

Abstract
We studied five individuals from three Jewish Bukharian families affected by an apparently autosomal-recessive form of hereditary spastic paraparesis accompanied by severe intellectual disability, fluctuating central hypoventilation, gastresophageal reflux disease, wake apnea, areflexia, and unique dysmorphic features. Exome sequencing identified one homozygous variant shared among all affected individuals and absent in controls: a 1 bp frameshift TECPR2 deletion leading to a premature stop codon and predicting significant degradation of the protein. TECPR2 has been reported as a positive regulator of autophagy. We thus examined the autophagy-related fate of two key autophagic proteins, SQSTM1 (p62) and MAP1LC3B (LC3), in skin fibroblasts of an affected individual, as compared to a healthy control, and found that both protein levels were decreased and that there was a more pronounced decrease in the lipidated form of LC3 (LC3II). siRNA knockdown of TECPR2 showed similar changes, consistent with aberrant autophagy. Our results are strengthened by the fact that autophagy dysfunction has been implicated in a number of other neurodegenerative diseases. The discovered TECPR2 mutation implicates autophagy, a central intracellular mechanism, in spastic paraparesis.
Main Text
Hereditary spastic paraparesis (HSP) is a diverse group of neurodegenerative disorders estimated to affect 9.6 out of 100,000 individuals. It is characterized by axonal degeneration of the corticospinal or pyramidal motor and sensory tracts that control the lower extremities and results in progressive spasticity and hyperreflexia of the lower limbs.1–4 The disorder exhibits clinical variability with an onset seen from early childhood through 70 years of age. Pure HSP is characterized by progressive spasticity and leg weakness and is often associated with autosomal-dominant inheritance, whereas complicated HSP involves lower-limb spasticity accompanied by other neurological symptoms and tends to be autosomal recessive5 (also see GeneReviews in Web Resources). Previously, 48 spastic-paraplegia (SPG) loci for different HSP types (including X-linked HSP) have been reported, and of these, 27 have been associated with identified genes (Table S1, available online).6 Because of the heterogeneity and the clinical overlap in HSP, genetic diagnosis and testing have been a daunting task.
We have encountered five individuals from three apparently unrelated Jewish Bukharian families, which were independently referred to the pediatric neurological clinic at the Sheba Medical Center in Israel. Affected individuals presented with what appeared to be autosomal-recessive HSP of the complicated form. Four of the five individuals were available for DNA-sequencing analysis (Figure 1A). The study was approved by the institutional review board at the Sheba Medical Center, and informed consent was obtained from all affected individuals and their family members. All affected individuals were presented to the clinic during the second year of life as a result of hypotonia and developmental delay. They had short stature, mild brachycephalic microcephaly, a round face, a low anterior hairline, dental crowding, a short broad neck, and a chubby appearance. The neurological phenotype included motor and cognitive delay followed by moderate to severe intellectual disability and hypotonia that, in four out of five individuals who developed independent walking, evolved until the end of the first decade of life into a spastic, rigid ataxic gait. Speech, when present, was dysarthric, their faces were hypomimic (although with a friendly disposition), and deep-tendon reflexes were absent. The more cooperative individuals were dysmetric in a finger-to-nose maneuver. All affected individuals had recurrent pulmonary infections due to gastresophageal-reflux disease (MIM 109350). Ongoing severe central-apnea episodes were characteristic of all individuals initially during sleep and evolved with age into the wake state. Four of the individuals had recurrent episodes of decreased alertness, aggravation of hypotonia, and inefficient respiration requiring mechanical ventilation, and the younger affected individuals experienced spontaneous remission to the baseline. The oldest affected individual (II-2 in family B), a 20-year-old female, did not recover from her last episode and currently requires mechanical ventilation. Some of the acute deterioration episodes were related to intercurrent infections, but there was no evidence of metabolic decompensation. The two siblings from family B (II-1 and II-2) had infrequent short generalized tonic-clonic seizures. Individual II-1 from family A had recurrent ankle pressure ulcers, and individual II-2 from family B had transient severe encephalopathy with intermittent elevation of transaminases. Extensive metabolic testing was normal for all. Individual II-1 from family B died at the age of 5.5 years as a result of aspiration. MRI scans from two individuals (II-1 in family A and II-2 in family C) showed a thin corpus callosum, as well as cerebral and cerebellar atrophy (mainly vermian) (Figures 1B–1E). Muscle biopsy from three individuals (II-1 and II-2 in family B and II-2 in family C) showed normal histology, and respiratory-chain studies revealed normal function. Polysomnograms showed very frequent central apneas (>90/hr) accompanied by hypoxemia and poor response to oxygen enrichment. Electroencephalography showed diffuse slowing with poor background organization and no epileptiform activity. Electromyography (EMG) and nerve-conduction studies were normal.
Figure 1.
