

Abstract
Cowchock syndrome (CMTX4) is a slowly progressive X-linked recessive disorder with axonal neuropathy, deafness, and cognitive impairment. The disease locus was previously mapped to an 11 cM region at chromosome X: q24-q26. Exome sequencing of an affected individual from the originally described family identified a missense change c.1478A>T (p.Glu493Val) in AIFM1, the gene encoding apoptosis-inducing factor (AIF) mitochondrion-associated 1. The change is at a highly conserved residue and cosegregated with the phenotype in the family. AIF is an FAD-dependent NADH oxidase that is imported into mitochondria. With apoptotic insults, a N-terminal transmembrane linker is cleaved off, producing a soluble fragment that is released into the cytosol and then transported into the nucleus, where it triggers caspase-independent apoptosis. Another AIFM1 mutation that predicts p.Arg201del has recently been associated with severe mitochondrial encephalomyopathy in two infants by impairing oxidative phosphorylation. The c.1478A>T (p.Glu493Val) mutation found in the family reported here alters the redox properties of the AIF protein and results in increased cell death via apoptosis, without affecting the activity of the respiratory chain complexes. Our findings expand the spectrum of AIF-related disease and provide insight into the effects of AIFM1 mutations.
Main Text
Charcot-Marie-Tooth disease (CMT) defines a clinically and genetically heterogeneous group of hereditary peripheral neuropathies characterized by chronic motor and sensory impairment. Charcot-Marie-Tooth disease is the most common hereditary neuromuscular disorder, with a prevalence of ∼1 in 2,500 people.1 Approximately 7%–10% of CMT is inherited as an X-linked trait (CMTX).2,3 Among the five known CMTX loci, the disease-causing mutations have been identified only for CMTX1 (MIM 304040)4 and CMTX5 (MIM 311850).5
In 1985, Cowchock et al. reported a large Italian-American family with a slowly progressive, childhood-onset, X-linked recessive axonal motor and sensory neuropathy associated with deafness and cognitive impairment.6 By linkage analysis, the disease locus was mapped to an 11 cM region at chromosome X: 116,500,000–130,400,000 (CMTX4 [MIM 310490]).7,8 We recently re-evaluated this family and identified a mutation in a candidate gene in this interval. The subjects were enrolled in an institutional review board-approved protocol of the National Institute of Neurological Disorders and Stroke (NINDS), and informed consent was obtained from all living subjects.
The most consistently observed clinical abnormalities were distal muscle wasting, weakness, and sensory loss, with lower limb greater than upper limb involvement. Only males displayed the phenotype, consistent with an X-linked recessive mode of inheritance (Figure 1A). As described previously,6 electrophysiological studies in affected individuals showed evidence of motor more than sensory axonal neuropathy. Bilateral sensorineural hearing loss was also present in three subjects. Diminished intellectual abilities had been noted in several affected individuals since childhood.
Figure 1.
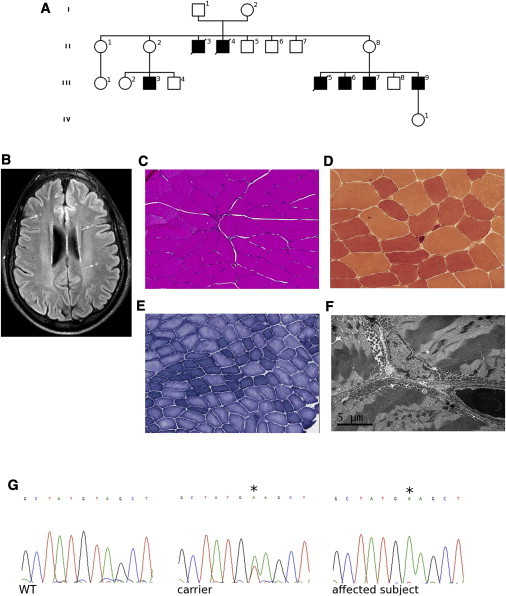
Pedigree, Brain Imaging, and Skeletal Muscle Analysis and Sanger Sequencing
(A) Pedigree of the family. White, unaffected; black, affected.
(B) Fluid-attenuated inversion recovery (FLAIR) magnetic resonance imaging on a transverse section of supratentorial brain of an affected individual showing multiple punctate hyperintensities in the white matter (arrows; subject III-6).
