

Abstract
Abnormalities in metabolite profiles are valuable indicators of underlying pathologic conditions at the molecular level. However, their interpretation relies on detailed knowledge of the pathways, enzymes, and genes involved. Identification and characterization of their physiological function are therefore crucial for our understanding of human disease: they can provide guidance for therapeutic intervention and help us to identify suitable biomarkers for monitoring associated disorders. We studied two individuals with 2-aminoadipic and 2-oxoadipic aciduria, a metabolic condition that is still unresolved at the molecular level. This disorder has been associated with varying neurological symptoms. Exome sequencing of a single affected individual revealed compound heterozygosity for an initiating methionine mutation (c.1A>G) and a missense mutation (c.2185G>A [p.Gly729Arg]) in DHTKD1. This gene codes for dehydrogenase E1 and transketolase domain-containing protein 1, which is part of a 2-oxoglutarate-dehydrogenase-complex-like protein. Sequence analysis of a second individual identified the same missense mutation together with a nonsense mutation (c.1228C>T [p.Arg410∗]) in DHTKD1. Increased levels of 2-oxoadipate in individual-derived fibroblasts normalized upon lentiviral expression of the wild-type DHTKD1 mRNA. Moreover, investigation of L-lysine metabolism showed an accumulation of deuterium-labeled 2-oxoadipate only in noncomplemented cells, demonstrating that DHTKD1 codes for the enzyme mediating the last unresolved step in the L-lysine-degradation pathway. All together, our results establish mutations in DHTKD1 as a cause of human 2-aminoadipic and 2-oxoadipic aciduria via impaired turnover of decarboxylation 2-oxoadipate to glutaryl-CoA.
Main Text
L-lysine degradation is a complex multistep mechanism involving mitochondrial, cytosolic, and peroxisomal enzymes. It converges with the degradative pathways of L-hydroxylysine and L-tryptophan at the levels of 2-aminoadipic-6-semialdehyde and 2-oxoadipate, respectively (Figure 1). Although the molecular causes of most inherited deficiencies of lysine degradation have been elucidated, those of 2-aminoadipic and 2-oxoadipic aciduria (MIM 204750) remain to be explained. Over 20 individuals with this metabolic disease are known. More than half of them are asymptomatic, whereas others have developed mild to severe intellectual disability, muscular hypotonia, developmental delay, ataxia, and epilepsy.1,2 A defect in 2-oxoadipate dehydrogenase was first suggested as an underlying cause, but there was no experimental evidence. Furthermore, it was shown that the 2-oxoglutarate-dehydrogenase complex (OGDHc) also has substrate specificity for 2-oxoadipate.3 Individuals with inherited 2-oxoglutarate-dehydrogenase deficiency usually present in the newborn period or infancy with severe neurological symptoms, as well as elevated 2-oxoglutarate and lactate.4 Fiermonte and coworkers suggested a deficiency of the human 2-oxoadipate mitochondrial carrier as the underlying cause.5
Figure 1.
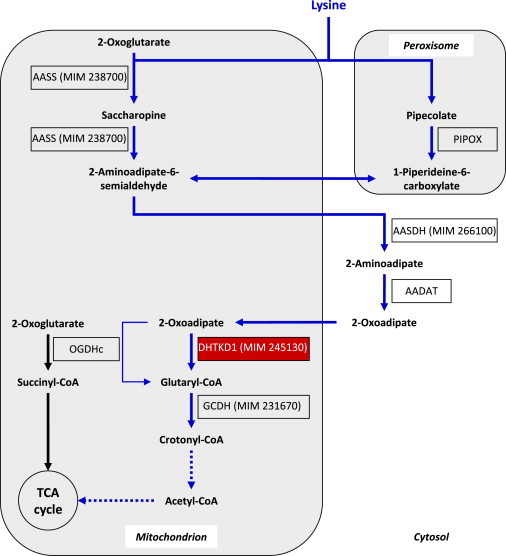
Proposed Multicompartmental Degradative Pathway of L-lysine
L-Lysine degradation is initiated by either peroxisomal α-deamination (pipecolate pathway) or mitochondrial ε-deamination (saccharopine pathway). Both pathways converge in 2-aminoadipate semialdehyde, which is subsequently metabolized to 2-aminoadipate and 2-oxoadipate. Our data strongly indicate that DHTKD1 is part of a 2-oxoglutarate-dehydrogenase-complex (OGDHc)-like enzyme and is involved in the decarboxylation of 2-oxoadipate to glutaryl-CoA. A minor route for this reaction via the tricarboxylic-acid (TCA)-cycle enzyme OGDHc might exist but does not compensate for the lack of DHTKD1. Succinyl-CoA and glutaryl-CoA are known to inhibit OGDHc (not shown). DTHKD1 might be an interesting drug target for glutaric aciduria type I, which is caused by inherited deficiency of glutaryl-CoA dehydrogenase (GCDH). MIM numbers of inherited diseases of the lysine degradative pathway are provided (e.g., MIM 245130 for 2-aminoadipic and 2-oxoadipic aciduria). Dotted lines indicate multiple enzymatic steps. The following abbreviations are used: AADAT, 2-aminoadipate transaminase; AASDH, 2-aminoadipate-6-semialdehyde dehydrogenase; AASS, 2-aminoadipate-6-semialdehyde synthetase; DHTKD1, proposed E1 subunit of an OGDHc-like complex; and PIPOX, pipecolate oxidase. Mitochondrial transporters are not shown in this figure.
