

Abstract
Cobblestone lissencephaly is a peculiar brain malformation with characteristic radiological anomalies. It is defined as cortical dysplasia that results when neuroglial overmigration into the arachnoid space forms an extracortical layer that produces agyria and/or a “cobblestone” brain surface and ventricular enlargement. Cobblestone lissencephaly is pathognomonic of a continuum of autosomal-recessive diseases characterized by cerebral, ocular, and muscular deficits. These include Walker-Warburg syndrome, muscle-eye-brain disease, and Fukuyama muscular dystrophy. Mutations in POMT1, POMT2, POMGNT1, LARGE, FKTN, and FKRP identified these diseases as alpha-dystroglycanopathies. Our exhaustive screening of these six genes, in a cohort of 90 fetal cases, led to the identification of a mutation in only 53% of the families, suggesting that other genes might also be involved. We therefore decided to perform a genome-wide study in two multiplex families. This allowed us to identify two additional genes: TMEM5 and ISPD. Because TMEM has a glycosyltransferase domain and ISPD has an isoprenoid synthase domain characteristic of nucleotide diP-sugar transferases, these two proteins are thought to be involved in the glycosylation of dystroglycan. Further screening of 40 families with cobblestone lissencephaly identified nonsense and frameshift mutations in another four unrelated cases for each gene, increasing the mutational rate to 64% in our cohort. All these cases displayed a severe phenotype of cobblestone lissencephaly A. TMEM5 mutations were frequently associated with gonadal dysgenesis and neural tube defects, and ISPD mutations were frequently associated with brain vascular anomalies.
Main Text
Cobblestone lissencephaly (cobblestone-LIS) is a peculiar brain malformation with characteristic radiological anomalies. It is defined as cerebral cortical dysplasia with an irregular limit between the white and gray matter, dysmyelination, severe dysplastic cerebellum with cysts, and brainstem hypoplasia.1 Cortical malformations are due to neuroglial overmigration into the arachnoid space, resulting in the formation of an extracortical neuroglial layer responsible for the agyria and/or an irregular, “cobblestone” surface of the brain and ventriculomegaly. The underlying mechanism is a disruption of the glia limitans, the outermost layer of the brain.2, 3, 4 Cobblestone-LIS is considered to be pathognomonic of a continuum of autosomal-recessive disorders resulting in ocular and muscular deficits. These disorders include Walker-Warburg syndrome (WWS, [MIM 236670]), muscle-eye-brain disease (MIM 253280), and Fukuyama muscular and cerebral dystrophy (MIM 253800). Walker-Warburg syndrome, also known by the HARD±E eponym (hydrocephalus, agyria, retinal dysplasia, ± encephalocele), is characterized by major neurological deficit, visual and muscular impairment, and a rapidly fatal outcome.5 Less severe features within the same spectrum include subtle eye abnormalities, a milder neurological deficit, and muscular dystrophy and can be observed in muscle-eye-brain disease, which was first described in a Finnish population,6 and in Fukuyama muscular and cerebral dystrophy, which is common in Japan.7, 8 Classically, these syndromes with cerebral ocular and muscular dystrophy are attributed to abnormal glycosylation of alpha-dystroglycan and are now designated as “muscular congenital alpha-dystroglycanopathy with brain and eye anomalies” (MDDGA). Aberrant post-translational modification of alpha-dystroglycan has been associated with mutations in at least six genes: POMT1 (MIM 607423), POMT2 (MIM 607439), POMGNT1 (MIM 606822), FKTN (MIM 607440), FKRP (MIM 606596), and LARGE (MIM 603590).9, 10, 11, 12, 13 Another group designated as having congenital muscular dystrophy-dystroglycanopathy with mental retardation (MDDGB) has been also associated with mutations of these six genes (MIM 613155, 613156, 613151, 613152, and 606612). Except for LARGE, these genes have also been implicated in the autosomal-recessive limb-girdle muscular dystrophies (LGMD, renamed MDDGC [MIM 609308, 613158, 613157, 611588, and 607155, respectively) characterized by variable age of onset, normal cognition, and no structural brain changes.14, 15, 16, 17, 18
Our molecular and neurohistopathological study of fetal cases of cobblestone-LIS demonstrated genetic heterogeneity and morphological diversity, along with some genotype-phenotype correlations.19, 20 In our growing cohort of 90 cobblestone-LIS families, 42 (47%) unrelated cases remained without any mutations in any of the six genes known to be implicated, suggesting that other genes might also be involved.
