

Abstract
Hereditary spastic paraplegia (HSP) is considered one of the most heterogeneous groups of neurological disorders, both clinically and genetically. The disease comprises pure and complex forms that clinically include slowly progressive lower-limb spasticity resulting from degeneration of the corticospinal tract. At least 48 loci accounting for these diseases have been mapped to date, and mutations have been identified in 22 genes, most of which play a role in intracellular trafficking. Here, we identified mutations in two functionally related genes (DDHD1 and CYP2U1) in individuals with autosomal-recessive forms of HSP by using either the classical positional cloning or a combination of whole-genome linkage mapping and next-generation sequencing. Interestingly, three subjects with CYP2U1 mutations presented with a thin corpus callosum, white-matter abnormalities, and/or calcification of the basal ganglia. These genes code for two enzymes involved in fatty-acid metabolism, and we have demonstrated in human cells that the HSP pathophysiology includes alteration of mitochondrial architecture and bioenergetics with increased oxidative stress. Our combined results focus attention on lipid metabolism as a critical HSP pathway with a deleterious impact on mitochondrial bioenergetic function.
Introduction
Hereditary spastic paraplegia (HSP), also known as Strümpell-Lorrain disease, is recognized as one of the most clinically and genetically heterogeneous groups of inherited neurodegenerative disorders. These disorders are mainly characterized by slowly progressive lower-limb spasticity that worsens over time. The symptoms are the consequence of corticospinal-tract degeneration.1–3 Affected subjects are clinically classified according to the absence (uncomplicated or pure HSP) or presence (complicated or complex HSP) of additional neurological or extraneurological signs. This clinical heterogeneity partially underlies the large genetic heterogeneity of this group of disorders; at least 48 loci have been mapped to date and account for all classical modes of inheritance.4,5 So far, mutations have been identified in 22 genes,5 most of which play a role in intracellular trafficking.6–8
Autosomal-recessive HSP (AR-HSP) is less common than the autosomal-dominant form, except in countries with a high rate of consanguinity.9,10 It is more often associated with clinically complex phenotypes, but pure forms of the disease can be due to mutations in SPG5/CYP7B1 (MIM 603711), SPG7/PGN (MIM 602783), and SPG30/KIF1A (MIM 601255) or can be linked to SPG28 (MIM 609340).11–15 Mutations in SPG11/KIAA1840 (MIM 610844) account for ∼20% of the autosomal-recessive (AR) forms,16 but many genes remain to be discovered given that 60% of AR-HSP is still genetically unexplained.5
Here, we report the identification of causative mutations in two genes after the use of next-generation sequencing focused on all exons of the SPG28 interval, as well as linkage mapping combined with systematic candidate-gene analysis in SPG49, identified in this study. These combined approaches enabled us to identify in individuals from three families four truncating mutations in DDHD1 (MIM 614603), encoding for a phosphatidic-acid (PA)-preferring phospholipase A1. In subjects from five different families, we also found five mutations in CYP2U1 (MIM 610670), encoding a P450 hydroxylase. We demonstrate in human cells that the pathophysiology of SPG28 and SPG49 includes alteration of mitochondrial architecture and bioenergetics with increased oxidative stress.
Subjects and Methods
Subjects
Ninety-nine index individuals from families affected by AR-HSP and without identified mutations in SPG11, SPG5, and SPG7, were included in this study and originated mainly from France, Italy, the Middle East, and North Africa. This study was approved by the local Bioethics Committee (approval number 03-12-07 from the Comité Consultatif pour la Protection des Personnes et la Recherche Biomédicale Paris-Necker to A. Durr and A. Brice). Written informed consent was signed by all index persons and by 39 additional participating members of the families before blood samples were collected for DNA extraction. All clinical evaluations included a full medical history and examination, estimation of the age of onset, observation of additional neurological signs, electroneuromyographic studies, and, when possible, brain MRI and/or computed-tomography (CT) scans.