To unravel the metabolic origin of 2-aminoadipic and 2-oxoadipic aciduria, we studied two individuals who presented with the characteristic metabolic profile of this disease. Both were born to nonconsanguineous parents after an uneventful pregnancy and delivery and had an unremarkable family history.
Individual 1, a girl, presented with moderately delayed psychomotor development (sitting alone at age 10 months and walking alone at age 25 months) in infancy, and this improved by physiotherapy and occupational therapy. Metabolic analysis at the age of 2 years showed elevated 2-oxoadipate (range = 10–120 mmol/mol creatinine) and 2-hydroxyadipate (range = 5–40 mmol/mol creatinine) in urine; these are usually not detected. The plasma concentration of 2-aminoadipate was also elevated (120 μmol/l). Moderate protein restriction was introduced but did not influence the disease course and was thus discontinued. Serial cranial magnetic-resonance-imaging studies did not reveal any morphological abnormalities, and neuropsychological testing at the age of 8 years revealed an intelligence quotient (IQ) of 117. At 11 years of age, she was diagnosed with attention deficit hyperactivity disorder. Symptoms improved after implementation of methylphenidate. At the age of 14 years, her health is in excellent condition, and she is attending regular school and is following a normal diet. Neurological examination was normal.
Individual 2, a girl, presented with microcephaly, mild motor developmental delay (sitting alone at the age of 9 months and walking alone at the age of 22 months), and predominant speech delay of unknown origin in early childhood. Sensorineural hearing loss was excluded. When she was 2 years old, metabolic analyses of urine and plasma revealed the characteristic biochemical profile of 2-aminoadipic and 2-oxoadipic aciduria (2-oxoadipate, 520–970 mmol/mol creatinine; 2-hydroxyadipate, 100–150 mmol/mol creatinine; 2-aminoadipate, elevated). Moderate protein restriction was transiently introduced but did not influence the disease course. At the age of 12 years, she still has significant problems with expressive and receptive speech (limited active and passive vocabulary, dyslalia, and dysgrammatism). Her IQ is 87. Except for mild muscular hypotonia, neurological examination was normal.
Written informed consent was obtained from all participants or their guardians at the recruiting center, and the study was approved by the ethical committee at the Technical University of Munich.
We performed exome sequencing as described previously6 to identify mutations responsible for 2-aminoadipic and 2-oxoadipic aciduria in individual 1. In brief, exonic sequences were enriched with the use of the SureSelect Human All Exon 50 Mb kit from Agilent and were subsequently sequenced as 100 bp paired-end runs on a Hiseq2000 system (Illumina). Data analysis included read alignment with the Burrows-Wheeler Aligner (version 0.5.8) and variant calling with SAMtools (version 0.1.7). We produced 8.98 Gb of mappable sequences with an average exome coverage of 102.8×, and >91% of the target was covered more than 20×.
We next applied additional filters under the assumption of autosomal-recessive inheritance and a low allele frequency of causal mutations in controls. Accordingly, we excluded variants with a minor allele frequency (MAF) > 0.4% in SNP databases (HapMap and 1000 Genomes) and in 1,458 exomes from individuals with unrelated phenotypes.