We therefore decided to use an SNP chip-based, genome-wide linkage approach combined with an exome study in two multiplex families (Figure S1 in the Supplemental Data available online). All fetal cases were included in the study on the basis of our diagnostic criteria of cobblestone-LIS as well as (1) neuroglial ectopia within the arachnoid space, (2) abnormal cortical lamination, and (3) brain stem and cerebellar dysplasia as described in Devisme et al.20 Informed consent was obtained from parents in accordance with French law and with the ethical standards of the national committee on human experimentation. Molecular screening was performed on genomic DNA (gDNA) extracted from frozen fetal tissues according to standard protocols. Genome-wide linkage studies were performed by Affymetrix Genome-wide Human SNP-Array 250K in two multiplex cobblestone-LIS families (F1 and F2) via Genotyping Console software (Affymetrix). Family F1 is consanguineous and includes four affected fetuses. Five runs of homozygosity (ROH) higher than 1 Mb were shared by three of the affected fetuses studied and were identified with the software PLINK21 in chromosomes 4 (two regions), 12, 15, and 20 (Table S1). The second multiplex family F2 is nonconsanguineous and includes three affected fetuses. Multipoint linkage analysis was performed with the software Merlin.22 Ten linkage regions that were more than 2 Mb and had an LOD score >1 were found on chromosomes 1, 6, 7 (two regions each), 10, 11, 17, 20, and 21 (Table S1). Illumina exome capture and next-generation sequencing on an Illumina platform were performed on samples from two affected fetuses of each of the F1 and F2 families. After regions were filtered on the basis of known variants, homozygosity for F1, two alleles mutated for F2, and location in the region of ROH (F1) or linkage regions (F2), a mutation in only one gene was found to segregate in the two siblings for each family (Table S2). In family F1, one homozygous variant, c.795delG (p.Arg266Glyfs∗8), was found in exon 5 of TMEM5 (NM_014254.1 [MIM 605862]) in the ROH region of chromosome 12. In family F2 an apparently homozygous c.638T>G p.Met213Arg missense mutation was found in exon 3 of ISPD (NM_001101426.3 MIM 614631]) in the largest linkage region on chromosome 7 (Table S1). Primers were designed in the flanking intronic region of both genes (Table S3) for Sanger sequencing with fluorescent di-deoxy capillary sequencing on an ABIPRISM 3130XL sequencer (Applied Biosystems). Sequence analysis was performed with Seqscape (Applied Biosystems) software. This analysis confirmed that all the F1 fetuses had inherited the homozygous c.795delG TMEM5 frameshift mutation from heterozygous parents. In the F2 family, the ISPD missense mutation was confirmed in siblings and was found in the heterozygous state in the mother only. Further molecular investigation by qPCR in exon 3 (data not shown) identified a paternally inherited deletion, confirming compound heterozygosity for a mutation and an intragenic deletion in family F2. Data from the SNP array in this family indicated the absence of any paternal contribution to the genotype of the affected fetuses for three intragenic SNPs (SNP_A-1826269, SNP_A-1948943, and SNP_A-1922943), which was consistent with hemizygosity from hg19: chromosome7:16,306,467 (intron 6) to 16,344,268 (intron 4). We therefore concluded that the size of the deletion in this family encompasses at least exon 3 to exon 6.
In order to confirm the involvement of TMEM5 and ISPD in cobblestone-LIS, we tested each coding exon and intron/exon junction of the two genes for mutations by direct gDNA SANGER sequencing in 40 cobblestone-LIS families that were part of our cohort but that did not have any mutation in the six genes known to be involved in alpha-dystroglycanopathies. We found pathogenic mutations in TMEM5 or ISPD in another eight families (Table 1). We identified a total of five distinct TMEM5 mutations, including three frameshift (c.795delG [p.Glu265fs∗8], c.1064_1091del [p.Asp355Valfs∗33], and c.279delA [p.Gly94Glufs∗33]) and two missense (c.1016A>G [p.Tyr339Cys] and c.1019_1020delinsTT [p. Arg340Leu]) mutations, in five families (Figure 1A). In ISPD, we identified nine distinct mutations, including two intragenic deletions, two nonsense mutations (c.256A>T [p.Arg86∗] and c.773C>A [p.Ser258∗]), one splice mutation (c.257+2T>G), and four missense mutations (c.466G>A [p.Asp156Asn], c.638T>G [p.Met213Arg], c.676T>C [p.Tyr226His], and c.713C>T [p.Thr238Ile]), in five families (Figure 1B). None of the missense variations were present in the DbSNP, 1000 Genomes, or Exome Variant Server data banks. All the amino acids concerned were conserved from humans to zebrafish. Furthermore, we used four different in silico prediction software packages (Polyphen, Panther, SIFT2, and Align GVGD) to evaluate the potential effect on protein function. All missense changes were predicted as being pathogenic by at least two out of four software packages (Table 1). Segregation of identified potentially disease-causing variants was performed in each available family member, and autosomal-recessive transmission was confirmed. In family F5, c.257+2T>G in ISPD appeared to be inherited de novo because it was not found in either parent's DNA. Paternity was confirmed by the Power Plex 16 system (Promega, Ca, USA), and allele-specific amplification was ruled out by PCR with two different pairs of primers. Two mutations in TMEM5 were found, in each case in two unrelated families: c.1016A>G (p.Tyr339Cys) in F7 and F9 and c.1064_1091del (p.Asp355Valfs∗33) in F8 and F9. We looked for a founder effect for these families by haplotype analysis with four microsatellite markers in the vicinity of TMEM5 and found a common haplotype for one allele for each of the two unrelated families, suggesting that these individuals could share the same ancestral chromosome segment. In the nonconsanguineous family F7, in which affected individuals had a homozygous TMEM5 mutation, parental DNAs were not available. We therefore confirmed homozygosity by qPCR, which excluded a heterozygous deletion.
Table 1.