Next-Generation Sequencing in Family FSP445
After exclusion of large genomic rearrangements in the SPG28 linkage interval by comparative genomic hybridization (CGH) in subjects FSP445-V.3 and FSP445-V.1 by chromosome-14-specific 385K NimbleGen arrays (data not shown), targeted enrichment and next-generation sequencing were performed on the DNA of individual FSP445-V.3 from the original SPG28-affected family.11 Enrichment of all 723 exons in chr14: 49,100,775–55,305,189 was performed by hybridization of shotgun fragment libraries to a custom Roche NimbleGen microarray according to the manufacturer’s recommendations. Three micrograms of amplified enriched DNA was used as input for massively parallel sequencing on a Roche 454-GS-FLX sequencer with Titanium reagents. Sequence data were then aligned with hg18 human genome as a reference. In total, 285,704 reads were obtained with an average length of 399 bp; 283,483 (99%) of those reads could be mapped, and 68% of them were on target. From the total of 723 enriched regions, 689 (95%) were covered entirely, and 99% of targeted bases were covered at least 10× (95% of all captured exons were covered entirely). The real coverage of enriched regions was 80×. Variants with fewer than three reads or accounting for less than 25% of all reads were excluded from the analysis. A total of 1,062 variations were identified. We excluded known validated SNPs present in dbSNP and the HapMap database. As expected, 88 of the remaining 239 variants were homozygous in this disease: 17 were intergenic, 63 were intronic, and 8, including 2 exonic variants, were in the mRNA.
Genome-wide Scan in Family FSP719
Exclusion of linkage to some of the most common AR-HSP-associated loci (SPG5, SPG15 [MIM 270700], SPG24 [MIM 607584], SPG28, SPG30, and SPG32 [MIM 611252]) and of mutations in SPG7 and SPG11 was performed prior to this study (data not shown).
A genome scan with 6,090 SNP markers (LINKAGE_V panel, Illumina) covering all chromosomes was performed on family FSP719. Genotypes were generated with BeadStudio software (Illumina) and analyzed with MERLIN 1.0,17 and all family members were considered. An AR inheritance, a penetrance of 95% without phenocopy, a disease allele frequency of 0.00001, equal allele frequencies for each marker, and equal male-female recombination rates were considered.
Linked and uninformative regions were explored by genotyping of 47 additional microsatellite markers in all family members. PCR amplicons were resolved on an ABI 3730 sequencer with the use of fluorescent primers, and the results were analyzed with GeneMapper 4 (Applied Biosystems). We manually constructed haplotypes by minimizing the number of recombination events. Genetic distances were taken from the MAP-O-MAT consortium.
Candidate-Gene Analysis
After exclusion of large genomic rearrangements by CGH in one person of SPG49-linked family FSP1015 (data not shown), all coding exons of genes in the SPG49 candidate interval, including flanking splicing sites and at least 50 bp of intronic sequence on each side, were analyzed by direct sequencing (primers are available from the authors upon request) with BigDye Chemistry (Applied Biosystems). The sequence products were run on an ABI 3730 sequencer, and electrophoretic profiles were compared with the reference hg18 sequence with the use of SeqScape 2.5 (Applied Biosystems).
Mutation screening of DDHD1 (RefSeq accession number NM_030637.2; hg19) and CYP2U1 (RefSeq NM_183075.2; hg19) was performed in 96 index cases by classical Sanger sequencing (primers and PCR conditions are available upon request).
The effects of mutations and amino acid conservation in other species were analyzed with ALAMUT 2.2 (Interactive Biosoftware), PolyPhen-2, and MutationTaster.18,19
Rearrangement Analysis
Exon deletions or duplications in CYP2U1 and DDHD1 were explored by quantitative multiplex PCR of short fragments (QMPSF; primers and PCR conditions are available from the authors upon request) and analyzed with GeneMapper 4.
