: MIUR-CNR Italian Flagship Project) for sharing genotyping data. L.N.C. is supported by National Institutes of Health grants NS050487 and NS060113 and the Parkinson’s Disease Foundation. The Glasgow-Malmo Hypertension study was funded by the British Heart Foundation (BHF) Chair CH/98001 (A.F.D.), the BHF program RG/07/005/23633 (A.F.D. and S.P.), the BHF Special Project grant SP/08/005/25115 (A.F.D. and S.P.), and the European Community Framework 6 Network of Excellence InGenious HyperCare. The Children’s Hospital of Philadelphia (CHOP) Control Copy Number Variation dataset was supplied by the Center for Applied Genomics at CHOP through dbGaP accession number phs000199.v1.p1. The Genetic Consortium for Late Onset Alzheimer’s Disease was provided through the Division of Neuroscience at the National Institute on Aging through dbGaP accession number phs000168.v1.p1. This study used DECHIPER-generated data, provided by the Wellcome Trust. A full list of contributing centers is available at http://decipher.sanger.ac.uk or by email to decipher@sanger.ac.uk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
DECIPHER, https://decipher.sanger.ac.uk/
ECARUCA, http://umcecaruca01.extern.umcn.nl:8080/ecaruca/ecaruca.jsp
GenePaint, http://www.genepaint.org/
Genome Reviews, http://www.ebi.ac.uk/GenomeReviews/
GUDMAP Genitourinary Database Molecular Anatomy Project, http://www.gudmap.org/
National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser Home, http://www.genome.ucsc.edu/
Accession Numbers
The dbGaP accession number for the SNP genotyping data presented in this paper is phs000565.v1.1.
References
- 1.Birth Defects Monitoring Program (BDMP)/Commission on Professional and Hospital Activities (CPHA) surveillance data, 1988-1991. Teratology. 1993;48:658–675. doi: 10.1002/tera.1420480608. [DOI] [PubMed] [Google Scholar]
- 2.Livera L.N., Brookfield D.S., Egginton J.A., Hawnaur J.M. Antenatal ultrasonography to detect fetal renal abnormalities: A prospective screening programme. BMJ. 1989;298:1421–1423. doi: 10.1136/bmj.298.6685.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Loane M., Dolk H., Kelly A., Teljeur C., Greenlees R., Densem J., EUROCAT Working Group Paper 4: EUROCAT statistical monitoring: Identification and investigation of ten year trends of congenital anomalies in Europe. Birth Defects Res. A Clin. Mol. Teratol. 2011;91(Suppl 1):S31–S43. doi: 10.1002/bdra.20778. [DOI] [PubMed] [Google Scholar]
- 4.Centers for Disease Control and Prevention (CDC) State-specific trends in chronic kidney failure—United States, 1990-2001. MMWR Morb. Mortal. Wkly. Rep. 2004;53:918–920. [PubMed] [Google Scholar]
- 5.Ardissino G., Daccò V., Testa S., Bonaudo R., Claris-Appiani A., Taioli E., Marra G., Edefonti A., Sereni F., ItalKid Project Epidemiology of chronic renal failure in children: Data from the ItalKid project. Pediatrics. 2003;111:e382–e387. doi: 10.1542/peds.111.4.e382. [DOI] [PubMed] [Google Scholar]
- 6.Sanna-Cherchi S., Ravani P., Corbani V., Parodi S., Haupt R., Piaggio G., Innocenti M.L., Somenzi D., Trivelli A., Caridi G. Renal outcome in patients with congenital anomalies of the kidney and urinary tract. Kidney Int. 2009;76:528–533. doi: 10.1038/ki.2009.220. [DOI] [PubMed] [Google Scholar]
- 7.Eccles M.R., Schimmenti L.A. Renal-coloboma syndrome: A multi-system developmental disorder caused by PAX2 mutations. Clin. Genet. 1999;56:1–9. doi: 10.1034/j.1399-0004.1999.560101.x. [DOI] [PubMed] [Google Scholar]