(C–E) Muscle biopsy showing variation in fiber size (hematoxylin and eosin staining, C), esterase-positive angular atrophic myofibers (esterase staining, D), and fiber grouping with some targetoid fibers (nicotinamide adenine dinucleotide-tetrazolium reductase, E) (subject III-9). (F) Electron microscopic image of muscle showing variation in shape and increase in number of mitochondria, mainly in the subsarcolemmal areas (subject III-9).
(G) Chromatograms of AIFM1 showing the c.1478A>T (p.Glu493Val) variant (asterisk) in an affected individual (hemizygote) and a carrier (heterozygote).
Laboratory investigations in two affected individuals (III-6 and III-9) showed elevation of serum transaminases (up to 2×), lactate dehydrogenase (up to 440 U/l, normal values 113–226), and creatine kinase (up to 1,169 U/l, normal values 52–386). Blood pyruvate and free and total carnitine were normal. Cranial MRI in two subjects (III-3 and III-9) showed multiple punctate T2 hyperintensities in the supratentorial white matter (Figure 1B). Muscle biopsy (left biceps; subject III-9) showed modest signs of neurogenic atrophy (Figures 1C–1E), with no ragged-red fibers by modified Gomori trichrome staining and no abnormalities in histochemical staining for cytochrome oxidase and succinate dehydrogenase (not shown). Electron microscopy showed an increased number of mitochondria mainly in the subsarcolemmal areas, with moderate abnormality of mitochondrial shape and internal structure (Figure 1F).
We performed exome capture on one affected subject by using SureSelect Human All Exome Kit v.1 (Agilent Technologies) and sequencing by using a Genome Analyzer IIx (Illumina). The exome sequencing covered 74.3% of targeted regions with a read depth >20×. Read alignment was performed to the human genome assembly hg19. We filtered variants to exclude HapMap SNPs that were present in dbSNP132 and that had an average heterozygosity greater than 0.02. This approach left as a single-candidate variant the substitution c.1478A>T (RefSeq NM_004208.3; GRCh37/hg19 chrX: 129,265,745T>A), within exon 14 of AIFM1 (MIM 300169), which predicts a p.Glu493Val change.
To confirm the AIFM1 mutation, we used Sanger sequencing to screen five affected (II-3, III-3, III-5, III-7, and III-9) and nine unaffected (I-1, I-2, II-1, II-2, II-5, II-8, III-2, and III-4) family members (Figure 1A). All clinically affected subjects were hemizygous and all mothers of affected individuals were heterozygous for the c.1478A>T (p.Glu493Val) variant (Figure 1G). None of the other unaffected subjects had this variant. Thus, the mutation in AIFM1 completely cosegregated with the CMT phenotype. The Glu493 residue is conserved across multiple species (Figure S1 available online) and the Val493 variant was absent in 712 control individuals in the ClinSeq cohort.9 No mutation in AIFM1 was found in 102 unrelated males with CMT of unknown genetic cause.
AIF, a phylogenetically conserved mitochondrial flavoprotein, is one of the key caspase-independent death effectors.10–12 The AIF precursor protein is a nuclear-encoded 67 kDa polypeptide, which, after cleavage of the N-terminal mitochondrial targeting sequence, is posttranslationally inserted into the inner mitochondrial membrane, with the N terminus facing the matrix and the C-terminal catalytic domain exposed to the intermembrane space.13 Upon apoptotic insult, the membrane linker of AIF is cleaved off, and a soluble 57 kDa C-terminal fragment is released into the cytosol and transported into the nucleus, where it triggers caspase-independent apoptosis with chromatin condensation and DNA fragmentation.10 Although the apoptotic function of AIF has been extensively investigated, the physiological role of the protein in normal mitochondria remains unclear. AIF has been proposed to function as a superoxide-generating NADH oxidase or electron transferase.14,15 Despite recent in vivo analyses of the metabolic changes caused by AIF deficiency or defects, neither the substrates nor the acceptor(s) of its redox activity have been identified thus far. When its cofactor FAD is incorporated, the protein has a 100-fold preference for NADH over NADPH and forms dimeric FADH−-NAD charge-transfer complexes (CTCs), which are inefficient for electron transfer. Formation of CTC reduces the susceptibility of the N terminus to proteolysis and weakens the AIF-DNA interaction, two events critical for initiation of caspase-independent apoptosis.16,17 Further studies have suggested that AIF may act as a redox sensor with interrelated physiologic and apoptotic functions.18 Insight into the normal function of AIF comes from the Harlequin (Hq) mouse, a spontaneous model of AIF deficiency resulting from retroviral insertion into Aifm1.19 The Hq model has 80% reduction in normal AIF protein levels and decreased complex I and III activities in affected tissues, suggesting that AIF may control the biogenesis and maintenance of the respiratory complexes.20,21 Recently, a c.del601_603 AIFM1 mutation predicted to cause p.Arg201del was reported in two male infants with severe progressive mitochondrial encephalomyopathy, multiple defects of the respiratory chain activities, and increased caspase-independent apoptosis.22 A second pathological AIFM1 mutation, c.923G>A, which predicts p.Gly308Glu, was associated with prenatal ventriculomegaly and infantile encephalomyopathy, primarily because of defects in the mitochondrial respiratory chain.23 Gly308 is located in a conserved NADH-binding region, suggesting that this function may be compromised by the p.Gly308Glu substitution.