Investigation of Mitochondrial Energetics and Oxidative Stress
The mitochondrial respiratory rate was measured in lymphocytes and fibroblasts grown in RPMI 1640 medium and DMEM, respectively, both of which contained 5 mM glucose and 2 mM glutamine (Glutamax) supplemented with 10% fetal bovine serum (PAA), 100 U/ml penicillin, and 100 U/ml of streptomycin. All experiments were performed with the same number of flask divisions in culture between cases and controls. For the lymphoblast cells (nonadherent), respiration was measured in real time on 1 × 106 cells per ml by polarography, with the use of the Hansatech Oxygraph system (Oxy 1, Hansatech), at 37°C in growth medium containing glucose and glutamine as energy substrates. We assessed respiratory coupling by adding oligomycin, a specific inhibitor of mitochondrial F1F0-ATPsynthase, in the cuvette of the oxygraph. The coupling of mitochondrial respiration with ADP phosphorylation was evaluated by the ratio of the routine respiratory rate to the rate measured in the presence of oligomycin (at steady state). Respiration was expressed as ngatom O per min per one million cells. Each cell culture was assayed at least three times (minimum of three different cultures according to the sample). For the fibroblasts, respiration was assayed on 96-well plates with the extracellular flux analyzer XF96 (Seahorse Bioscience).
The amount of mitochondria was compared on protein extracts from lymphoblastoid cell lines by immunoblot analysis according to standard procedures20 and by labeling of actin C4 (Abcam, 1/1,000) and TOM20 (BD Biosciences, 1/1,000). The total cellular ATP content and the mitochondrial contribution to cellular ATP production were measured by bioluminescence with the ATP Roche assay Kit II on 1 × 106 cells as described elsewhere.21 The concentration of reactive oxygen species was measured with the CMH2DCFDA fluorescent probe on a fluorometer (SAFAS) as previously reported.21
The measurement of respiratory-chain-enzyme activities was performed spectrophotometrically with standardized techniques as described previously.22 In brief, fibroblasts and lymphoblasts of affected and control subjects were grown in DMEM or RPMI and harvested at 70% confluency. We performed cell- and mitochondrial-membrane disruption by freezing the cells in liquid nitrogen, thawing them at room temperature, and subsequent sonication. Total protein content was measured with the bicinchoninic acid assay, and maximal activities of respiratory-chain complex I (NADH: ubiquinone oxidoreductase), complex II (succinate dehydrogenase), complex III (ubiquinol cytochrome c reductase), and complex IV (cytochrome c oxydase) were assayed at 30°C and expressed as nmol/min/mg of protein. Mitochondrial transmembrane electric potential (Δψ) was measured on a fluorometer (SAFAS Xenius) with the potentiometric dye tetramethylrhodamine methyl esther (TMRM) obtained from Invitrogen as previously detailed.23
mRNA-Expression Analysis
Total RNA was extracted from various tissues and different development stages of four wild-type c57bl6 mice with the RNeasy Mini kit (QIAGEN) and was used for generating cDNA with the Superscript III First-Strand Synthesis SuperMix for quantitative RT-PCR (Invitrogen). Real-time quantitative expression analysis of DDHD1, CYP2U1, and RAD51 (control [MIM 179617]) was performed on a Roche LC480-1536 with QIAGEN Quantitect primer assays in triplicate. Expression levels relative to PGK1 (MIM 311800) or PP1A, used as reference genes, were determined with qbase Plus software (Biogazelle) and were then compared with the Pearson test.
In lymphoblasts from individuals FSP445-4 and FSP445-6, we analyzed the effect on mRNA splicing of a variant affecting the last codon of exon 7 of DDHD1 and predicted to alter splicing by the Splice Site Prediction by Neural Network. This was done on polyA+ RNA that was reversely transcribed with the 1st Strand cDNA Synthesis Kit (Roche) according to the manufacturer’s random primer protocol and was followed by subcloning and direct sequencing of the PCR products with specific primers (available upon request).