This analysis identified eight candidate genes carrying predicted compound heterozygous or homozygous variants (Table 1). No mutation was detected in ODC, mainly excluding a dysfunctional 2-oxoadipate mitochondrial carrier as the underlying cause.5 Of the eight candidates, three (ADAMTS10, AIRE, and C8orf38) had been previously linked to unrelated phenotypes and were therefore considered to be not involved. Among the remaining five genes (DHTKD1, OVOS2, CAMTA2, LPPR3, and RADIL), only one, DHTKD1 (RefSeq accession number NM_018706.5), carried two probably pathogenic variants as predicted by in-silico-analysis software (PolyPhen-2 and MutationTaster7). The first mutation, c.1A>G (p.Met1?), affects the start codon and might cause out-of-frame usage of the next AUG triplet 160 nucleotides downstream; it is therefore expected to produce a nonfunctional protein. The second mutation, c.2185G>A, is predicted to cause an amino acid exchange, p.Gly729Arg, affecting an amino acid residue evolutionarily conserved from Homo sapiens to Caenorhabditis elegans and Drosophila melanogaster. This mutation is listed in dbSNP (rs117225135) and was found in the National Heart, Lung, and Blood Institute (NHLBI) Exome Variant Server with a MAF of 0.17%, corresponding to 19 heterozygous but no homozygous carriers among 5,379 individuals. Investigation of the carrier status of the parents showed that the c.1A>G mutation is present on the maternal allele but that the c.2185G>A mutation is absent from both the maternal and the paternal allele (Figure 2).
Table 1.
Variants Identified in an Individual with 2-Aminoadipic and 2-Oxoadipic Aciduria
| Number of Variants | |
|---|---|
| Synonymous | 11,229 |
| Nonsynonymous | 10,252 |
| Rare nonsynonymous | 313 |
| Genes with ≥ two nonsynonymous variants | 8 |
“Rare” indicates a frequency < 0.4% in 1,458 control exomes, HapMap, and the 1000 Genomes Project. Nonsynonymous variants include missense, nonsense, stoploss, and splice-site mutations, as well as insertions and deletions.
Figure 2.
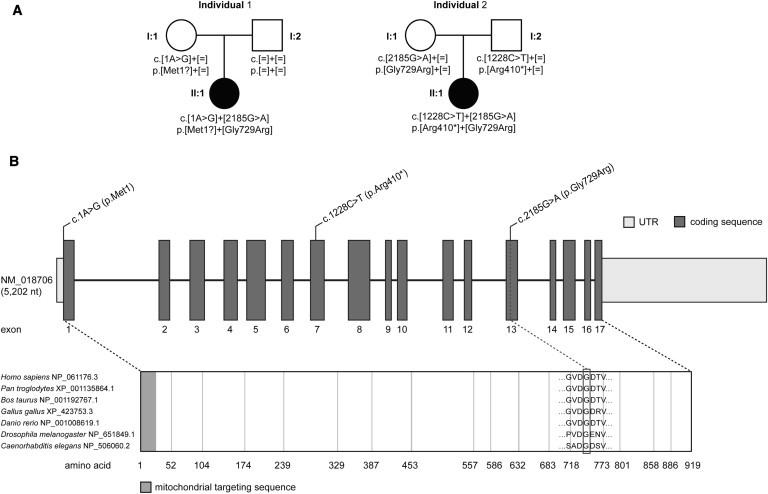
Pedigrees of Investigated Families and DHTKD1 Structure and Conservation of Identified Mutations
(A) Mutation status of affected (closed symbols) and unaffected (open symbols) family members.
(B) Structure of DHTKD1 with localization and conservation of affected amino acid residues of identified mutations.
Sanger sequencing of the DHTKD1 coding regions from individual 2 with the use of intronic primers (sequences are available upon request) identified compound heterozygosity for an exon 7 nucleotide transition, c.1228C>T, which is predicted to generate a stop codon (p.Arg410∗), as well as heterozygous mutation c.2185G>A, also observed in individual 1. Both parents were found to carry one mutation each in the heterozygous state. Identification of DHTKD1 mutations in a second individual with the same metabolic profile supports a causal role for these mutations in the pathogenesis of 2-aminoadipic and 2-oxoadipic aciduria. A summary of the genetic, biochemical, and clinical phenotype of individuals 1 and 2 is provided in Table 2.
Table 2.