The negatively charged residue Glu493 mutated in the family reported here is not directly involved in FAD or NADH binding but is only 10 Å away from the isoalloxazine ring that is the redox moiety of FAD. In oxidized AIF, Glu493 is buried and assists in folding of the C-terminal regulatory insertion by mediating water-bridged contacts between the 532–534 and 581–584 peptides (Figures 2A and 2B). The Glu493Val substitution may weaken these interactions and lead to a more unstable protein conformation. In NADH-reduced AIF, the region surrounding Glu493 becomes exposed to solvent.16 As seen in Figure 2C, this surface is highly charged, with several acidic residues clustering near the pyrimidine portion of the isoalloxazine ring. This charge distribution is known to define the redox potential of FAD and, consequently, the flavoprotein reactivity. The X-ray structure of oxidized p.Glu493Val AIF shows that this substitution at position 493 does not affect protein folding but perturbs the electrostatic surface potential near the redox center and changes the surface profile (Figures 2D–2F). This, in turn, could increase solvent accessibility of FAD and affect the redox reactivity of AIF.
Figure 2.
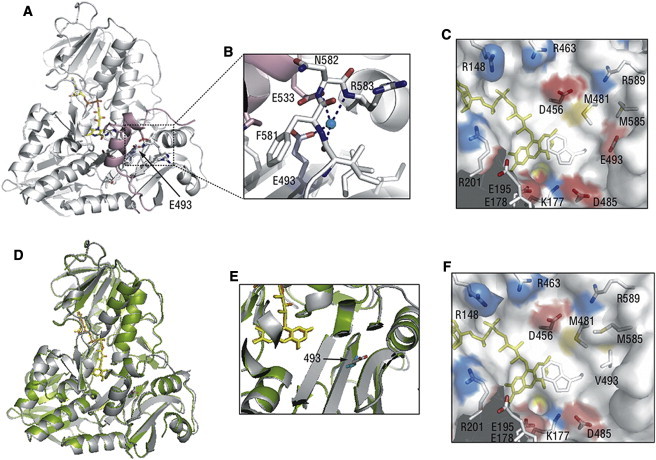
Structural Comparison of the Wild-Type and p.Glu493Val Mutant of AIF
The p.Glu493Val substitution was introduced into the human AIF1 expression plasmid by Stratagene QuikChange kit. Expression, purification, and removal of the C-terminal six-histidine affinity tag were carried out as described previously.18
(A) Position of Glu493 (indicated by an arrow) in oxidized human AIFwt (PDB code 1M6I). FAD is shown in yellow and a partially disordered regulatory peptide is in pink.
(B) Water-bridged contacts involving Glu493 hold the 512–533 helical fragment of the regulatory peptide (pink) close to the active site.
(C) Charge distribution near the flavin cofactor. Positively and negatively charged residues are depicted in blue and red, respectively. Glu493 is 10 Å away from FAD and is part of an acidic cluster adjacent to the isoalloxazine ring.
(D) Superposition of the structures of the wild-type (gray, PDB code 1M6I) and Glu493Val mutant of human AIF (green, PDB code 4FDC). The structure of AIFE493V was solved at 2.4 Å resolution. Data collection and refinement statistics are given in Table S1. The valine side chain is shown in cyan.
(E) A magnified view at the region of AIF near the site of the p.Glu493 residue demonstrating that the substitution does not alter the structure near the active site of AIF.
(F) The Glu493Val substitution changes both the surface profile and electrostatic potential, which may affect solvent accessibility and redox properties of FAD.