Overexpression Analysis
HeLa cells used for transient transfection of the CYP2U1 cDNA fused to green fluorescent protein (GFP) (Origen) were maintained in DMEM (Invitrogen) supplemented with fetal calf serum and antibiotics (100 UI/ml penicillin and 100 μg/ml streptomycin). Cells (10,000 cells per well) were transfected with the use of Fugene Xtreme Gene according to the manufacturer’s instructions (Promega). After 48 hr, cells were fixed with 4% paraformaldehyde, washed, permeabilized with 0.25% Triton/PBS, blocked for 1 hr with 10% normal goat serum (NGS) before being incubated for 1 hr with primary antibodies at room temperature in 3% NGS/PBS, and then washed in PBS and incubated for 45 min with secondary antibodies in 3% NGS/PBS. Cultures were mounted with Vectashield HardSet Mounting Medium (Eurobio) or counterstained with DAPI (1 μg/ml, Sigma) and mounted with Fluoromount-G (Southern Biotech). Immunocytochemistry was performed with the following primary antibodies: rabbit anti-SDHA (Abcam), anti-Calregulin (Santa Cruz Biotechnology), and mouse anti-FLAG (Sigma). Secondary antibodies were alexa-fluor 568 and 647 (Invitrogen) anti-mouse and anti-rabbit. Images of CYP2U1 overexpression were acquired with a Zeiss VivaTome microscope with a 60× objective. AxioVision software (Zeiss) was used for analyzing one stack for visualizing the colocalization. For quantitative colocalization analysis of CYP2U1 with mitochondria or endoplasmic reticulum (ER), all images were processed identically with MBF Image J software and were quantified with Intensity Correlation Analysis plug-in (ICA) as defined elsewhere.24 For each stack, similar background signal was subtracted, and images were analyzed as a z-series projection taken in intervals 0.2 mm deep. Two coefficients were calculated for each cell: (1) Rr is the Pearson’s correlation coefficient and ranges from −1 to +1 (a value of 1 represents perfect correlation, −1 represents perfect exclusion, and 0 represents random localization) and (2) ICQ is the intensity correlation quotient in which colocalization is defined as the synchronous increase or decrease in fluorescence intensities.24,25 The ICQ is based on the nonparametric sign-test analysis of the PDM (product of the differences from the mean) values and is equal to the ratio of the number of positive PDM values to the total number of pixel values. The ICQ values are distributed between −0.5 and +0.5 (−0.5 represents a random staining, and +0.5 represents perfect overlap). Each experiment was performed on three independent cells preparations, and 25–30 cells were quantified for each condition.
Fibroblasts were transfected with a pCMV/mito/GFP vector (Invitrogen) in which the Myc epitope was deleted26 with the use of the Neon system (Invitrogen) according to the manufacturer‘s instructions. Twenty four hours after transfection, live cells were observed on a Zeiss Axiovert 200 inverted video-microscope with a 63× oil objective. Metamorph 7.7.7.0 software was used for acquiring the image.
Results
Identification of Mutations in SPG28/DDHD1
We previously described a consanguineous Moroccan family (with AR inheritance) in which we mapped the disease locus, SPG28, to chromosome 14q.11 We sequenced all exons of the SPG28 interval in one affected subject (FSP445-V.3) from the original SPG28-affected kindred by using a custom Roche-NimbleGen Capture assay and massively parallel sequencing on a Roche 454-GS-FLX sequencer. From 239 variants identified, we focused our analysis on the two homozygous exonic variants not reported as polymorphisms: (1) the c.427G>T (p.Ala143Ser) transversion in SAMD4A (MIM 610747; RefSeq NM_015589.4), encoding SMAUG1, an RNA-binding protein involved in mRNA silencing and deadenylation in postsynaptic densities, and (2) the c.1766G>A (p.Arg589Gln) variant located in the last nucleotide of exon 7 of DDHD1 (encoding an enzyme of lipid metabolism) and affecting correct DDHD1 mRNA splicing (Figure 1A, Figures S1 and S2, and Table S1, available online). These two variants cosegregated with the disease and were absent in controls. The deleterious nature of the DDHD1 mutation let us focus on this gene. Further screening of all DDHD1 coding exons by Sanger sequencing and QMPSF in 96 index cases affected by HSP with an unknown genetic cause identified mutations absent in controls in two individuals (Figures 1B and 1C, Figure S2, and Table S1). One simplex French case (FSP375-II.1) harbored heterozygous mutations c.1249C>T (p.Gln417∗) and c.2438-1G>T in exon 4 and intron 11, respectively. DNA of relatives or mRNA was not available, so we could not exclude that the mutations were in cis, although this is unlikely given their rarity (Table S1). One Turkish boy and his affected brother harbored the homozygous c.1874del (p.Leu625∗) mutation, which was also present at the heterozygous state in their healthy parents.