Summary of the Pathogenic TMEM5 (NM_014254.1) and ISPD (NM_001101426.3) Mutations Detected in This Study
| Family Number, Number of Sibs | Consanguinity | Gene | Zygosity | Nucleotide Variant | Amino Acid Alteration | Exon(s) Affected | Type of Mutation | Panther Prediction pdeleterious | Align GVGD Class | PolyPhen-2 | SIFT |
|---|---|---|---|---|---|---|---|---|---|---|---|
| F1, 4 fetuses | yes | TMEM5 | homozygous | c.795 del G | p.Arg266Glyfs∗8 | exon 5 | frameshift | NA | NA | NA | NA |
| F7, 2 fetuses | no | TMEM5 | homozygous | c.1016A>G | p.Tyr339Cys | exon 6 | missense | 0.82 | C65 | probably damaging | affected protein (0.00) |
| F8, 1 fetus | no | TMEM5 | heterozygous | c. 1019_1020del insTT | p.Arg340Leu | exon 6 | missense | 0.57 | C65 | probably damaging | affected protein (0.00) |
| c.1064_1091del | p.Asp355Valfs∗33 | exon 6 | nonsense | NA | NA | NA | NA | ||||
| F9, 1 fetus | no | TMEM5 | heterozygous | c.1016A>G | p.Tyr339Cys | exon 6 | missense | 0.82 | C65 | probably damaging | affected protein (0.00) |
| c.1064_1091del | p.Asp355Valfs∗33 | exon 6 | frameshift | NA | NA | NA | NA | ||||
| F10, 1 fetus | ? | TMEM5 | homozygous | c.279 delA | p.Gly94Glufs∗33 | exon 2 | frameshift | NA | NA | NA | NA |
| F2, 3 fetuses | no | ISPD | heterozygous | c.638T>G | p.Met213Arg | exon 3 | missense | 0.67 | C65 | probably damaging | affected protein (0.03) |
| heterozygous | deletion | deletion | exons 3 to 6 at least | gene deletion | NA | NA | NA | NA | |||
| F3, 2 fetuses | yes | ISPD | homozygous | c.466G>A | p.Asp156Asn | exon2 | Missense | 0.64 | C15 | probably damaging | affected protein (0.00) |
| F4, 1 fetus | no | ISPD | heterozygous | c.713C>T | p.Thr238Ile | exon4 | Missense | 0.22 | C65 | probably damaging | affected protein (0.01) |
| c.256A>T | p.Arg86∗ | exon1 | Nonsense | NA | NA | NA | NA | ||||
| F5, 1 fetus | no | ISPD | heterozygous | c.257+2T>G | ∗ | exon1 | Splicing defect | NA | NA | NA | NA |
| c.773C>A | p.Ser258 | exon4 | Nonsense | NA | NA | NA | NA | ||||
| F6, 1 fetus | no | ISPD | heterozygous | c.676T>C | p.Tyr226His | exon3 | Missense | 0.45 | C65 | probably damaging | tolerated (0.43) |
| deletion | deletion | Ex4 to ex6 | Gene deletion | NA | NA | NA | NA |
Panther: the probability that a given variant will cause a deleterious effect is estimated by pdeleterious such that a subPSEC score (substitution position-specific evolutionary conservation score based on an alignment of evolutionarily related proteins) of −3 corresponds to a pdeleterious value of 0.5. Phenotype prediction for Align GVGD class C65 corresponds to "most likely interferes with function." PolyPhen-2 extracts various sequences and structure-based features of the substitution site and feeds them into a probabilistic classifier. For the SIFT score, the amino acid substitution is predicted to be harmful if the score is ≤0.05 and is predicted to be tolerated if the score is >0.05. NA = not applicable.
Figure 1.
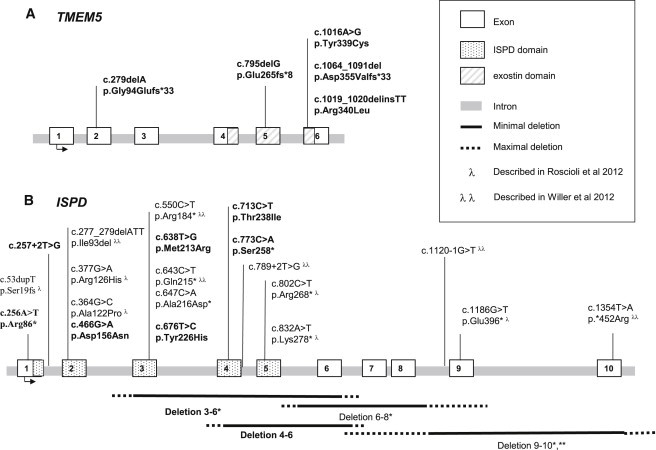
Diagram of the ISPD and TMEM5 Exon-Intron Gene Structure
All the pathogenic changes identified in this study (bold) and in two previous ISPD studies, Roscioli et al., 201230 (λ) and Willer et al., 201231 (λ λ) are included.
Both the genes we identified in this study seem to be functionally related to glycosylation. TMEM5 encodes a 443 amino acid, type II transmembrane protein of unknown function. The protein contains a signal anchor for a type II membrane protein encoded by amino acids 10–30, a cytoplasmic domain encoded by amino acids 1–9, and an extracellular domain encoded by amino acids 31–443.23 Interestingly, TMEM5 contains a domain that has some similarities to exostosin (amino acids 218–353), which is a domain found in EXT1. EXT1 codes for a glycosyltransferase required for the biosynthesis of heparan sulfate24 and is a putative tumor suppressor gene involved in multiple type-I exostoses (MIM 133700).25 A BlastP analysis identified a region of 19% identity with EXT1, which contains an aspartic acid-any amino acid-aspartic acid (DXD) motif that is present in many glycosyltransferases and is critical for catalytic function thought to be essential for binding the UDP-sugar donor through the coordination of a divalent cation, Mn2+ 26, 27 (Figure S2). Furthermore, EXT1 contains three other conserved DXD motifs, but substitutions (Arg340Ser, Arg340His, or Arg340Leu; and Arg280Gly or Arg280Ser) that have been shown to result in defective heparan sulfate biosynthesis cluster around the second DXD motif in this region of homology with TMEM528, 29 (Figure S2). Interestingly, three out of the five TMEM5 mutations identified in our study are also located in the exostosin domain (Figures 1A and 2). TMEM5 is therefore thought to be a transmembrane protein with a glycosyltransferase function.
Figure 2.
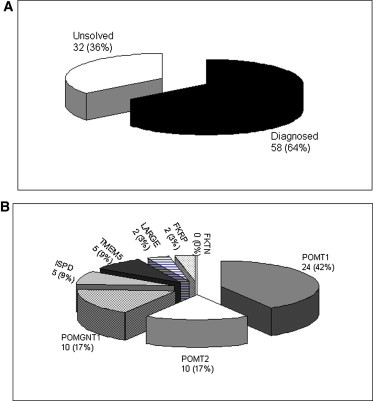
Results of Molecular Diagnosis of Cobblestone-LIS Fetuses
(A) Proportion of cases diagnosed after this study in our cohort of 90 cobblestone-LIS fetuses.
(B) Contribution of alpha-dystroglycanopathy gene mutations in our 58 diagnosed cobblestone-LIS fetuses.