To better understand the pathogenicity of the p.Glu493Val substitution identified in the family with CMTX, several properties of the recombinant mutant protein were analyzed and compared to those of the wild-type AIF and the previously reported p.Arg201del variant.22 The absorbance spectrum, extinction coefficient, and midpoint redox potential of AIF were not affected by the p.Glu493Val substitution. Similar to the wild-type AIF, p.Glu493Val AIF has high affinity for NADH, binds the cofactor tightly and in a stoichiometric fashion, and forms CTC upon reduction (Figure 3A). This implies that the NADH binding site in p.Glu493Val AIF is preserved. Nevertheless, a subtle difference in circular dichroism spectra, slightly higher flavin fluorescence, and a distinct proteolytic pattern of p.Glu493Val AIF were observed (Figures S2A–S2C), indicating structural dissimilarities between wild-type and mutant proteins.
Figure 3.
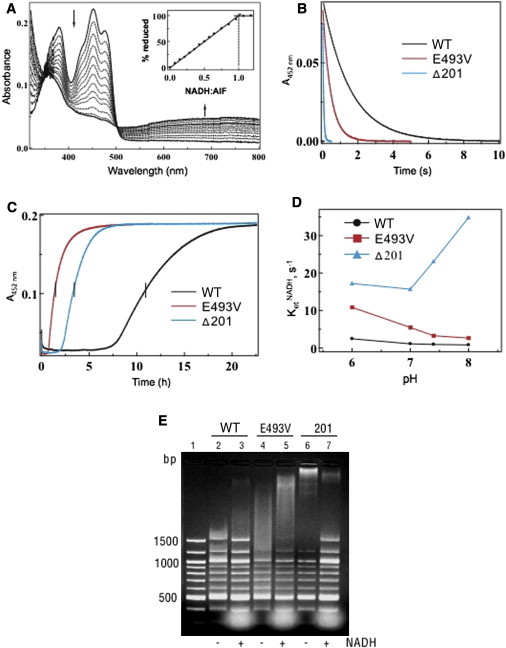
Comparison of the Redox and Molecular Properties of the Wild-Type and p.Glu493Val and p.Arg201del Mutants of AIF
(A) Anaerobic titration of AIFE493V with NADH. Similar to the wild-type protein, the p.Glu493Val variant binds NADH tightly and produces a FADH−-NAD charge-transfer complex (CTC) absorbing in the long-wavelength region. Inset shows that an equimolar amount of NADH is required to fully reduce FAD.
(B) Kinetics of AIF reduction with NADH. Proteins were mixed with NADH in a stopped flow spectrophotometer, and reduction of FAD was monitored at 452 nm. The derived kinetic parameters are given in Table 1.
(C) Kinetics of oxidation of NADH-reduced AIF. Proteins were reduced with a 4-fold excess of NADH before exposure to atmospheric oxygen. Flavin oxidation was monitored at 452 nm.
(D) Effect of pH on the kinetics of AIF reduction with NADH.
(E) Comparison of DNA binding by wild-type and mutant AIF. Equal amounts of protein (100 μg) were incubated for 15 min with 250 ng of 100 bp DNA ladder (New England Biolabs) in the absence and presence of a 20-fold excess of NADH. After separation on a 2% agarose gel, DNA was visualized with ethidium bromide. Lane 1 is a control (DNA only).
The most notable changes caused by the p.Glu493Val substitution were observed in electron transfer ability. First, p.Glu493Val AIF has lower KM for NADH (by 20%) than does wild-type AIF and becomes reduced by NADH 4-fold faster (Figure 3B and Table 1). Second, the dimeric CTC produced by wild-type AIF reacts with oxygen slowly and is relatively stable,18 whereas the half-life (t1/2) of CTC produced by p.Glu493Val AIF is one eighth that produced by the wild-type protein (Figure 3C and Table 1). Because p.Glu493Val AIF retains its ability to dimerize upon reduction with NADH (Figure S2D), an effect of the p.Glu493Val substitution on redox-linked dimer formation can be ruled out. Finally, p.Glu493Val AIF more efficiently transfers electrons to small electron-accepting molecules and cytochrome c (compare the kcat/KMNADH values in Table 1, which reflect the enzyme efficiency), but these redox reactions are partially uncoupled (∼10% for p.Glu493Val AIF versus 1%–2% for wild-type AIF).
Table 1.