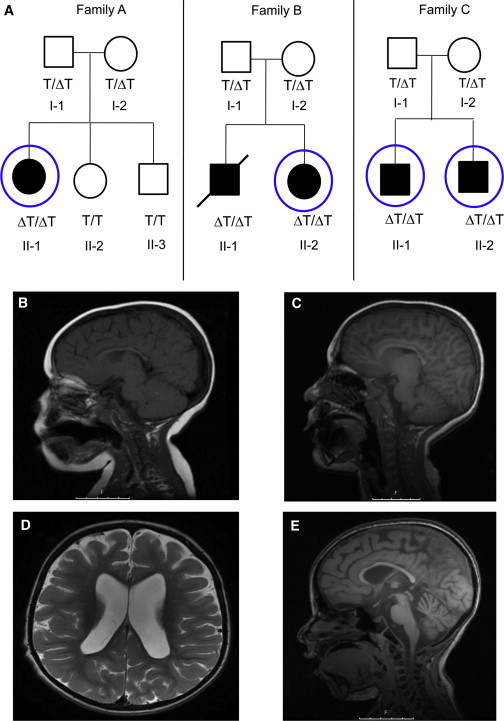
Pedigrees and Neuroimaging Phenotypes
(A) Pedigrees of the three Jewish Bukharian families. Filled symbols represent affected individuals, roman numerals indicate generations, and numbers are serial within the family. The slash represents the deceased individual with DNA available from a remaining muscle biopsy. TECPR2 genotype status for the c.3416delT variant is indicated by ΔT/ΔT for a homozygous mutation, ΔT/T for a heterozygous mutation, and T/T for a wild-type form. Circles indicate individuals who underwent exome sequencing.
(B) A T1 sagittal MRI from individual II-1 in family A shows a thin corpus callosum at the age of 3 years.
(C) A T1 sagittal MRI from individual II-1 in family A shows progressive cerebellar vermian atrophy at the age of 10 years.
(D) A T2 axial MRI from individual II-2 in family C at the age of 7 years shows enlarged lateral ventricles and deep sulci, indicating cerebral atrophy.
(E) A T2 sagittal MRI from individual II-2 in family C at the age of 7 years shows deep cerebral sulci (indicating cerebral atrophy), a thin corpus callosum, and vermian atrophy.
We performed exome sequencing in the four available individuals. On the basis of previous studies indicating that Bukhara Jewish families are distantly related7 and of the inferred autosomal-recessive inheritance in all three families, we expected a founder mutation and thus focused on homozygous variants shared among all affected individuals. Targeted exonic regions were captured by the SureSelect Human All Exon kit (Agilent Technologies). Samples were subsequently sequenced with a Genome Analyzer IIx (Illumina). The resulting sequence reads (113 bp) were aligned to the reference genome (NCBI human genome assembly build 36) with the Burrows-Wheeler Aligner.8 The average coverage for these four samples was 86×, and each sample had >96% of the bases covered. Genetic differences with respect to the reference genome were identified with the SAMtools variant-calling program,9 which identifies both single-nucleotide variants and small indels. We focused on variants annotated as functional, i.e., nonsynonymous, essential splice-site, stop-gain, stop-loss, and intron-exon-boundary variants, as well as frame-disrupting indels, by using SequenceVariantAnalyzer software.10 We selected variants that were rare (<3% minor allele frequency [MAF]) in dbSNP132 and in 909 available control samples previously subjected to exome and whole-genome sequencing as described11 (Table S2). The latter group did not include any Jewish Bukharian individuals or any known relatives of the three families and was not enriched with abnormal neuronal phenotypes.
One homozygous variant was shared among all four affected individuals and had a MAF = 0 in controls. This was a single-base deletion of T in exon 16 within TECPR2 (KIAA0329), transcript ENST00000359520 (c.3416delT, RefSeq accession number NM_014844.3) (Figure S1). The deletion causes a frameshift (p.Leu1139Argfs∗75) leading to a premature stop codon, resulting in the loss of four out of six TECPR domains (Figure 2). Genotyping of the c.3416delT variant by the ABI prism 3100 Genetic Analyzer (Life Technologies, Carlsbad, CA, USA) showed perfect cosegregation in all three families (Figure 1A). This included DNA from a muscle biopsy available for the deceased fifth individual (II-1 in family B), also found to be homozygous for the TECPR2 mutation. Subsequent application of PLINK analysis12 to all high-quality exome sequence variants showed that a 1 Mb region of homozygosity encompassing TECPR2 at chromosomal region 14q: 102,356,475–103,388,999 (corresponding to rs1904298–rs2295828 according to NCBI build 37/hg19) was shared among all four sequenced affected individuals. This analysis further increased our confidence in the TECPR2 mutation. We note, however, that such support is not fool proof and that, although unlikely, the culprit could be elsewhere in the region, including in an untranslated sequence.
Figure 2.