Concerning ISPD, two recent independent studies simultaneously reported mutations in ISPD in postnatal WWS cases (MDDGA7 [MIM 614631]).30, 31 ISPD encodes an isoprenoid-synthase-domain-containing protein and belongs to the super-family of nucleotide diP-sugar transferases (amino acids 47–278). In plants, protozoa, and some bacteria, ISPD is part of the methylerythritol phosphate (MEP) pathway that leads to the polyisoprenoid alcohols (dolichols and polyphenols).32 This MEP pathway involving ISPD is missing in animals, which use an alternative ISPD-independent mevalonate pathway for isoprenoid synthesis.33, 34 The specific role of ISPD in humans is therefore unclear, but it is likely that ISPD synthesizes a new nucleotide sugar that could be incorporated into alpha-dystroglycan O-glycan.30 Assays on fibroblasts derived from ISPD-mutated WWS subjects complemented with wild-type ISPD restored alpha-dystroglycan glycosylation.31 Furthermore, the knockdown presentation in zebrafish reflected the WWS semiology, including hydrocephalus, reduced eye size, and muscle degeneration.30 Glycosylation of alpha-dystroglycan was reduced in zebrafish embryos injected with an dose of ISPD antisense morpholino oligonucleotides (MOs) sufficient to knock down ISPD isoforms. Coinjection with subeffective doses of FKTN or FKRP MO reduced glycosylation more than injecting subeffective doses of ISPD, FKTN, or FKRP MO alone.30 These findings support a cooperative interaction between ISPD and both FKTN and FKRP in alpha-dystroglycan glycosylation.
The ISPD mutations identified here are newly described, apart from the deletion of exons 3 to 6, which was previously described by Roscioli et al..30 Figure 1B shows a recapitulation including previously published mutations. Interestingly, the deletion of exons 3 to 6 in ISPD is located in a known CNV (hapMap variation_2683: four observed losses in 270 control samples, corresponding to a carrier frequency of 1.5%). The frequency of the CNV does not exclude its having a role in a rare disease, but it is possible that homozygosity leads to embryonic lethality, similar to that known for another glycosylation-related disease, PMM2-CDG (MIM 212065), and to frequent p.Arg141His mutation,35 which could explain the discrepancy between the relatively high frequency of this variation and the low frequency of cobblestone-LIS.
So far, only three out of the six genes mutated in alpha-dystroglycanopathies have a clearly established function; these are POMT1, POMT2, and POMGNT1, the genes encoding the three glycosyltransferases involved in early steps of alpha-dystroglycan O-mannosylation. The function of Fukutin-related protein (FKRP) and fukutin (FKTN) remains unknown. Both are Golgi-resident proteins, and FKRP is required for the posttranslational modification of alpha-dystroglycan in the Golgi apparatus.36 Fukutin-related protein resides in the Golgi cisternae of skeletal muscle fibers and forms disulfide-linked homodimers via an N-terminal interaction.37 Fukutin seems to bind the core area of alpha-dystroglycan during glycosylation.38 The exact function of LARGE is also unknown, but the overexpression of LARGE restores O-glycosylation of alpha-dystroglycan in cells of individuals with POMT1, POMGNT1, LARGE, or FKTN mutations.39 LARGE is involved in a phosphorylation process of the O-glycosyl alpha-dystroglycan, which is required for laminin binding and occurs on a specific Thr317 or Thr319 of the mucin-like domain of alpha-dystroglycan.40, 41 It has also been shown that LARGE induces the functional glycans in a DPM2- and O-mannosylation-dependent manner.42 Like the other genes involved in alpha-dystroglycanopathies, the two genes identified in this study are thought to be glycosyl or nucleotide diP-sugar transferases, on the basis of their similarities with known related domains.
In our previous study, we proposed a new classification of cobblestone-LIS in three subtypes depending on the severity of cortical malformations. In our classification, the most severe form, called cobblestone-LIS(A), is usually linked to mutations in POMT1 (34%) and POMT2 (8%); the least severe, cobblestone-LIS(C), is linked to POMGNT1 mutations; and an intermediate type, cobblestone-LIS(B), is linked to LARGE mutations.20 TMEM5 and ISPD mutations were found in cobblestone-LIS(A), associated with occipital neural-tube defects, facial clefts, visceral malformations, and gonadal dysplasia (Table 2). It is noteworthy that neural-tube defects, visceral malformations, and gonadal dysplasia were frequently observed in TMEM5-mutated cases (5/7 cases) but were absent in ISPD-mutated fetuses. In our previous study, we showed that visceral malformations were observed in almost half of the cases with POMT1 mutations (9/22) and in most of cases with POMT2 mutations (4/5).20 POMT1 was frequently associated with cleft lip and cleft palate.20, 43 POMT2 and TMEM5 were frequently associated with gonadal dysgenesis, found in 3/5 of the POMT2 cases20 and 5/8 of the TMEM5 cases. It is noteworthy that in two cases with ISPD mutations, the neuropathological study revealed severe vascular dysplasia and perfusion impairment, leading to focal brain parenchyma necrosis.
Table 2.