Redox Properties and Activities of the Wild-Type and Mutants of Mature Human AIF
| WT | p.Glu493Val | p.Arg201del | |
|---|---|---|---|
| ketNADH (s−1)a | 0.98 ± 0.02 | 3.9 ± 0.1 | 23 ± 0.4 |
| KMNADH (mM) | 1.53 ± 0.06 | 1.23 ± 0.05 | 0.23 ± 0.02 |
| t1/2CTC (hr)b | 11 | 1.4 | 3.4 |
| koxNADH (min−1)c | 0.015 ± 0.002 | 0.12 ± 0.02 | 0.06 ± 0.01 |
| DCIP | |||
| kcat (min−1) | 137 ± 18 | 335 ± 32 | 394 ± 28 |
| KMDCIP (μM) | 202 ± 36 | 97 ± 20 | 16 ± 3 |
| KMNADH (mM) | 0.74 ± 0.12 | 0.16 ± 0.04 | 0.14 ± 0.02 |
| kcat/ KMNADH (mM−1 min−1) | 185 | 2,094 | 2,814 |
| K3Fe(CN)6 | |||
| kcat (min−1) | 314 ± 25 | 745 ± 30 | 1,464 ± 92 |
| KMK3Fe(CN)6 (μM) | 28 ± 5 | 26 ± 5 | 68 ± 6 |
| KMNADH (mM) | 0.61 ± 0.10 | 0.26 ± 0.05 | 0.20 ± 0.03 |
| kcat/KMNADH (mM−1 min−1) | 515 | 2,865 | 7,320 |
| Cytochrome c | |||
| kcat (min−1) | 54 ± 15 | 35 ± 5 | 39 ± 2 |
| KMcyt c (μM) | 26 ± 13 | 28 ± 8 | 94 ± 10 |
| KMNADH (mM) | 0.27 ± 0.02 | 0.15 ± 0.02 | 0.07 ± 0.01 |
| kcat/ KMNADH (mM−1 min−1) | 200 | 233 | 557 |
All activities were determined in 50 mM phosphate buffer (pH 7.4) at 25°C.
The rate constant for the AIF reduction by NADH was determined by stopped flow spectrophotometry at 25°C.
The half-life of the FADH2-NAD charge-transfer complex (CTC) was determined at 25°C in the presence of 15 μM AIF and 60 μM NADH.
The rate of NADH oxidation by AIF was determined at 25°C.
Another distinct feature of p.Glu493Val AIF is the pH dependence of its reaction with NADH. Unlike wild-type AIF, which accepts the hydride atom from NADH in a pH-independent manner, the rate of p.Glu493Val AIF reduction is notably affected and increases by 6-fold at higher pH (Figure 3D). Such pH dependence indicates that reduction of p.Glu493Val AIF is associated with proton consumption and may lead to the formation of a neutral FAD hydroquinone (FADH2). The latter redox species is less thermodynamically stable than an anionic hydroquinone (FADH−) formed by wild-type AIF, which may be the reason for faster oxidation of p.Glu493Val AIF by electron acceptors. Based on these experimental data, we conclude that the Glu493 residue controls proton accessibility to the flavin moiety and modulates the redox reactivity of AIF.
Importantly, the destabilization and faster oxidation of the p.Glu493Val AIF-NADH complexes in vivo can determine the accumulation of AIF monomers that are more prone to N-terminal proteolysis and release of the soluble, apoptogenic fragment.16,18 Similar to p.Arg201del AIF,22 the truncated apoptogenic form of p.Glu493Val AIF has higher affinity for DNA than the wild-type protein (Figure 3E). The lack of an inhibiting effect of NADH on the p.Glu493Val AIF-DNA interaction, which is observed for wild-type AIF,18 was probably due to rapid and complete oxidation of p.Glu493Val AIF before the gel retardation experiment was over.
We then investigated the consequences of the p.Glu493Val substitution in cultured skin fibroblasts and skeletal muscle derived from an affected individual. We first showed that the levels of mutant AIF in fibroblasts from subject III-3 were similar to those of wild-type AIF in control cells (Figure S3). Next, in order to test whether the p.Glu493Val AIF variant is directly linked to impaired oxidative phosphorylation, we analyzed the activity of the respiratory chain complexes in mutant fibroblast cell lines grown for 48 hr in a galactose-rich (5 mM), glucose-free medium, a condition that forces cells to rely for energy on mitochondrial respiration rather than glycolysis. The activity of respiratory chain complexes in fibroblasts derived from an affected individual was not reduced compared to carrier and controls (not shown). Likewise, no impairment in the respiratory chain activities was detected in a muscle biopsy from subject III-9 (not shown). Mitotracker-based visualization (Invitrogen) was used to assess fragmentation of the mitochondrial network, which is a well-documented alteration related to reduced fusion of mitochondrial membranes. No difference could be detected in galactose-grown mutant fibroblasts compared to control, supporting the hypothesis that the mutation does not grossly perturb mitochondrial function (not shown).