Genetic, Biochemical, and Clinical Phenotype of Individuals with DHTKD1 Mutations
| ID | Sex |
DHTKD1 Mutations Identified |
Biochemical Investigations |
Clinical Features |
||||
|---|---|---|---|---|---|---|---|---|
| cDNA (RefSeq NM_018706.5) | Protein (RefSeq NP_061176) | 2-OAa | 2-OHAa | 2-AAa | Age of Diagnosis | Clinical Presentation | ||
| 1 | female | c.1A>G c.2185G>A |
p.Met1? p.Gly729Arg |
10–120 mmol/mol creatinine | 5–40 mmol/mol creatinine | 120 μmol/l | 2 years | moderate psychomotor developmental delay and attention deficit hyperactivity syndrome |
| 2 | female | c.1228C>T c.2185G>A |
p.Arg410∗ p.Gly729Arg |
520–970 mmol/mol creatinine | 100–150 mmol/mol creatinine | above normal | 2 years | slight psychomotor developmental delay, predominant speech delay, and mild muscular hypotonia |
The following abbreviations are used: 2-OA, 2-oxoadipate; 2-OHA, 2-hydroxyadipate; and 2-AA, 2-aminoadipate.
The reference range is <5.
Although both individuals carried one predicted loss-of-function allele, the effects of the shared missense mutation on DHTKD1 activity were unclear. In order to confirm the detrimental functional consequences of the c.2185G>A (p.Gly729Arg) mutation and to verify the causal role of the identified variants in DHTKD1, we performed complementation experiments in fibroblast cell lines established from both individuals 1 and 2. By using a feline-immunodeficiency-virus-based lentiviral system, we transduced an expression vector (Genecopoeia) containing the cDNA of DHTKD1 and a neomycin resistance gene as described previously.8 Immunoblotting with an antibody against DHTKD1 (Sigma-Aldrich) on mitochondria-enriched samples confirmed higher protein levels after transduction (Figure 3). Next, we cultivated primary fibroblasts, as well as fibroblasts expressing wild-type DHTKD1, from individuals 1 and 2 and from a healthy control in MEM medium (PAA) containing 10% fetal calf serum (GIBCO, Life Technologies) and penicillin and streptomycin (PAA) for 4 days. Afterward, 2-oxoadipate levels were detected in cells and media with the use of gas chromatography/mass spectrometry (GC/MS) according to a previously described method9 (fragment ion m/z 302). Compared to the control cell line (NHDF-neo, Lonza), both cell lines of the affected individuals showed increased levels of 2-oxoadipate in cells and media. Expressions of the wild-type DHTKD1 in fibroblasts of individuals 1 and 2 led to a strong decrease in intracellular and extracellular 2-oxoadipate concentrations to the levels of those in metabolically competent control fibroblasts (Figure 3). To pinpoint the role of DHTKD1 in lysine metabolism, we cultivated primary fibroblasts, as well as fibroblasts expressing the wild-type DHTKD1, in MEM medium as described above, but we supplemented them with 4 mmol/l 4,4,5,5-d4-lysine (CDN Isotope). The production of deuterium-labeled 2-oxoadipate was detected by GC/MS9 (fragment ion m/z 306). Indeed, 4,4,5,5-d4-labeled 2-oxoadipate could be shown in medium and cells of individuals 1 and 2, whereas its levels were strongly reduced when the wild-type DHTKD1 was expressed in these cells (Figure 4). In control fibroblast cells, 4,4,5,5-d4-labeled 2-oxoadipate was not detected. All together, our data provide strong evidence that reduced DHTKD1 activity is responsible for 2-aminoadipic and 2-oxoadipic aciduria in the investigated individuals.
Figure 3.
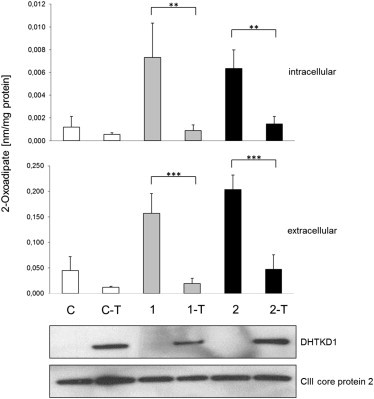
Metabolic Function of DHTKD1
Primary fibroblasts of several controls (C) and of individuals 1 and 2, as well as fibroblasts expressing wild-type DHTKD1 from individuals 1 and 2 (1-T and 2-T, respectively) and a healthy control (C-T) were cultivated in MEM medium for 4 days. Fibroblasts from the affected individuals showed increased intracellular and extracellular levels of 2-oxoadipate. Expressions of wild-type DHTKD1 in these cells reduced intracellular and extracellular 2-oxoadipate concentrations to the levels seen in metabolically competent control fibroblasts. Data are presented as the mean of five independent experiments ± the standard deviation (∗∗p < 0.01, ∗∗∗p < 0.001). Immunoblotting of the same cell lines was performed on mitochondria-enriched samples. The antibody against DHTKD1 (Sigma-Aldrich, SAB 1400619, dilution 1:1,000) demonstrated increased levels of DHTKD1. An antibody against the complex III core protein 2 (Abcam, ab14745, dilution 1:1,000) was used as a loading control.