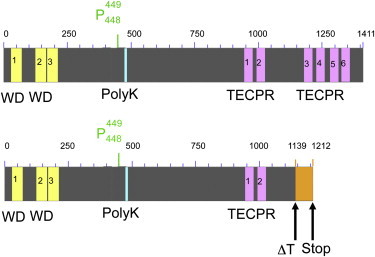
Protein Attributes of TECPR2
Domain structure and mutation according to UniProt and NCBI conserved domains.
Top: Conserved domains in the wild-type TECPR2 according to UniProt. Yellow represents WD (tryptophan-aspartic-acid dipeptide) repeats, purple indicates TECPR domains, and blue represents the polylysin tract.
Bottom: The effect of the mutation. Orange indicates the modified protein sequence resulting from a frameshift (at position 1,139) leading to a premature stop codon (at position 1,212).
We also interrogated the exomes of these affected individuals to determine whether copy-number variants (CNVs) could explain the observed idiosyncratic HSP phenotype. We therefore looked for exons that had very low or zero coverage and that would suggest homozygous (CNV) deletions, but we found none. In addition, we applied CoNIFER,13 an algorithm that attempts to detect deletions and duplications in exome data, but did not detect any CNVs shared by all affected individuals. Although CNV detection from exome data is known to be prone to error,14 this significantly decreases the probability that the causative variation is a genomic deletion or duplication.
To better estimate the frequency of the c.3416delT variant in healthy individuals, we genotyped 1,098 additional DNA samples from healthy controls that were not ethnically matched, and we found that none were carriers (the specific primers used are reported in Table S3). Thus, overall, this truncation was not found in any of the 2,007 non-Bukharian controls that we checked. We also genotyped 150 Jewish Bukharian controls and found the mutation in a heterozygous form in 4 out of the 300 chromosomes, accounting for an allele frequency of 0.013 in this ethnic group. This implies that the disease could be seen in 1.7 out of 10,000 endogamy couples, consistent with our identification of three affected families by an admittedly incomplete screen in a community of 150,000 Israeli Bukharian Jews with likely partial endogamy. To test the effect of the mutation, we derived full-length cDNA from the human kidney cell line HEK293 and used PCR-directed mutagenesis to generate the c.3416delT form of the gene. C-terminal or N-terminal FLAG-tagged TECPR2 was made by two-step PCR-directed mutagenesis. We then transfected the constructs into monkey kidney cell line COS-7, human kidney cell line HEK293, and human epithelial cell line HeLa; derived total cDNA; and visualized it by semiquantitative RT-PCR (the specific primer sets used are reported in Table S3). Transcripts were seen for both the normal and mutated forms (Figure 3A). We then used FLAG M2 monoclonal antibody (Sigma Aldrich), human TECPR2 polyclonal antibody (Sigma Aldrich), or GAPDH monoclonal antibody (Cell Signaling Technology) to detect TECPR2 forms. TECPR2 was detected by anti-FLAG and anti-TECPR2 immunoblotting only in the wild-type, but not in the mutant, transfection (Figures 3B and 3C). This suggests that the mistranslated and truncated protein was degraded in these cells. Inhibiting the proteasome degradation pathway by MG132 and lactacystin rescued the mutated protein (Figures 3D and 3E and Figure S2). This indicates that the mutated protein was targeted for proteasome degradation.
Figure 3.
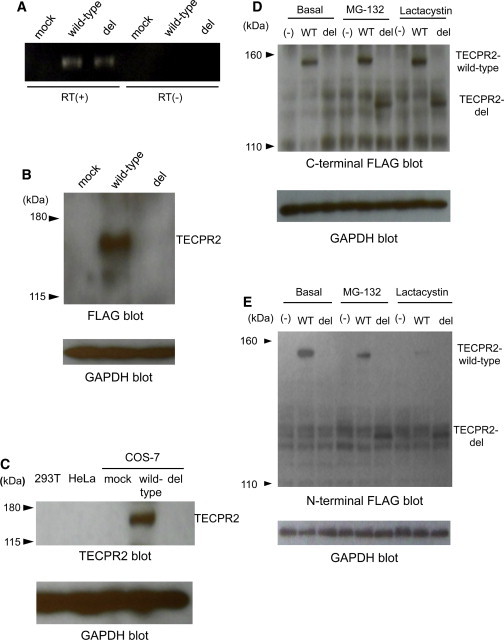
Effect of TECPR2 Mutation
(A) Semiquantatative RT-PCR analysis of the fate of TECPR2 mRNA expression in COS-7 transfectants for the wild-type and c.3416delT (“del”) forms of the transcript. “Mock” indicates the empty vector, “RT(+)” is with reverse transcriptase, and “RT(−)” is without reverse transcriptase. The data reveal no effect of the deletion on PCR-amplified mRNA levels.
(B) Immunoblotting of TECPR2 levels with FLAG monoclonal antibody. Notations are the same as in (A). GAPDH was used as a loading control. The data reveal a major disappearance of p. Leu1139Argfs∗75 TECPR2. Wild-type TECPR2 is seen at the expected molecular weight (154 kDa).