Clinical Data of ISPD- and TMEM5-Mutated Cobblestone-Lissencephaly Fetal Cases Identified in This Study
| Gene | Fetus |
Devisme et al., 201220 |
NTD | Limb Deformations | Gonadal Dysgenesis | Visceral Malformation | Brain Vascular Anomalies |
Cerebellar Dysplasia |
Retinal Dysplasia |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ECL/CP |
Extension |
Severity |
Extension |
Severity |
|||||||||||||
| Family Number | Fetus Number | Diffuse | Focal | Major | Minor | Diffuse | Focal | Major | Minor | ||||||||
| ISPD | F2-1 | F26 | 35 | + | - | - | - | >1 | + | + | + | + | |||||
| ISPD | F2-2 | F26 | 67 | + | + | + | + | >1 | + | + | + | + | |||||
| ISPD | F2-3 | F26 | 68 | + | - | - | + | >1 | + | + | + | + | |||||
| ISPD | F3-1 | F23 | 29 | - | - | - | - | + | >1 | + | + | + | + | ||||
| ISPD | F3-2 | F23 | 30 | - | + | - | - | >1 | + | + | NE | NE | NE | NE | |||
| ISPD | F4 | F25 | 34 | - | + | - | - | >1 | + | + | + | + | |||||
| ISPD | F5 | F55 | 69 | + | - | - | - | + | >1 | + | + | NE | NE | NE | NE | ||
| ISPD | F6 | / | / | - | - | - | - | NE | NE | NE | NE | NE | NE | NE | |||
| TMEM5 | F1-1 | F1 | 1 | + | - | + | + | >1 | + | + | NE | NE | NE | NE | |||
| TMEM5 | F1-2 | F1 | 2 | - | - | + | + | >1 | + | + | + | + | |||||
| TMEM5 | F1-3 | F1 | 3 | + | - | - | - | >1 | + | + | + | + | |||||
| TMEM5 | F1-4 | / | / | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | NE | |
| TMEM5 | F7-1 | F24 | 31 | + | - | + | + | >1 | + | + | + | + | |||||
| TMEM5 | F7-2 | F24 | 32 | + | - | - | - | >1 | + | + | + | + | |||||
| TMEM5 | F8 | F27 | 36 | - | - | + | - | >1 | + | + | + | + | |||||
| TMEM5 | F9 | F50 | 62 | + | + | + | + | >1 | + | + | + | + | |||||
| TMEM5 | F10 | F54 | 66 | - | - | - | - | >1 | + | + | NE | NE | NE | NE | |||
Abbreviations are as follows: NTD = neural-tube defect; ECL/CP = extracortical layer/cortical plate; and NE = not evaluated.
An overview of mutated alpha-dystroglycanopathy genes in our cohort of 90 cobblestone-LIS families is provided in Figure 2. Altogether, mutations in POMT1 (n = 24, 42%), POMT2 (n = 10, 17%), POMGNT1 (n = 10, 17%), TMEM5 (n = 5, 9%), ISPD (n = 5, 9%), LARGE (n = 2, 3%), and FKRP (n = 2, 3%) (Figure 2B) have now been identified in 58 cobblestone-LIS families. TMEM5 and ISPD each accounted for almost 10% of the mutated cases, which is consistent with the previously published data for ISPD in postnatal WWS cases.30, 31 Both genes were involved three times more frequently than LARGE in our series of cobblestone-LIS families and may therefore be considered to be two additional important genes with regard to this pathology. Our study raises the mutational rate to 64% in our cohort and once more highlights the importance of fetal pathological examination, not only for accurate diagnosis and subsequent genetic counseling but also for the classification of homogeneous groups and genotype-phenotype correlations that considerably accelerate molecular screening.
More recently, four other genes related to alpha-dystroglycanopathies were reported, although they were found in only a few individuals with mild myopathy. Three of these genes are related to glycosylation, and the fourth is the dystroglycan gene DAG1 itself (MIM 128239). One individual with dilated cardiomyopathy and a low but normal IQ was found to have a mutation in DPM3 (MIM 605951);44 two siblings with CMD and mental retardation were found to harbor mutations in DPM2 (MIM 603564);45, 46 and four families with dilated cardiomyopathy were reported to have DOLK (MIM 610746) mutations.47 Finally, two individuals were found to carry a mutation in DAG1; one carried a heterozygous deletion of a 2 MB region including DAG1,48 and the other, with LGMD and cognitive impairment, had a homozygous missense mutation.49 Considering the numbers of individuals reported to date, these four genes should be considered to be minor alpha-dystroglycanopathy genes.
The molecular basis of cobblestone-LIS remains unexplained in more than 36% (32/90) of our families (Figure 2A), suggesting either that some of the mutations of these genes were not detected or that other genes remain to be discovered. We studied all the genes by sequencing and genomic assays with CGH array analysis or qPCR to exclude intragenic rearrangements. Large-scale DNA rearrangements have now been identified in five genes associated with dystroglycanopathies (these genes are LARGE, POMT1, POMT2, POMGnT1, and ISPD).30, 50 In some cases, such as a recent LGMD case in which an alteration in the POMGNT1 promoter was reported,51 mutations might be located in noncoding sequences; in other cases, there might be a deep intronic mutation, and so cDNA analysis by RT-PCR will need to be performed.
In conclusion, TMEM5 and ISPD are two additional important genes involved in cobblestone-LIS. They are thought to be glycosyl or nucleotide diP-sugar transferases and are responsible for the severe type A. Genotype-phenotype correlation improves the efficiency of genetic counseling through better orientation of molecular screening on the basis of the clinical severity of the cobblestone-LIS type. Our results do indeed suggest a molecular diagnostic strategy pointing through POMT1, POMT2, TMEM5, and ISPD in severe cobblestone-LIS type A. Because six other genes have been shown to be involved in the wide clinical spectrum of alpha-dystroglycanopathies, ISPD and TMEM5 will also be studied in less severe clinical cases presenting a spectrum of defects ranging from congenital muscular dystrophy (CMD) associated with mental retardation and cerebellar malformation to mild limb girdle muscular dystrophy (LGMD) with or without mental retardation. The same combination of an SNP array and exome sequencing in multiplex or consanguineous families is now underway in the remaining unexplained cases and can be expected to lead to the identification of so-far-unknown genes that contribute to cobblestone-LIS.
Acknowledgments
This study was supported by a grant from the GIS Institute for rare diseases. Exome sequencing was performed at the IGBMC (Institut de Génétique et de Biologie Moléculaire, Illkirch). The CGH and SNP analyses were performed by the genomic platform of Groupement Hospitalier Universitaire-Nord with grants from the Direction de L'Hospitalization et de l'Organisation des Soins (OPRC 207/35) for rare diseases and by the Necker genomic platform with funding from Association IMAGE to T.A.-B.
Published online: December 6, 2012
Footnotes
Supplemental data include two figures and three tables and can be found with this article online at http://www.cell.com/AJHG.
Contributor Information
Sandrine Vuillaumier-Barrot, Email: sandrine.vuillaumier@bch.aphp.fr.