Figure 4.
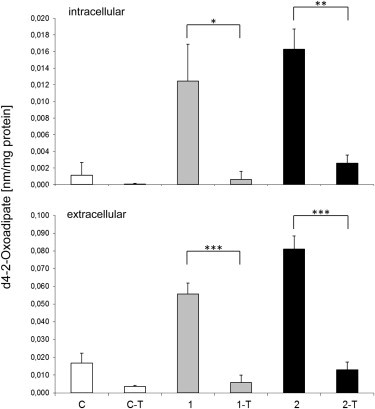
d4-Oxoadipate Production by Fibroblasts
Several control (C) fibroblasts and fibroblasts from individuals 1 and 2 with compound heterozygous DHTKD1 mutations were cultivated in MEM medium supplemented with 4 mM 4,4,5,5-d4-lysine. Detection of d4-oxoapidate in cells and media showed an overproduction of d4-oxoadipate in fibroblasts of individuals 1 and 2; this overproduction was reduced by the expression of wild-type DHTKD1 (C-T, 1-T, and 2-T). Data are presented as the mean of four independent measurements ± the standard deviation. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
We applied an unbiased exome-sequencing-based approach to identify DHTKD1 mutations as an underlying molecular-genetic cause of a rare metabolic disorder, 2-aminoadipic and 2-oxoadipic aciduria. Although this metabolic profile is extremely rare, elucidation of the genetic defect and subsequent characterization of enzymatic activity of the gene product have provided new insights into a basic cellular pathway of amino acid degradation.
DHTKD1 is one of two known isoforms of the E1 subunit of the OGDHc. It has been speculated that the 2-oxoglutarate-dehydrogenase-like proteins have different substrate affinities.10 The OGDHc catalyzes the decarboxylation of 2-oxoglutarate to succinyl-CoA within the citrate cycle. Our data suggest that DHTKD1 is part of an OGDHc-like supercomplex responsible for the decarboxylation of 2-oxoadipate to glutaryl-CoA within the common degradative pathways of L-lysine, L-hydroxylysine, and L-tryptophan.
In line with these findings, defective DHTKD1 activities resulted in increased levels of 2-oxoadipate and 2-oxoaminoadipate. In contrast to individuals who have other inherited disorders of amino acid degradation and who present with accumulating toxic metabolites and a severe clinical phenotype, such as phenylketonuria (MIM 261600), maple syrup urine disease (MIM 248600), tyrosinemia type I (MIM 276700), nonketotic hyperglycinemia (MIM 605899), and glutaric aciduria type I (MIM 231670), the individuals investigated in this study displayed rather mild and unspecific phenotypes. Of note, both individuals carried the same missense mutation, which might allow residual protein function. Therefore, it can be speculated that either complete loss of function leads to a more severe clinical phenotype or the mild clinical phenotype is explained by the low toxicity of accumulating metabolites. DHTKD1 is located one enzymatic step above glutaryl-CoA dehydrogenase in the lysine degradative pathway. Deficiency of glutaryl-CoA dehydrogenase causes glutaric aciduria type I, a cerebral organic acidemia that causes a severe neurological phenotype if untreated. Pharmacologic inhibition of DHTKD1 might be an interesting new drug target for glutaric aciduria type I in that it converts this disease into 2-aminoadipate and 2-oxoadipate aciduria, which has a less severe neurological phenotype. Interestingly, it has been reported that an apparent 2-aminoadipic aciduria also occurred during an antiepilectic treatment with vigabatrin and was probably caused by the inhibition of the metabolizing transaminase.11
Acknowledgments
We thank the affected individuals and their families for participation in the study. We acknowledge the technical support of Evelyn Botz and Carola Fischer. T.M. and H.P. were supported by the Systems Biology of Metabotypes grant (SysMBo 0315494A) funded by the German Federal Ministry of Education and Research, the German Network for Mitochondrial Disorders (mitoNET 01GM1113c), and the E-Rare project GENOMIT (01GM1207). T.M. and T.M.S. were supported by the European Commission 7th Framework Program (N. 261123), the Genetic European Variation in Disease Consortium, and the German Ministry for Education and Research (01GR0804-4).