(C) Immunoblotting with the TECPR2 antibody. 293T and HeLa are unmodified cell lines; endogenous TECPR2 is not detectable. Labels are the same as in (A) and (B). Wild-type TECPR2, but not the p.Leu1139Argfs∗75 form, is observed in transfected COS-7 cells.
(D) Effect of proteasome inhibition with MG132 and lactacystin. Immunoblotting with FLAG monoclonal antibody for COS-7 transfectants of C-terminal FLAG-tagged TECPR2 is shown. Rescue of the altered TECPR2 and enhancement of wild-type TECPR2 are revealed upon proteasome inhibition. The following abbreviations are used: (−), mock transfected; WT, wild-type; del, variant; and GAPDH, loading control.
(E) Effect of proteasome inhibition with MG132 and lactacystin. Immunoblotting with FLAG monoclonal antibody for COS-7 transfectants of N-terminal FLAG-tagged TECPR2 is shown. Notations are the same as in (D).
The protein encoded by TECPR2 has recently been shown by immunoprecipitation-based proteomic analysis to interact with the six human Atg8 homologs, including the MAP1LC3 group (LC3). In parallel, it was shown to be a positive regulator of autophagosome accumulation by a TECPR2 siRNA approach.15 Autophagy is the major protein degradation system responsible for the turnover of bulky cellular constituents (protein aggregates and organelles), contributing to cellular homeostasis and survival.16 Dysfunction of autophagy has been proposed as an underlying mechanism for several neurodegenerative and muscle diseases.17,18 We also note that TECPR2 is highly expressed in the human brain, especially in the prefrontal cortex,19,20 providing a potential basis for the organ-specific outcome of the deleterious TECPR2 mutation.
We thus hypothesized that the truncated TECPR2 leads to modifications of autophagy and thus potentially results in the observed phenotype.
To test this, we derived skin fibroblasts from an affected individual (II-2 in family B) and from an unrelated healthy control, and we examined the effect of the mutation on the amount and cellular disposition of two endogenous autophagy-related proteins. The first is the cargo-recruiting polyubiquitin-binding SQSTM1 (p62), which, together with cargo proteins, is delivered by autophagosomes to lysosomal degradation.21,22 The second is the ubiquitin-like autophagy-initiation MAP1LC3B (LC3), for which starvation-induced autophagy leads to a transition from the cytosolic form (LC3I) to the autophagosome-associated, phosphatidylethanolamine-conjugated form (LC3II).23,24 We induced autophagy by starvation in nutrient-poor medium for 6 hr and employed bafilomycin A, an inhibitor of autophagosome-lysosome fusion, reported to enhance p62 and LC3II levels. The variously treated fibroblast lysates were subjected to SDS-PAGE followed by immunoblotting as described.25 The TECPR2 antibody used in Figure 3C was ineffective in detecting endogenous protein in the present experiment. The presence of autophagosomes in the starvation and bafilomicyn A condition was evidenced by transmission electron microscopy (Figure 4C), preformed as previously described.26 In general, whereas starvation alone led to a slight diminution in the amount of both proteins, the addition of bafilomycin A led to a considerable augmentation of the amounts of p62 and LC3II in the control.
Figure 4.
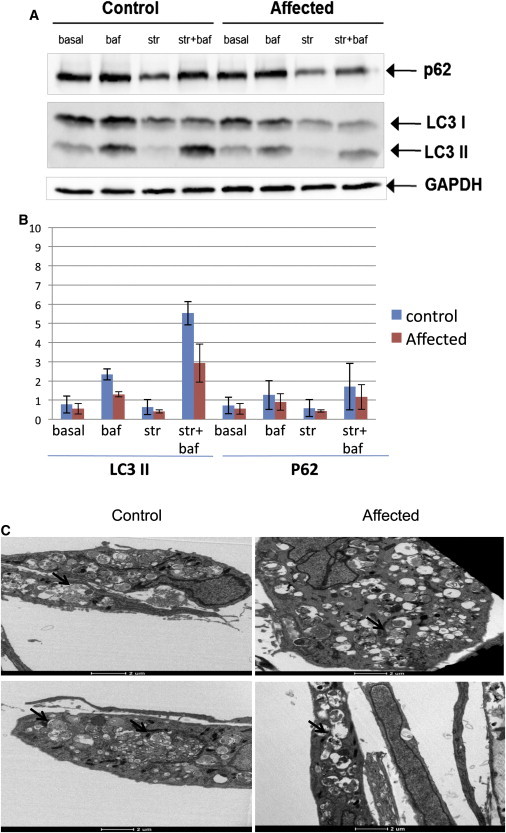
Effect of the Mutation on Autophagy Markers
(A) Immunoblotting with p62 antibody and LC3 antibody for skin-fibroblast lysates of both the affected individual and the control. Cell lines were used at an early passage, and there were a maximum of seven passages. LC3I is the cytoplasmic form, and LC3II is the phosphatidylethanolamine-conjugated form. GAPDH was used as a loading control. The following abbreviations are used: basal, rich medium; str, starvation; and baf, with the lysosomal inhibitor bafilomycin A.