In order to detect DNA fragmentation, which is a hallmark of apoptosis, we performed TUNEL assay in muscle biopsy. Numerous apoptotic cells were present in the sample from the affected individual, whereas little to no staining was detected in healthy control muscle (Figure 4A).
Figure 4.
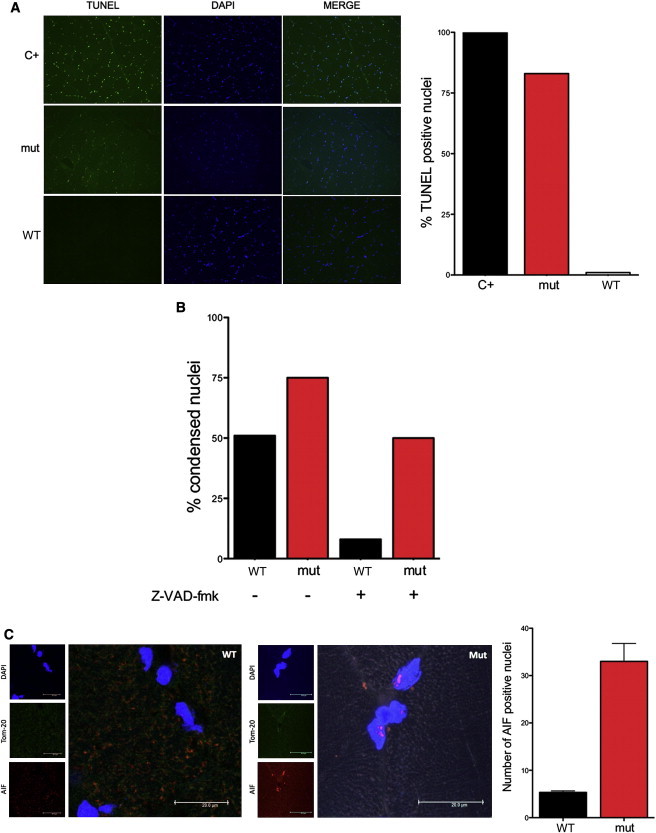
Caspase-Independent Apoptosis and Nuclear Localization of p.Glu493Val AIF
(A) TUNEL and DAPI staining of skeletal muscle nuclei from an affected subject (mut) and a control (WT). A normal biopsy treated with DNase was used a positive control (C+). Numerous TUNEL-positive nuclei are present in the affected subject, whereas no TUNEL-positive nuclei are seen in the control. Quantification of TUNEL-positive nuclei (n = 100) on the right.
(B) Quantification of altered nuclei in control fibroblasts (WT) and in fibroblasts from an affected individual (mut) after treatment with staurosporine (1 μM for 2 hr) in presence or absence of Z-VAD-fmk (100 μM for 0.5 hr).
(C) AIFM1 (1:1,000; Millipore, AB16501) immunostaining of cross section of muscle biopsy showing nuclear localization of mutant AIF in a subject hemizygous for p.Glu493Val (mut) compared to control (WT). Nuclei and mitochondria were counterstained with DAPI and mouse Tom20 antibody (BD Transduction Laboratory), respectively. AIF-positive nuclei were counted and normalized to the total number of DAPI-stained nuclei from three random and nonoverlapping fields (mean; SEM; p < 0.01).
Because AIF is a caspase-independent death effector, we treated p.Glu493Val AIF mutant and wild-type AIF control fibroblasts with staurosporine, a protein-kinase C inhibitor that induces cell death through the intrinsic, mitochondria-mediated pathway, at 1 μM for 2 hr, enough to trigger some of the early changes of apoptotic execution like the compaction of nuclear chromatin without resulting in cell death (Figure S4). We observed a much higher number of morphologically altered nuclei in staurosporine-treated p.Glu493Val AIF fibroblasts as compared to wild-type AIF cells (Figure 4B). In cells pretreated with the general caspase inhibitor z-VAD-FMK (100 μM for 0.5 hr), the staurosporine-induced nuclear abnormalities dropped to less than 10% in control fibroblasts, but remained as high as 50% in p.Glu493Val AIF mutant cells (Figure 4B), suggesting prominent activation of the caspase-independent, AIF-specific cell death pathway.
In order to gain insight into p.Glu493Val AIF localization in vivo, 8 μm muscle slices from subject III-9 and a healthy control were immunostained for AIF. Interestingly, more AIF-positive nuclear inclusions were detected, indicating that mutant p.Glu493Val AIF has higher propensity to translocate into the nucleus (Figure 4C).