Céline Bouchet-Séraphin, Email: celine.bouchet@bch.aphp.fr.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
Align GVGD, http://agvgd.iarc.fr/agvgd_input.php
BlastP (protein-protein BLAST), http://blast.ncbi.nlm.nih.gov/
dbSNP build 131, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?build_id%BC131
Exome Variant Server, evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
PolyPhen, http://genetics.bwh.harvard.edu/pph2/
Supplemental Data
References
- 1.van der Knaap M.S., Smit L.M., Barth P.G., Catsman-Berrevoets C.E., Brouwer O.F., Begeer J.H., de Coo I.F., Valk J. Magnetic resonance imaging in classification of congenital muscular dystrophies with brain abnormalities. Ann. Neurol. 1997;42:50–59. doi: 10.1002/ana.410420110. [DOI] [PubMed] [Google Scholar]
- 2.Nakano I., Funahashi M., Takada K., Toda T. Are breaches in the glia limitans the primary cause of the micropolygyria in Fukuyama-type congenital muscular dystrophy (FCMD)? Pathological study of the cerebral cortex of an FCMD fetus. Acta Neuropathol. 1996;91:313–321. doi: 10.1007/s004010050431. [DOI] [PubMed] [Google Scholar]
- 3.Barkovich A.J., Kuzniecky R.I., Jackson G.D., Guerrini R., Dobyns W.B. Classification system for malformations of cortical development: Update 2001. Neurology. 2001;57:2168–2178. doi: 10.1212/wnl.57.12.2168. [DOI] [PubMed] [Google Scholar]
- 4.Barkovich A.J., Guerrini R., Kuzniecky R.I., Jackson G.D., Dobyns W.B. A developmental and genetic classification for malformations of cortical development: Update 2012. Brain. 2012;135:1348–1369. doi: 10.1093/brain/aws019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dobyns W.B., Pagon R.A., Armstrong D., Curry C.J., Greenberg F., Grix A., Holmes L.B., Laxova R., Michels V.V., Robinow M., et al. Diagnostic criteria for Walker-Warburg syndrome. Am. J. Med. Genet. 1989;32:195–210. doi: 10.1002/ajmg.1320320213. [DOI] [PubMed] [Google Scholar]
- 6.Santavuori P., Somer H., Sainio K., Rapola J., Kruus S., Nikitin T., Ketonen L., Leisti J. Muscle-eye-brain disease (MEB) Brain Dev. 1989;11:147–153. doi: 10.1016/s0387-7604(89)80088-9. [DOI] [PubMed] [Google Scholar]
- 7.Fukuyama Y., Osawa M., Suzuki H. Congenital progressive muscular dystrophy of the Fukuyama type—Clinical, genetic and pathological considerations. Brain Dev. 1981;3:1–29. doi: 10.1016/s0387-7604(81)80002-2. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi K., Nakahori Y., Miyake M., Matsumura K., Kondo-Iida E., Nomura Y., Segawa M., Yoshioka M., Saito K., Osawa M., et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature. 1998;394:388–392. doi: 10.1038/28653. [DOI] [PubMed] [Google Scholar]
- 9.Beltrán-Valero de Bernabé D., Currier S., Steinbrecher A., Celli J., van Beusekom E., van der Zwaag B., Kayserili H., Merlini L., Chitayat D., Dobyns W.B., et al. Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker-Warburg syndrome. Am. J. Hum. Genet. 2002;71:1033–1043. doi: 10.1086/342975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Reeuwijk J., Janssen M., van den Elzen C., Beltran-Valero de Bernabé D., Sabatelli P., Merlini L., Boon M., Scheffer H., Brockington M., Muntoni F., et al. POMT2 mutations cause alpha-dystroglycan hypoglycosylation and Walker-Warburg syndrome. J. Med. Genet. 2005;42:907–912. doi: 10.1136/jmg.2005.031963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Godfrey C., Clement E., Mein R., Brockington M., Smith J., Talim B., Straub V., Robb S., Quinlivan R., Feng L., et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain. 2007;130:2725–2735. doi: 10.1093/brain/awm212. [DOI] [PubMed] [Google Scholar]
- 12.Taniguchi K., Kobayashi K., Saito K., Yamanouchi H., Ohnuma A., Hayashi Y.K., Manya H., Jin D.K., Lee M., Parano E., et al. Worldwide distribution and broader clinical spectrum of muscle-eye-brain disease. Hum. Mol. Genet. 2003;12:527–534. doi: 10.1093/hmg/ddg043. [DOI] [PubMed] [Google Scholar]
- 13.Clement E., Mercuri E., Godfrey C., Smith J., Robb S., Kinali M., Straub V., Bushby K., Manzur A., Talim B., et al. Brain involvement in muscular dystrophies with defective dystroglycan glycosylation. Ann. Neurol. 2008;64:573–582. doi: 10.1002/ana.21482. [DOI] [PubMed] [Google Scholar]
- 14.Balci B., Uyanik G., Dincer P., Gross C., Willer T., Talim B., Haliloglu G., Kale G., Hehr U., Winkler J., Topaloğlu H. An autosomal recessive limb girdle muscular dystrophy (LGMD2) with mild mental retardation is allelic to Walker-Warburg syndrome (WWS) caused by a mutation in the POMT1 gene. Neuromuscul. Disord. 2005;15:271–275. doi: 10.1016/j.nmd.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 15.Biancheri R., Falace A., Tessa A., Pedemonte M., Scapolan S., Cassandrini D., Aiello C., Rossi A., Broda P., Zara F., et al. POMT2 gene mutation in limb-girdle muscular dystrophy with inflammatory changes. Biochem. Biophys. Res. Commun. 2007;363:1033–1037. doi: 10.1016/j.bbrc.2007.09.066. [DOI] [PubMed] [Google Scholar]
- 16.Clement E.M., Godfrey C., Tan J., Brockington M., Torelli S., Feng L., Brown S.C., Jimenez-Mallebrera C., Sewry C.A., Longman C., et al. Mild POMGnT1 mutations underlie a novel limb-girdle muscular dystrophy variant. Arch. Neurol. 2008;65:137–141. doi: 10.1001/archneurol.2007.2. [DOI] [PubMed] [Google Scholar]