(B) Summary of three biological replicates (Figure S3), each in duplicate, for experiments as shown in (A) (with the same notations) with the use of immunoblot scan quantitation and normalization by GAPDH control. Error bars are the SD for the four replicates. The data show across-the-board diminution of both LC3II and p62 amounts in the affected individual as compared to the control.
(C) Skin fibroblasts of an affected individual (right) and a healthy control (left) were incubated under starvation conditions in the presence of bafilomycin A for 6 hr, and thin sections were visualized by transmission electron microscopy as described.26 Arrows show representative autophagosomes or autolysosomes and a possible slight diminution of organelle accumulation within autophagic bodies in the affected fibroblasts.
In comparison, the affected-individual sample showed an across-the-board diminution of p62 and LC3II levels under all conditions (Figures 4A and 4B). Furthermore, we performed siRNA knockdown of TECPR2 in HeLa cells transfected by DharmaFect 1 (Dharmacon) with siRNA SMARTpools (50 nM siRNA SMARTpool), consisting of four RNA duplexes all targeting TECPR2; we then compared them to the nontargeting siRNA control and examined their autophagy-related phenotype. For validating the knockdown effect on TECPR2, quantitative real-time PCR was carried out with platinum SYBR Green qPCR SuperMix-UDG with ROX (Invitrogen, Life Technologies), and HPRT was used as the quantification control. The primer sequences used can be found in Table S3. By 72 hr, a minimal level of TECPR2 could be detected in comparison to the nontargeting siRNA control (Figure 5A). In cells in which TECPR2 was knocked down, we observed a major reduction of the bafilomycin-induced LC3II immunoreactivity. Of note, p62 amounts were much less affected by the mutation (Figure 5B). Using confocal immunofluorescence analysis as described,26 we confirmed these observations for both proteins (Figures 5C and 5D and Figure S4).
Figure 5.
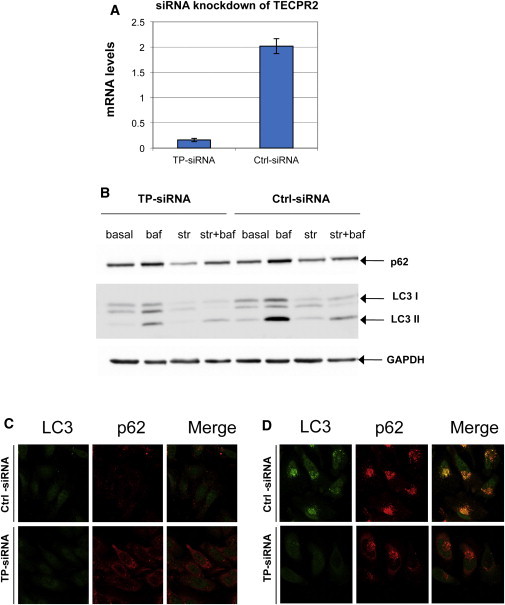
TECPR2 Knockdown in HeLa Cells
(A) Real-time PCR amplification of cDNA from transfected HeLa cells with either nontargeting siRNA (Ctrl-siRNA) or a pool of four TECPR2 siRNAs (TP-siRNA) with the use of primers specific to TECPR2 (exons 16 and 17).
(B) Immunoblotting with p62 antibody and LC3 antibody for HeLa cell lysates transfected with either nontargeting siRNA (Ctrl-siRNA) or a pool of four TECPR2 siRNAs (TP-siRNA). Notation is the same as in Figure 4A. LC3II levels are mostly affected by the knockdown, whereas the effect is less pronounced for p62.
(C) Basal levels of LC3II and p62 in HeLa cells in which TECPR2 is knocked down by siRNA are viewed by immunofluorescence confocal microscopy. Notation is the same as in (B). The following colors are used: green, LC3 antibody; red, p62 antibody; and yellow, merger of both signals.
(D) LC3II and p62 levels in the same cells under the conditions of starvation + bafilomycin. For nontarget siRNA, punctate perinuclear structures, stained for both p62 and LC3II, are most likely autophagosomes. TECPR2 siRNA knockdown shows strongly attenuated LC3II signal, whereas the p62 signal is only slightly diminished.
These combined results suggest that the presumed knockdown of TECPR2 in the HSP subjects brings upon a decreased accumulation of LC3II-labeled autophagosomes and attenuates the delivery of LC3II and p62 to lysosomal degradation. This indicates that the autophagy pathway is impaired, but not completely eliminated, given that we observed a lower impact on p62 protein levels. Of note, p62 is selectively recruited into autophagosomes, and therefore, even partial autophagic activity might be sufficient to deliver this protein to the lysosome.