Web Resources
The URLs for data presented herein are as follows:
MutationTaster, http://www.mutationtaster.org
NHLBI Exome Sequencing Project Exome Variant Server, http://evs.gs.washington.edu/EVS
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2
References
- 1.Przyrembel H., Bachmann D., Lombeck I., Becker K., Wendel U., Wadman S.K., Bremer H.J. Alpha-ketoadipic aciduria, a new inborn error of lysine metabolism; biochemical studies. Clin. Chim. Acta. 1975;58:257–269. doi: 10.1016/0009-8981(75)90445-3. [DOI] [PubMed] [Google Scholar]
- 2.Duran M., Beemer F.A., Wadman S.K., Wendel U., Janssen B. A patient with alpha-ketoadipic and alpha-aminoadipic aciduria. J. Inherit. Metab. Dis. 1984;7:61. doi: 10.1007/BF01805803. [DOI] [PubMed] [Google Scholar]
- 3.Sauer S.W., Opp S., Hoffmann G.F., Koeller D.M., Okun J.G., Kölker S. Therapeutic modulation of cerebral L-lysine metabolism in a mouse model for glutaric aciduria type I. Brain. 2011;134:157–170. doi: 10.1093/brain/awq269. [DOI] [PubMed] [Google Scholar]
- 4.Rustin P., Bourgeron T., Parfait B., Chretien D., Munnich A., Rötig A. Inborn errors of the Krebs cycle: A group of unusual mitochondrial diseases in human. Biochim. Biophys. Acta. 1997;1361:185–197. doi: 10.1016/s0925-4439(97)00035-5. [DOI] [PubMed] [Google Scholar]
- 5.Fiermonte G., Dolce V., Palmieri L., Ventura M., Runswick M.J., Palmieri F., Walker J.E. Identification of the human mitochondrial oxodicarboxylate carrier. Bacterial expression, reconstitution, functional characterization, tissue distribution, and chromosomal location. J. Biol. Chem. 2001;276:8225–8230. doi: 10.1074/jbc.M009607200. [DOI] [PubMed] [Google Scholar]
- 6.Mayr J.A., Haack T.B., Graf E., Zimmermann F.A., Wieland T., Haberberger B., Superti-Furga A., Kirschner J., Steinmann B., Baumgartner M.R. Lack of the mitochondrial protein acylglycerol kinase causes Sengers syndrome. Am. J. Hum. Genet. 2012;90:314–320. doi: 10.1016/j.ajhg.2011.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 8.Danhauser K., Iuso A., Haack T.B., Freisinger P., Brockmann K., Mayr J.A., Meitinger T., Prokisch H. Cellular rescue-assay aids verification of causative DNA-variants in mitochondrial complex I deficiency. Mol. Genet. Metab. 2011;103:161–166. doi: 10.1016/j.ymgme.2011.03.004. [DOI] [PubMed] [Google Scholar]
- 9.Sahm F., Capper D., Pusch S., Balss J., Koch A., Langhans C.D., Okun J.G., von Deimling A. Detection of 2-hydroxyglutarate in formalin-fixed paraffin-embedded glioma specimens by gas chromatography/mass spectrometry. Brain Pathol. 2012;22:26–31. doi: 10.1111/j.1750-3639.2011.00506.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bunik V.I., Degtyarev D. Structure-function relationships in the 2-oxo acid dehydrogenase family: Substrate-specific signatures and functional predictions for the 2-oxoglutarate dehydrogenase-like proteins. Proteins. 2008;71:874–890. doi: 10.1002/prot.21766. [DOI] [PubMed] [Google Scholar]
- 11.Vallat C., Rivier F., Bellet H., Magnan de Bornier B., Mion H., Echenne B. Treatment with vigabatrin may mimic alpha-aminoadipic aciduria. Epilepsia. 1996;37:803–805. doi: 10.1111/j.1528-1157.1996.tb00655.x. [DOI] [PubMed] [Google Scholar]