- 8.Thomas R., Sanna-Cherchi S., Warady B.A., Furth S.L., Kaskel F.J., Gharavi A.G. HNF1B and PAX2 mutations are a common cause of renal hypodysplasia in the CKiD cohort. Pediatr. Nephrol. 2011;26:897–903. doi: 10.1007/s00467-011-1826-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weber S., Moriniere V., Knüppel T., Charbit M., Dusek J., Ghiggeri G.M., Jankauskiené A., Mir S., Montini G., Peco-Antic A. Prevalence of mutations in renal developmental genes in children with renal hypodysplasia: Results of the ESCAPE study. J. Am. Soc. Nephrol. 2006;17:2864–2870. doi: 10.1681/ASN.2006030277. [DOI] [PubMed] [Google Scholar]
- 10.Mefford H.C., Clauin S., Sharp A.J., Moller R.S., Ullmann R., Kapur R., Pinkel D., Cooper G.M., Ventura M., Ropers H.H. Recurrent reciprocal genomic rearrangements of 17q12 are associated with renal disease, diabetes, and epilepsy. Am. J. Hum. Genet. 2007;81:1057–1069. doi: 10.1086/522591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ulinski T., Lescure S., Beaufils S., Guigonis V., Decramer S., Morin D., Clauin S., Deschênes G., Bouissou F., Bensman A., Bellanné-Chantelot C. Renal phenotypes related to hepatocyte nuclear factor-1beta (TCF2) mutations in a pediatric cohort. J. Am. Soc. Nephrol. 2006;17:497–503. doi: 10.1681/ASN.2005101040. [DOI] [PubMed] [Google Scholar]
- 12.Sanna-Cherchi S., Caridi G., Weng P.L., Dagnino M., Seri M., Konka A., Somenzi D., Carrea A., Izzi C., Casu D. Localization of a gene for nonsyndromic renal hypodysplasia to chromosome 1p32-33. Am. J. Hum. Genet. 2007;80:539–549. doi: 10.1086/512248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weng P.L., Sanna-Cherchi S., Hensle T., Shapiro E., Werzberger A., Caridi G., Izzi C., Konka A., Reese A.C., Cheng R. A recessive gene for primary vesicoureteral reflux maps to chromosome 12p11-q13. J. Am. Soc. Nephrol. 2009;20:1633–1640. doi: 10.1681/ASN.2008111199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashraf S., Hoskins B.E., Chaib H., Hoefele J., Pasch A., Saisawat P., Trefz F., Hacker H.W., Nuernberg G., Nuernberg P. Mapping of a new locus for congenital anomalies of the kidney and urinary tract on chromosome 8q24. Nephrol. Dial. Transplant. 2010;25:1496–1501. doi: 10.1093/ndt/gfp650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khaja R., Zhang J., MacDonald J.R., He Y., Joseph-George A.M., Wei J., Rafiq M.A., Qian C., Shago M., Pantano L. Genome assembly comparison identifies structural variants in the human genome. Nat. Genet. 2006;38:1413–1418. doi: 10.1038/ng1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stefansson H., Rujescu D., Cichon S., Pietiläinen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., GROUP Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vacic V., McCarthy S., Malhotra D., Murray F., Chou H.H., Peoples A., Makarov V., Yoon S., Bhandari A., Corominas R. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elia J., Glessner J.T., Wang K., Takahashi N., Shtir C.J., Hadley D., Sleiman P.M., Zhang H., Kim C.E., Robison R. Genome-wide copy number variation study associates metabotropic glutamate receptor gene networks with attention deficit hyperactivity disorder. Nat. Genet. 2012;44:78–84. doi: 10.1038/ng.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brunetti-Pierri N., Berg J.S., Scaglia F., Belmont J., Bacino C.A., Sahoo T., Lalani S.R., Graham B., Lee B., Shinawi M. Recurrent reciprocal 1q21.1 deletions and duplications associated with microcephaly or macrocephaly and developmental and behavioral abnormalities. Nat. Genet. 2008;40:1466–1471. doi: 10.1038/ng.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw-Smith C., Pittman A.M., Willatt L., Martin H., Rickman L., Gribble S., Curley R., Cumming S., Dunn C., Kalaitzopoulos D. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat. Genet. 2006;38:1032–1037. doi: 10.1038/ng1858. [DOI] [PubMed] [Google Scholar]