In conclusion, we found that a missense mutation in AIFM1 causes Cowchock syndrome, a form of X-linked recessive axonal motor and sensory neuropathy with sensorineural hearing loss and cognitive impairment. According to our analysis, p.Glu493Val AIF has slight structural changes and an abnormal propensity to NADH reduction and O2 oxidation, probably because of changes in the FAD protonation state. Faster oxidation of p.Glu493Val AIF could lead to accumulation of AIF monomers, which translocate into the nucleus, where they trigger caspase-independent apoptosis. Interestingly, compared to the previously described p.Arg201del substitution,22 the variant reported here alters the structure and redox properties of AIF to a milder extent (Figures 3B–3D and Table 1), which is consistent with a less severe clinical phenotype. Also, whereas oxidative phosphorylation (OXPHOS) failure is thought to play a major role in the pathophysiology of the p.Arg201del AIF-related syndrome,22 the mechanism of damage in CMTX4 appears to be OXPHOS independent and primarily mediated by increased caspase-independent apoptosis.
Our identification of AIFM1 as a gene mutated in CMT considerably broadens the phenotypic spectrum associated with AIFM1 mutations. The results presented here also shed new light on the multiple pathogenic mechanisms in AIF-related diseases.
Analysis of additional AIFM1 mutations may help to further unravel the pathophysiology of AIF defects, to identify proteins dependent on AIF activity, and to determine the exact role of AIF in mitochondria. Considering the wide range and importance of the cellular processes regulated by AIF, better understanding of AIF functions may have broad and unexpected implications in both medicine and cell biology.
Acknowledgments
We are grateful to the family members for their participation and to Justin Y. Kwan, at NINDS, for performing the muscle biopsy. This work was supported in part by NINDS and NHGRI intramural research funding; National Institutes of Health Grant GM67637; the Pierfranco and Luisa Mariani Foundation, Italy; Fondazione Telethon grants GGP11011 and GPP10005; CARIPLO grant 2011/0526; and the Italian Association of Mitochondrial Disease Patients and Families (Mitocon).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
Accession Numbers
The dbSNP accession numbers for the AIFM1 sequences reported in this paper is ss539209971.
References
- 1.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth’s disease. Clin. Genet. 1974;6:98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 2.Pareyson D., Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009;8:654–667. doi: 10.1016/S1474-4422(09)70110-3. [DOI] [PubMed] [Google Scholar]
- 3.Boerkoel C.F., Takashima H., Garcia C.A., Olney R.K., Johnson J., Berry K., Russo P., Kennedy S., Teebi A.S., Scavina M. Charcot-Marie-Tooth disease and related neuropathies: mutation distribution and genotype-phenotype correlation. Ann. Neurol. 2002;51:190–201. doi: 10.1002/ana.10089. [DOI] [PubMed] [Google Scholar]
- 4.Bergoffen J., Scherer S.S., Wang S., Scott M.O., Bone L.J., Paul D.L., Chen K., Lensch M.W., Chance P.F., Fischbeck K.H. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 5.Kim H.J., Sohn K.M., Shy M.E., Krajewski K.M., Hwang M., Park J.H., Jang S.Y., Won H.H., Choi B.O., Hong S.H. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (cmtx5) Am. J. Hum. Genet. 2007;81:552–558. doi: 10.1086/519529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cowchock F.S., Duckett S.W., Streletz L.J., Graziani L.J., Jackson L.G. X-linked motor-sensory neuropathy type-II with deafness and mental retardation: a new disorder. Am. J. Med. Genet. 1985;20:307–315. doi: 10.1002/ajmg.1320200214. [DOI] [PubMed] [Google Scholar]
- 7.Fischbeck K.H., ar-Rushdi N., Pericak-Vance M., Rozear M., Roses A.D., Fryns J.P. X-linked neuropathy: gene localization with DNA probes. Ann. Neurol. 1986;20:527–532. doi: 10.1002/ana.410200414. [DOI] [PubMed] [Google Scholar]
- 8.Priest J.M., Fischbeck K.H., Nouri N., Keats B.J. A locus for axonal motor-sensory neuropathy with deafness and mental retardation maps to Xq24-q26. Genomics. 1995;29:409–412. doi: 10.1006/geno.1995.9987. [DOI] [PubMed] [Google Scholar]