For a group of progressive neurodegenerative diseases such as Huntington disease (MIM 143100), Alzheimer disease (MIM 104300), Parkinson disease (MIM 168600), spinocerebellar ataxias, and amyotrophic lateral sclerosis, mutant proteins become aggregate prone and hence less accessible to proteasome degradation; they thus become more dependent on autophagy for their clearance.18,27,28 These studies have shown that constitutive autophagy is essential to the survival of motor neurons and that failure to induce autophagosome formation results in cytosolic persistence of unsequestered cargo; this could promote aggregation of intracellular components leading to cytotoxicity and could disrupt neural function.29,30 Alternatively, malformation of autophagosomes could lead to malfunction at downstream cellular pathways, suggesting that subsequent autophagic steps are modified.
The core neuropathology of HSP is distal degeneration of the lateral corticospinal tract.31 Many biochemical mechanisms, including mitochondrial mechanisms, endoplasmic-reticulum shaping, endosomal trafficking, and microtubule stability, contribute to axonal function within the corticospinal tracts and affect anterograde and retrograde axonal transport.32 Another form of recessive, early-onset complicated HSP, SPG20 (Troyer syndrome [MIM 275900]), shows some similarity to the presently described SPG49: both syndromes include short stature and dismorphic features but no eye involvement or EMG abnormalities. Interestingly, Troyer syndrome involves a homozygous frameshift mutation, c.1110delA (p.Lys370Asnfs∗30), in SPG20 (MIM 607111, RefSeq NM_015087.4), leading to degradation of the spartin protein implicated in endosomal trafficking,33,34 an allied intracellular-membrane-transport pathway, thus supporting the implication of autophagy on the cellular mechanism of spastic paraparesis.
TECPR2 has a relatively high molecular mass (1,411 amino acids), hinting at a molecular linker function. It is unique in having both WD (tryptophan-aspartic-acid repeat) domains and TECPR domains, both implicated in protein-protein interactions involved in diseases.35,36 In its domain disposition, TECPR2 appears to be specific to mammals in that lower organisms show orthologs with either WD or TECPR domains, but not both (Figure S5). TECPR2 shows relatively weak but significant similarity (by BLAST via PairsDB37) to two other human proteins (Figure S5). One is TECPR1, implicated in selective recruitment of bacteria into autophagosomes38–40 and in autophagosome assembly.41 The other is HPS5, underlying a specific type of Hermansky-Pudlak syndrome (MIM 614074), which involves dysfunction of lysosome-related organelles.42 The latter information further strengthens the functional classification of TECPR2 and its disease affiliation.
Our observation of three independent Jewish Bukharian families affected by a unique spectrum of symptoms resulted in the identification of a different form of complicated HSP caused by a variation in TECPR2. We thus propose calling the present disease SPG49 and adding it to the SPG list (Table S1)5,6 (also see GeneReviews in Web Resources). Although the phenotype described here is quite remarkable, it remains to be established whether the involvement of an autophagic gene is unique to this disease subtype and ethnicity.
It would be useful to look in the future for other potential variants in the same or related genes, especially ones with evidence of peripheral and autonomic involvement, in other affected individuals with complicated HSP.
In summary, the results presented here highlight the importance and diagnostic value of accurate phenotyping combined with exome and whole-genome sequencing in deciphering the disease-causing mutation, even in relatively isolated cases with very few unrelated affected individuals. Identification of additional families in the same specific community, or outside it (potentially with different mutations), would provide further evidence implicating TECPR2 in this added form of HSP.