- 17.Brockington M., Yuva Y., Prandini P., Brown S.C., Torelli S., Benson M.A., Herrmann R., Anderson L.V., Bashir R., Burgunder J.M., et al. Mutations in the fukutin-related protein gene (FKRP) identify limb girdle muscular dystrophy 2I as a milder allelic variant of congenital muscular dystrophy MDC1C. Hum. Mol. Genet. 2001;10:2851–2859. doi: 10.1093/hmg/10.25.2851. [DOI] [PubMed] [Google Scholar]
- 18.Vuillaumier-Barrot S., Quijano-Roy S., Bouchet-Seraphin C., Maugenre S., Peudenier S., Van den Bergh P., Marcorelles P., Avila-Smirnow D., Chelbi M., Romero N.B., et al. Four Caucasian patients with mutations in the fukutin gene and variable clinical phenotype. Neuromuscul. Disord. 2009;19:182–188. doi: 10.1016/j.nmd.2008.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Bouchet C., Gonzales M., Vuillaumier-Barrot S., Devisme L., Lebizec C., Alanio E., Bazin A., Bessières-Grattagliano B., Bigi N., Blanchet P., et al. Molecular heterogeneity in fetal forms of type II lissencephaly. Hum. Mutat. 2007;28:1020–1027. doi: 10.1002/humu.20561. [DOI] [PubMed] [Google Scholar]
- 20.Devisme L., Bouchet C., Gonzalès M., Alanio E., Bazin A., Bessières B., Bigi N., Blanchet P., Bonneau D., Bonnières M., et al. Cobblestone lissencephaly: Neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain. 2012;135:469–482. doi: 10.1093/brain/awr357. [DOI] [PubMed] [Google Scholar]
- 21.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 23.Yokoyama-Kobayashi M., Yamaguchi T., Sekine S., Kato S. Selection of cDNAs encoding putative type II membrane proteins on the cell surface from a human full-length cDNA bank. Gene. 1999;228:161–167. doi: 10.1016/s0378-1119(99)00004-9. [DOI] [PubMed] [Google Scholar]
- 24.Lind T., Tufaro F., McCormick C., Lindahl U., Lidholt K. The putative tumor suppressors EXT1 and EXT2 are glycosyltransferases required for the biosynthesis of heparan sulfate. J. Biol. Chem. 1998;273:26265–26268. doi: 10.1074/jbc.273.41.26265. [DOI] [PubMed] [Google Scholar]
- 25.Ahn J., Lüdecke H.J., Lindow S., Horton W.A., Lee B., Wagner M.J., Horsthemke B., Wells D.E. Cloning of the putative tumour suppressor gene for hereditary multiple exostoses (EXT1) Nat. Genet. 1995;11:137–143. doi: 10.1038/ng1095-137. [DOI] [PubMed] [Google Scholar]
- 26.Wiggins C.A., Munro S. Activity of the yeast MNN1 alpha-1,3-mannosyltransferase requires a motif conserved in many other families of glycosyltransferases. Proc. Natl. Acad. Sci. USA. 1998;95:7945–7950. doi: 10.1073/pnas.95.14.7945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedersen L.C., Tsuchida K., Kitagawa H., Sugahara K., Darden T.A., Negishi M. Heparan/chondroitin sulfate biosynthesis. Structure and mechanism of human glucuronyltransferase I. J. Biol. Chem. 2000;275:34580–34585. doi: 10.1074/jbc.M007399200. [DOI] [PubMed] [Google Scholar]
- 28.Cheung P.K., McCormick C., Crawford B.E., Esko J.D., Tufaro F., Duncan G. Etiological point mutations in the hereditary multiple exostoses gene EXT1: A functional analysis of heparan sulfate polymerase activity. Am. J. Hum. Genet. 2001;69:55–66. doi: 10.1086/321278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zak B.M., Crawford B.E., Esko J.D. Hereditary multiple exostoses and heparan sulfate polymerization. Biochim. Biophys. Acta. 2002;1573:346–355. doi: 10.1016/s0304-4165(02)00402-6. [DOI] [PubMed] [Google Scholar]
- 30.Roscioli T., Kamsteeg E.J., Buysse K., Maystadt I., van Reeuwijk J., van den Elzen C., van Beusekom E., Riemersma M., Pfundt R., Vissers L.E., et al. Mutations in ISPD cause Walker-Warburg syndrome and defective glycosylation of α-dystroglycan. Nat. Genet. 2012;44:581–585. doi: 10.1038/ng.2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Willer T., Lee H., Lommel M., Yoshida-Moriguchi T., de Bernabe D.B., Venzke D., Cirak S., Schachter H., Vajsar J., Voit T., et al. ISPD loss-of-function mutations disrupt dystroglycan O-mannosylation and cause Walker-Warburg syndrome. Nat. Genet. 2012;44:575–580. doi: 10.1038/ng.2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Richard S.B., Bowman M.E., Kwiatkowski W., Kang I., Chow C., Lillo A.M., Cane D.E., Noel J.P. Structure of 4-diphosphocytidyl-2-C- methylerythritol synthetase involved in mevalonate- independent isoprenoid biosynthesis. Nat. Struct. Biol. 2001;8:641–648. doi: 10.1038/89691. [DOI] [PubMed] [Google Scholar]
- 33.Hunter W.N. The non-mevalonate pathway of isoprenoid precursor biosynthesis. J. Biol. Chem. 2007;282:21573–21577. doi: 10.1074/jbc.R700005200. [DOI] [PubMed] [Google Scholar]
- 34.Miziorko H.M. Enzymes of the mevalonate pathway of isoprenoid biosynthesis. Arch. Biochem. Biophys. 2011;505:131–143. doi: 10.1016/j.abb.2010.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schollen E., Kjaergaard S., Legius E., Schwartz M., Matthijs G. Lack of Hardy-Weinberg equilibrium for the most prevalent PMM2 mutation in CDG-Ia (congenital disorders of glycosylation type Ia) Eur. J. Hum. Genet. 2000;8:367–371. doi: 10.1038/sj.ejhg.5200470. [DOI] [PubMed] [Google Scholar]