- 23.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanders S.J., Ercan-Sencicek A.G., Hus V., Luo R., Murtha M.T., Moreno-De-Luca D., Chu S.H., Moreau M.P., Gupta A.R., Thomson S.A. Multiple recurrent de novo CNVs, including duplications of the 7q11.23 Williams syndrome region, are strongly associated with autism. Neuron. 2011;70:863–885. doi: 10.1016/j.neuron.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conrad D.F., Pinto D., Redon R., Feuk L., Gokcumen O., Zhang Y., Aerts J., Andrews T.D., Barnes C., Campbell P., Wellcome Trust Case Control Consortium Origins and functional impact of copy number variation in the human genome. Nature. 2010;464:704–712. doi: 10.1038/nature08516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galasso C., Lo-Castro A., Lalli C., Nardone A.M., Gullotta F., Curatolo P. Deletion 2q37: An identifiable clinical syndrome with mental retardation and autism. J. Child Neurol. 2008;23:802–806. doi: 10.1177/0883073808314150. [DOI] [PubMed] [Google Scholar]
- 28.Potocki L., Bi W., Treadwell-Deering D., Carvalho C.M., Eifert A., Friedman E.M., Glaze D., Krull K., Lee J.A., Lewis R.A. Characterization of Potocki-Lupski syndrome (dup(17)(p11.2p11.2)) and delineation of a dosage-sensitive critical interval that can convey an autism phenotype. Am. J. Hum. Genet. 2007;80:633–649. doi: 10.1086/512864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schedl A. Renal abnormalities and their developmental origin. Nat. Rev. Genet. 2007;8:791–802. doi: 10.1038/nrg2205. [DOI] [PubMed] [Google Scholar]
- 30.Burtey S. 22q11.2 microdeletion syndrome is a common cause of renal tract malformations. Nat. Clin. Pract. Nephrol. 2008;4:E1. doi: 10.1038/ncpneph0906. [DOI] [PubMed] [Google Scholar]
- 31.Shprintzen R.J. Velocardiofacial syndrome and DiGeorge sequence. J. Med. Genet. 1994;31:423–424. doi: 10.1136/jmg.31.5.423-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bingham C., Hattersley A.T. Renal cysts and diabetes syndrome resulting from mutations in hepatocyte nuclear factor-1beta. Nephrol. Dial. Transplant. 2004;19:2703–2708. doi: 10.1093/ndt/gfh348. [DOI] [PubMed] [Google Scholar]
- 33.Moreno-De-Luca D., Mulle J.G., Kaminsky E.B., Sanders S.J., Myers S.M., Adam M.P., Pakula A.T., Eisenhauer N.J., Uhas K., Weik L., SGENE Consortium. Simons Simplex Collection Genetics Consortium. GeneSTAR Deletion 17q12 is a recurrent copy number variant that confers high risk of autism and schizophrenia. Am. J. Hum. Genet. 2010;87:618–630. doi: 10.1016/j.ajhg.2010.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nagamani S.C., Erez A., Shen J., Li C., Roeder E., Cox S., Karaviti L., Pearson M., Kang S.H., Sahoo T. Clinical spectrum associated with recurrent genomic rearrangements in chromosome 17q12. Eur. J. Hum. Genet. 2010;18:278–284. doi: 10.1038/ejhg.2009.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stone E.M., Lotery A.J., Munier F.L., Héon E., Piguet B., Guymer R.H., Vandenburgh K., Cousin P., Nishimura D., Swiderski R.E. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat. Genet. 1999;22:199–202. doi: 10.1038/9722. [DOI] [PubMed] [Google Scholar]
- 36.Marmorstein L.Y., McLaughlin P.J., Peachey N.S., Sasaki T., Marmorstein A.D. Formation and progression of sub-retinal pigment epithelium deposits in Efemp1 mutation knock-in mice: A model for the early pathogenic course of macular degeneration. Hum. Mol. Genet. 2007;16:2423–2432. doi: 10.1093/hmg/ddm199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.