Acknowledgments
We thank the individuals and their families, as well as the following individuals for contributing control samples: J. Burke, C. Hulette, K. Welsh-Bohmer, F.J. McMahon, N. Akula, J. Hoover-Fong, N.L. Sobreira, D. Valle, M.C. Manzini, A. Poduri, N. Calakos, David H. Murdock, The MURDOCK Study Community Registry and Biorepository, J.P. McEvoy, A. Need, J. Silver, M. Silver, E.J. Holtzman, G. Cavalleri, N. Delanty, C. Depondt, S. Sisodiya, W.B. Gallentine, E.L. Heinzen, A.M. Husain, K.N. Linney, M.A. Mikati, K. Pelak, R.A. Radtke, S.R. Sinha, N.M. Walley, D. Koltai Attix, O. Chiba-Falek, E.T. Cirulli, V. Dixon, J. McEvoy, K. Schmader, S. McDonald, H.K. White, M. Yanamadala, the Carol Woods and Crosdaile Retirement Communities, V. Shashi, P. Lugar, W.L. Lowe, S.M. Palmer, D. Marchuk, D. Levy, Z. Farfel, D. Lancet, E. Pras, Y. Jiang, Q. Zhao, J. Milner, D. Daskalakis, R. Gbadegesin, M. Winn, A. Holden, E. Behr, R.H. Brown, Jr., S. Kerns, and H. Oster. We thank the Weizmann Institute Electron Microscopy unit. This study was funded by the American Recovery and Reinvestment Act; Bryan Alzheimer’s Disease Research Center funded by the National Institute on Aging; National Institute of Mental Health; Ellison Medical Foundation New Scholar award; Division of Intramural Research at the National Institute of Allergy and Infectious Diseases (NIAID) (National Institutes of Health) and Center for HIV/AIDS Vaccine Immunology under NIAID (to D.B.G.); Crown Human Genome Center and Benoziyo Center of the Weizmann Institute of Science (to D.L.); and Legacy Heritage Fund, Israeli Science Foundation, and German Israeli Foundation (to Z.E).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GeneCards, http://www.genecards.org/
GeneReviews, Fink, J.K. (1993). Hereditary Spastic Paraplegia Overview, http://www.ncbi.nlm.nih.gov/books/NBK1509/
NCBI Conserved Domains Database, http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd.shtml
NCBI dbSNP, http://www.ncbi.nlm.nih.gov/snp/
Online Mendelian Inheritance in Man (OMIM), http://omim.org/
PairsDB, http://pairsdb.csc.fi
UCSC Genome Browser, http://genome.ucsc.edu/
References
- 1.Stevanin G., Santorelli F.M., Azzedine H., Coutinho P., Chomilier J., Denora P.S., Martin E., Ouvrard-Hernandez A.M., Tessa A., Bouslam N. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat. Genet. 2007;39:366–372. doi: 10.1038/ng1980. [DOI] [PubMed] [Google Scholar]
- 2.Roşulescu E., Stănoiu C., Buteică E., Stănoiu B., Burada F., Zăvăleanu M. Hereditary spastic paraplegia. Rom. J. Morphol. Embryol. 2009;50:299–303. [PubMed] [Google Scholar]
- 3.Schüle R., Schöls L. Genetics of hereditary spastic paraplegias. Semin. Neurol. 2011;31:484–493. doi: 10.1055/s-0031-1299787. [DOI] [PubMed] [Google Scholar]
- 4.Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 2012;35:25–47. doi: 10.1146/annurev-neuro-062111-150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Depienne C., Stevanin G., Brice A., Durr A. Hereditary spastic paraplegias: An update. Curr. Opin. Neurol. 2007;20:674–680. doi: 10.1097/WCO.0b013e3282f190ba. [DOI] [PubMed] [Google Scholar]
- 6.Salinas S., Proukakis C., Crosby A., Warner T.T. Hereditary spastic paraplegia: Clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- 7.Blumen S.C., Korczyn A.D., Lavoie H., Medynski S., Chapman J., Asherov A., Nisipeanu P., Inzelberg R., Carasso R.L., Bouchard J.P. Oculopharyngeal MD among Bukhara Jews is due to a founder (GCG)9 mutation in the PABP2 gene. Neurology. 2000;55:1267–1270. doi: 10.1212/wnl.55.9.1267. [DOI] [PubMed] [Google Scholar]
- 8.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ge D., Ruzzo E.K., Shianna K.V., He M., Pelak K., Heinzen E.L., Need A.C., Cirulli E.T., Maia J.M., Dickson S.P. SVA: Software for annotating and visualizing sequenced human genomes. Bioinformatics. 2011;27:1998–2000. doi: 10.1093/bioinformatics/btr317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Need A.C., McEvoy J.P., Gennarelli M., Heinzen E.L., Ge D., Maia J.M., Shianna K.V., He M., Cirulli E.T., Gumbs C.E. Exome sequencing followed by large-scale genotyping suggests a limited role for moderately rare risk factors of strong effect in schizophrenia. Am. J. Hum. Genet. 2012;91:303–312. doi: 10.1016/j.ajhg.2012.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krumm N., Sudmant P.H., Ko A., O’Roak B.J., Malig M., Coe B.P., Quinlan A.R., Nickerson D.A., Eichler E.E., NHLBI Exome Sequencing Project Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525–1532. doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alkan C., Kidd J.M., Marques-Bonet T., Aksay G., Antonacci F., Hormozdiari F., Kitzman J.O., Baker C., Malig M., Mutlu O. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behrends C., Sowa M.E., Gygi S.P., Harper J.W. Network organization of the human autophagy system. Nature. 2010;466:68–76. doi: 10.1038/nature09204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weidberg H., Shvets E., Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu. Rev. Biochem. 2011;80:125–156. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- 17.Levine B., Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Menzies F.M., Rubinsztein D.C. Broadening the therapeutic scope for rapamycin treatment. Autophagy. 2010;6:286–287. doi: 10.4161/auto.6.2.11078. [DOI] [PubMed] [Google Scholar]