Figure 1.
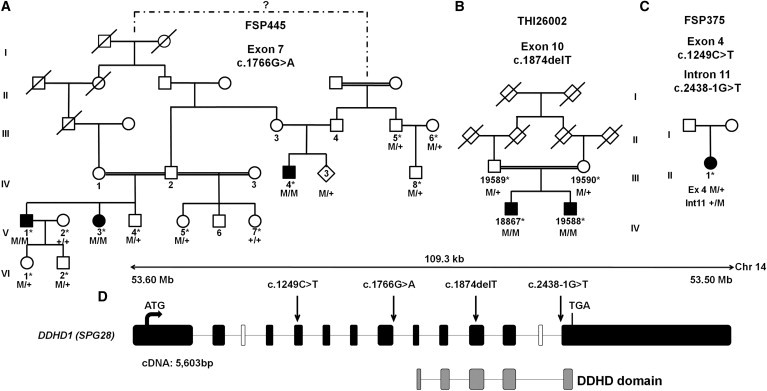
Mutations in DDHD1
(A–C) Family trees and segregation analysis of the mutations identified in families FSP445 (A), THI26002 (B) and FSP375 (C). Squares represent males, circles represent females, diamonds indicate anonymized subjects, filled symbols represent affected individuals, and a double line indicates consanguinity. The following abbreviations are used: M, mutation; +, wild-type; and ∗, sampled individuals. The electrophoregrams are shown in Figure S2.
(D) A graphical representation of DDHD1 (RefSeq NM_030637.2) on chromosome 14 indicates the DDHD domain (gray boxes) and the location of the mutations (c.1249C>T [p.Gln417∗], c.1766G>A [p.Arg589Gln; r.spl?], c.1874delT [p.Leu625∗], and c.2438-1G>T [r.spl?]) (arrows). Black and white boxes represent coding and noncoding exons, respectively. The DDHD1 transcript is 5,603 bp long and is composed of 12 exons that encode an 872 amino acid protein. There are three known isoforms of DDHD1; isoforms b and c are longer than isoform a because they include an alternate in-frame coding exon (white box). The sequence contains a lipase consensus domain and also includes a putative coiled-coil-forming region and a DDHD domain between residues 611 and 858 of isoform a; the four conserved residues that can form a metal binding site are seen in phosphoesterase domains. This domain is found in retinal degeneration B proteins, as well as in a family of probable phospholipases.
Clinical data from the three additional SPG28-affected subjects identified in this study extended the clinical spectrum of this disease compared to the pure phenotype originally described.11 When examined at 30 years of age, the two Turkish brothers showed progressive spastic gait (which had been developing since adolescence), and one had a cerebellar oculomotor disturbance with saccadic eye pursuit. No other signs were noticed, and brain and spine MRI, sensory-evoked potentials (tibial nerve and median nerve), and nerve-conduction studies were normal. The French SPG28-affected woman, aged 62 years at her latest examination, had suffered since infancy from HSP with axonal neuropathy but had unremarkable spinal and brain MRI.