- 9.Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F., NISC Comparative Sequencing Program The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–1674. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Susin S.A., Lorenzo H.K., Zamzami N., Marzo I., Snow B.E., Brothers G.M., Mangion J., Jacotot E., Costantini P., Loeffler M. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 11.Joza N., Susin S.A., Daugas E., Stanford W.L., Cho S.K., Li C.Y., Sasaki T., Elia A.J., Cheng H.Y., Ravagnan L. Essential role of the mitochondrial apoptosis-inducing factor in programmed cell death. Nature. 2001;410:549–554. doi: 10.1038/35069004. [DOI] [PubMed] [Google Scholar]
- 12.Lorenzo H.K., Susin S.A., Penninger J., Kroemer G. Apoptosis inducing factor (AIF): a phylogenetically old, caspase-independent effector of cell death. Cell Death Differ. 1999;6:516–524. doi: 10.1038/sj.cdd.4400527. [DOI] [PubMed] [Google Scholar]
- 13.Otera H., Ohsakaya S., Nagaura Z., Ishihara N., Mihara K. Export of mitochondrial AIF in response to proapoptotic stimuli depends on processing at the intermembrane space. EMBO J. 2005;24:1375–1386. doi: 10.1038/sj.emboj.7600614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miramar M.D., Costantini P., Ravagnan L., Saraiva L.M., Haouzi D., Brothers G., Penninger J.M., Peleato M.L., Kroemer G., Susin S.A. NADH oxidase activity of mitochondrial apoptosis-inducing factor. J. Biol. Chem. 2001;276:16391–16398. doi: 10.1074/jbc.M010498200. [DOI] [PubMed] [Google Scholar]
- 15.Maté M.J., Ortiz-Lombardía M., Boitel B., Haouz A., Tello D., Susin S.A., Penninger J., Kroemer G., Alzari P.M. The crystal structure of the mouse apoptosis-inducing factor AIF. Nat. Struct. Biol. 2002;9:442–446. doi: 10.1038/nsb793. [DOI] [PubMed] [Google Scholar]
- 16.Sevrioukova I.F. Redox-linked conformational dynamics in apoptosis-inducing factor. J. Mol. Biol. 2009;390:924–938. doi: 10.1016/j.jmb.2009.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sevrioukova I.F. Apoptosis-inducing factor: structure, function, and redox regulation. Antioxid. Redox Signal. 2011;14:2545–2579. doi: 10.1089/ars.2010.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Churbanova I.Y., Sevrioukova I.F. Redox-dependent changes in molecular properties of mitochondrial apoptosis-inducing factor. J. Biol. Chem. 2008;283:5622–5631. doi: 10.1074/jbc.M709147200. [DOI] [PubMed] [Google Scholar]
- 19.Klein J.A., Longo-Guess C.M., Rossmann M.P., Seburn K.L., Hurd R.E., Frankel W.N., Bronson R.T., Ackerman S.L. The harlequin mouse mutation downregulates apoptosis-inducing factor. Nature. 2002;419:367–374. doi: 10.1038/nature01034. [DOI] [PubMed] [Google Scholar]
- 20.Vahsen N., Candé C., Brière J.J., Bénit P., Joza N., Larochette N., Mastroberardino P.G., Pequignot M.O., Casares N., Lazar V. AIF deficiency compromises oxidative phosphorylation. EMBO J. 2004;23:4679–4689. doi: 10.1038/sj.emboj.7600461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Apostolova N., Cervera A.M., Victor V.M., Cadenas S., Sanjuan-Pla A., Alvarez-Barrientos A., Esplugues J.V., McCreath K.J. Loss of apoptosis-inducing factor leads to an increase in reactive oxygen species, and an impairment of respiration that can be reversed by antioxidants. Cell Death Differ. 2006;13:354–357. doi: 10.1038/sj.cdd.4401776. [DOI] [PubMed] [Google Scholar]
- 22.Ghezzi D., Sevrioukova I., Invernizzi F., Lamperti C., Mora M., D’Adamo P., Novara F., Zuffardi O., Uziel G., Zeviani M. Severe X-linked mitochondrial encephalomyopathy associated with a mutation in apoptosis-inducing factor. Am. J. Hum. Genet. 2010;86:639–649. doi: 10.1016/j.ajhg.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berger I., Ben-Neriah Z., Dor-Wolman T., Shaag A., Saada A., Zenvirt S., Raas-Rothschild A., Nadjari M., Kaestner K.H., Elpeleg O. Early prenatal ventriculomegaly due to an AIFM1 mutation identified by linkage analysis and whole exome sequencing. Mol. Genet. Metab. 2011;104:517–520. doi: 10.1016/j.ymgme.2011.09.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.