- 19.Middleton F.A., Strick P.L. Anatomical evidence for cerebellar and basal ganglia involvement in higher cognitive function. Science. 1994;266:458–461. doi: 10.1126/science.7939688. [DOI] [PubMed] [Google Scholar]
- 20.Schmahmann J.D. Disorders of the cerebellum: Ataxia, dysmetria of thought, and the cerebellar cognitive affective syndrome. J. Neuropsychiatry Clin. Neurosci. 2004;16:367–378. doi: 10.1176/jnp.16.3.367. [DOI] [PubMed] [Google Scholar]
- 21.Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 22.Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shpilka T., Weidberg H., Pietrokovski S., Elazar Z. Atg8: An autophagy-related ubiquitin-like protein family. Genome Biol. 2011;12:226. doi: 10.1186/gb-2011-12-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakatogawa H., Ichimura Y., Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130:165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 25.Weidberg H., Shpilka T., Shvets E., Abada A., Shimron F., Elazar Z. LC3 and GATE-16 N termini mediate membrane fusion processes required for autophagosome biogenesis. Dev. Cell. 2011;20:444–454. doi: 10.1016/j.devcel.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 26.Weidberg H., Shvets E., Shpilka T., Shimron F., Shinder V., Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010;29:1792–1802. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasquali L., Longone P., Isidoro C., Ruggieri S., Paparelli A., Fornai F. Autophagy, lithium, and amyotrophic lateral sclerosis. Muscle Nerve. 2009;40:173–194. doi: 10.1002/mus.21423. [DOI] [PubMed] [Google Scholar]
- 28.Knaevelsrud H., Simonsen A. Fighting disease by selective autophagy of aggregate-prone proteins. FEBS Lett. 2010;584:2635–2645. doi: 10.1016/j.febslet.2010.04.041. [DOI] [PubMed] [Google Scholar]
- 29.Moreau K., Luo S., Rubinsztein D.C. Cytoprotective roles for autophagy. Curr. Opin. Cell Biol. 2010;22:206–211. doi: 10.1016/j.ceb.2009.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong E., Cuervo A.M. Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci. 2010;13:805–811. doi: 10.1038/nn.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wharton S.B., McDermott C.J., Grierson A.J., Wood J.D., Gelsthorpe C., Ince P.G., Shaw P.J. The cellular and molecular pathology of the motor system in hereditary spastic paraparesis due to mutation of the spastin gene. J. Neuropathol. Exp. Neurol. 2003;62:1166–1177. doi: 10.1093/jnen/62.11.1166. [DOI] [PubMed] [Google Scholar]
- 32.Henson B.J., Zhu W., Hardaway K., Wetzel J.L., Stefan M., Albers K.M., Nicholls R.D. Transcriptional and post-transcriptional regulation of SPAST, the gene most frequently mutated in hereditary spastic paraplegia. PLoS ONE. 2012;7:e36505. doi: 10.1371/journal.pone.0036505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Patel H., Cross H., Proukakis C., Hershberger R., Bork P., Ciccarelli F.D., Patton M.A., McKusick V.A., Crosby A.H. SPG20 is mutated in Troyer syndrome, an hereditary spastic paraplegia. Nat. Genet. 2002;31:347–348. doi: 10.1038/ng937. [DOI] [PubMed] [Google Scholar]
- 34.Bakowska J.C., Jupille H., Fatheddin P., Puertollano R., Blackstone C. Troyer syndrome protein spartin is mono-ubiquitinated and functions in EGF receptor trafficking. Mol. Biol. Cell. 2007;18:1683–1692. doi: 10.1091/mbc.E06-09-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li D., Roberts R. WD-repeat proteins: Structure characteristics, biological function, and their involvement in human diseases. Cell. Mol. Life Sci. 2001;58:2085–2097. doi: 10.1007/PL00000838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith T.F. Diversity of WD-repeat proteins. Subcell. Biochem. 2008;48:20–30. doi: 10.1007/978-0-387-09595-0_3. [DOI] [PubMed] [Google Scholar]
- 37.Heger A., Korpelainen E., Hupponen T., Mattila K., Ollikainen V., Holm L. PairsDB atlas of protein sequence space. Nucleic Acids Res. 2008;36(Database issue):D276–D280. doi: 10.1093/nar/gkm879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogawa M., Yoshikawa Y., Kobayashi T., Mimuro H., Fukumatsu M., Kiga K., Piao Z., Ashida H., Yoshida M., Kakuta S. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell Host Microbe. 2011;9:376–389. doi: 10.1016/j.chom.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 39.Levine B., Mizushima N., Virgin H.W. Autophagy in immunity and inflammation. Nature. 2011;469:323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rubinsztein D.C., Shpilka T., Elazar Z. Mechanisms of autophagosome biogenesis. Curr. Biol. 2012;22:R29–R34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 41.Chen D., Fan W., Lu Y., Ding X., Chen S., Zhong Q. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol. Cell. 2012;45:629–641. doi: 10.1016/j.molcel.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei M.L. Hermansky-Pudlak syndrome: A disease of protein trafficking and organelle function. Pigment Cell Res. 2006;19:19–42. doi: 10.1111/j.1600-0749.2005.00289.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.