- 36.Esapa C.T., Benson M.A., Schröder J.E., Martin-Rendon E., Brockington M., Brown S.C., Muntoni F., Kröger S., Blake D.J. Functional requirements for fukutin-related protein in the Golgi apparatus. Hum. Mol. Genet. 2002;11:3319–3331. doi: 10.1093/hmg/11.26.3319. [DOI] [PubMed] [Google Scholar]
- 37.Alhamidi M., Kjeldsen Buvang E., Fagerheim T., Brox V., Lindal S., Van Ghelue M., Nilssen O. Fukutin-related protein resides in the Golgi cisternae of skeletal muscle fibres and forms disulfide-linked homodimers via an N-terminal interaction. PLoS ONE. 2011;6:e22968. doi: 10.1371/journal.pone.0022968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yamamoto T., Kawaguchi M., Sakayori N., Muramatsu F., Morikawa S., Kato Y., Shibata N., Kobayashi M. Intracellular binding of fukutin and alpha-dystroglycan: relation to glycosylation of alpha-dystroglycan. Neurosci. Res. 2006;56:391–399. doi: 10.1016/j.neures.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 39.Barresi R., Michele D.E., Kanagawa M., Harper H.A., Dovico S.A., Satz J.S., Moore S.A., Zhang W., Schachter H., Dumanski J.P., et al. LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat. Med. 2004;10:696–703. doi: 10.1038/nm1059. [DOI] [PubMed] [Google Scholar]
- 40.Yoshida-Moriguchi T., Yu L., Stalnaker S.H., Davis S., Kunz S., Madson M., Oldstone M.B., Schachter H., Wells L., Campbell K.P. O-mannosyl phosphorylation of alpha-dystroglycan is required for laminin binding. Science. 2010;327:88–92. doi: 10.1126/science.1180512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hara Y., Kanagawa M., Kunz S., Yoshida-Moriguchi T., Satz J.S., Kobayashi Y.M., Zhu Z., Burden S.J., Oldstone M.B., Campbell K.P. Like-acetylglucosaminyltransferase (LARGE)-dependent modification of dystroglycan at Thr-317/319 is required for laminin binding and arenavirus infection. Proc. Natl. Acad. Sci. USA. 2011;108:17426–17431. doi: 10.1073/pnas.1114836108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hu Y., Li Z.F., Wu X., Lu Q. Large induces functional glycans in an O-mannosylation dependent manner and targets GlcNAc terminals on alpha-dystroglycan. PLoS ONE. 2011;6:e16866. doi: 10.1371/journal.pone.0016866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vajsar J., Baskin B., Swoboda K., Biggar D.W., Schachter H., Ray P.N. Walker-Warburg Syndrome with POMT1 mutations can be associated with cleft lip and cleft palate. Neuromuscul. Disord. 2008;18:675–677. doi: 10.1016/j.nmd.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 44.Lefeber D.J., Schönberger J., Morava E., Guillard M., Huyben K.M., Verrijp K., Grafakou O., Evangeliou A., Preijers F.W., Manta P., et al. Deficiency of Dol-P-Man synthase subunit DPM3 bridges the congenital disorders of glycosylation with the dystroglycanopathies. Am. J. Hum. Genet. 2009;85:76–86. doi: 10.1016/j.ajhg.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Godfrey C., Foley A.R., Clement E., Muntoni F. Dystroglycanopathies: coming into focus. Curr. Opin. Genet. Dev. 2011;21:278–285. doi: 10.1016/j.gde.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Messina S., Tortorella G., Concolino D., Spanò M., D’Amico A., Bruno C., Santorelli F.M., Mercuri E., Bertini E. Congenital muscular dystrophy with defective alpha-dystroglycan, cerebellar hypoplasia, and epilepsy. Neurology. 2009;73:1599–1601. doi: 10.1212/WNL.0b013e3181c0d47a. [DOI] [PubMed] [Google Scholar]
- 47.Lefeber D.J., de Brouwer A.P., Morava E., Riemersma M., Schuurs-Hoeijmakers J.H., Absmanner B., Verrijp K., van den Akker W.M., Huijben K., Steenbergen G., et al. Autosomal recessive dilated cardiomyopathy due to DOLK mutations results from abnormal dystroglycan O-mannosylation. PLoS Genet. 2011;7:e1002427. doi: 10.1371/journal.pgen.1002427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frost A.R., Böhm S.V., Sewduth R.N., Josifova D., Ogilvie C.M., Izatt L., Roberts R.G. Heterozygous deletion of a 2-Mb region including the dystroglycan gene in a patient with mild myopathy, facial hypotonia, oral-motor dyspraxia and white matter abnormalities. Eur. J. Hum. Genet. 2010;18:852–855. doi: 10.1038/ejhg.2010.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hara Y., Balci-Hayta B., Yoshida-Moriguchi T., Kanagawa M., Beltrán-Valero de Bernabé D., Gündeşli H., Willer T., Satz J.S., Crawford R.W., Burden S.J., et al. A dystroglycan mutation associated with limb-girdle muscular dystrophy. N. Engl. J. Med. 2011;364:939–946. doi: 10.1056/NEJMoa1006939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vuillaumier-Barrot S., Bouchet-Seraphin C., Chelbi M., Eude-Caye A., Charluteau E., Besson C., Quentin S., Devisme L., Le Bizec C., Landrieu P., et al. Intragenic rearrangements in LARGE and POMGNT1 genes in severe dystroglycanopathies. Neuromuscul. Disord. 2011;21:782–790. doi: 10.1016/j.nmd.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 51.Raducu M., Baets J., Fano O., Van Coster R., Cruces J. Promoter alteration causes transcriptional repression of the POMGNT1 gene in limb-girdle muscular dystrophy type 2O. Eur. J. Hum. Genet. 2012;20:945–952. doi: 10.1038/ejhg.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.