Mapping of an AR-HSP-Associated Locus, SPG49
A genome scan with the Illumina LINKAGE Mapping V SNP Set was performed in family FSP719, composed of two nuclear consanguineous kindreds from Saudi Arabia. Multipoint linkage analysis (Figure S3A) identified on chromosomes 4, 19, and X three genomic regions with uninformative multipoint LOD scores between 0 and +1; these regions were later excluded with the use of 23 additional markers (data not shown). A fourth region of 34 consecutive SNPs on chromosome 4 presented a significant multipoint LOD score of +3.97 between markers rs1528381 and rs1525760. Further genotyping of 24 additional microsatellite markers and haplotype reconstructions restricted the region and confirmed a 5.9 Mb (6.29 cM) region of homozygosity by descent between markers D4S3256 and D4S2309 in all four affected individuals of the family (Figure S4). A maximum and significant multipoint LOD score of +4.76 was then reached (Figure S3B). This locus was named SPG49 according to the Human Genome Organization. A second Saudi Arabian AR-HSP-affected family was found to be linked to this locus with a multipoint LOD score of +2.4. Both families shared a portion of the same homozygous haplotype, suggesting a common ancestry and narrowing down the SPG49 candidate interval to the 3.8 Mb (4.42 cM) between markers D4S3256 and D4S2940 (Figure S4).
Identification of Mutations in SPG49/CYP2U1
In all coding exons of the 23 genes assigned to the SPG49 region, we identified a single CYP2U1 variant, c.947A>T (p.Asp316Val), which segregated with the disease at the homozygous state in the two SPG49-affected families (Figure 2A and Figure S2) and was not detected in healthy controls (Table S1). This variant affected an amino acid highly conserved during evolution among orthologs of CYP2U1 (Figure 2B) but also among other cytochrome P450 proteins (data not shown), including CYP7B1 (SPG5).15 The mutation was localized in the cytochrome P450 functional domain and was predicted to be damaging.18,19 Sanger sequencing and QMPSF analysis of all exons of CYP2U1 in 94 index cases affected by HSP with an undetermined genetic basis identified four mutations predicted to be damaging and absent in controls (Figures 2C–2E, Figure S2, and Table S1). A homozygous missense mutation affecting a conserved amino acid (c.1139A>G [p.Glu380Gly]) was detected in an Italian simplex case (ITAP9-II.1) with suspected consanguinity. A homozygous 13 bp deletion (in exon 1) leading to a frameshift (c.61_73del [p.Leu21Trpfs∗19]) segregated with the disease in two Egyptian brothers (from family HSP1363). Finally, two heterozygous mutations (c.784T>C [p.Cys262Arg] and c.1462C>T [p.Arg488Trp]) segregated in trans in two siblings with Spanish and Vietnamese ancestry.
Figure 2.
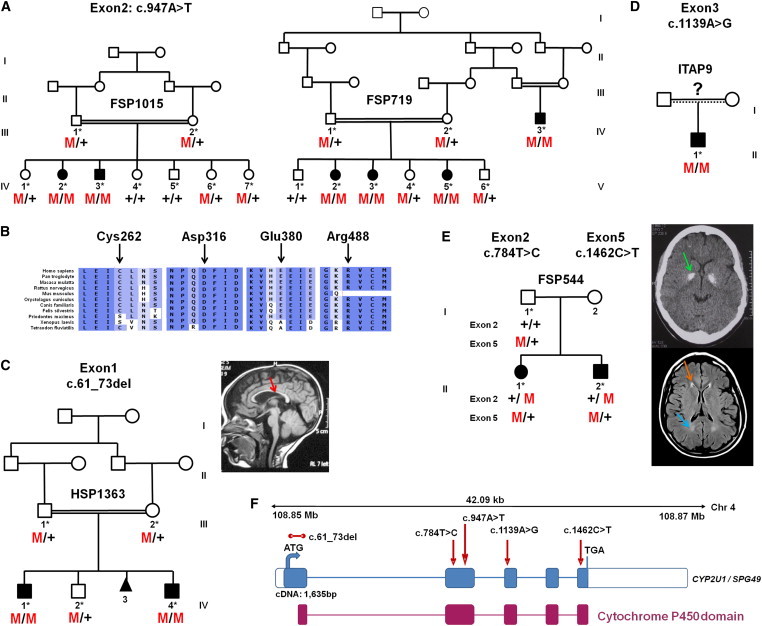
CYP2U1 Mutations in SPG49-Affected Subjects
(A) Segregation analysis of the c.947A>T mutation in two Saudi Arabian families. Their corresponding electrophoregrams are in Figure S2.
(B) Conservation of the amino acids affected by missense variations with the use of ALAMUT software.
(C–E) Pedigrees and segregation of mutations in AR-HSP-affected families HSP1363 (C), ITAP9 (D), and FSP544 (E). A T1-weighted sagittal cerebral MRI from individual HSP1363-IV.4 shows a thin corpus callosum (C, red arrow). A CT scan of individual FSP544-III.1 (E, top view) shows bilateral globus pallidus calcifications (E, green arrow), and a brain MRI (E, bottom view) shows white-matter abnormalities (E, blue arrow), including the “ear of the lynx” aspect of frontal horns of the lateral ventricles (E, orange arrow).
(F) Schematic representation of CYP2U1 (coding exons are represented by blue boxes). The locations of the identified mutations (c.61_73del [p.Leu21Trpfs∗19], c.784T>C [p.Cys262Arg], c.947A>T [p.Asp316Val], c.1139A>G [p.Glu380Gly], and c.1462C>T [p.Arg488Trp]) are shown with red arrows, and the cytochrome P450 domain is indicated by red boxes. The five exons of CYP2U1 (RefSeq NM_183075.2) cover 1,635 bp and encode a 544 amino acid protein with potential transmembrane domains and a heme binding site in the cytochrome P450 domain.
Phenotype Associated with the SPG49 Clinicogenetic Entity
The overall SPG49 phenotype (Table 1) was an early-onset (2.5 ± 2.5 SD years; range = birth to 8 years) spastic paraplegia frequently involving the upper limbs (7/11 cases) and rarely associated with dystonic postures (n = 2) and cognitive alterations (n = 3). Among eight tested subjects, all had normal conduction velocities of the median and peroneal nerves, but the facts that five of them had a slight reduction in the amplitude of compound muscle action potentials and that three of them had a reduction in the amplitude of sensory-nerve action potential indicate the presence of infraclinical axonal neuropathy, predominantly in the lower limbs (data not shown). On brain MRI, thinning of the corpus callosum (n = 1) and white-matter lesions (3/8) were observed but were not inaugural MRI features in two affected subjects (Figure S5). Interestingly, globus pallidus hypointensities were detected during follow-up analysis of two siblings and were confirmed as calcifications by CT scans; they were reminiscent of the familial idiopathic basal-ganglia calcification (Fahr syndrome) (see GeneReviews in Web Resources). Therefore, SPG49 can be added to the growing number of mixed forms (pure and complex) of HSP, such as SPG5 or SPG7.12,13 The severity of symptoms varied widely among affected subjects, even in the same family, and independently of the nature of the mutation; two individuals (FSP719-V.5 and HSP1363-IV.4) never walked, and a third (FSP719-IV.3) was limited in his running capacities, although he had otherwise fully conserved autonomy at 30 years of age.
Table 1.
Clinical Phenotypes in 11 SPG49-Affected Individuals
|
SPG49-Affected Subject |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| FSP719-IV.3 | FSP719-V.2 | FSP719-V.3 | FSP719-V.5 | FSP1015-IV.2 | FSP1015-IV.3 | HSP1363-IV.1 | HSP1363-IV.4 | ITAP9-II.1 | FSP544-II-1 | FSP544-II-2 | |
| Gender | male | female | female | female | female | male | male | male | male | female | male |
| Age (years) | 33 | 21 | 18 | 11 | 25 | 21 | 12 | 5 | 31 | 29 | 27 |
| Duration of follow up (years) | 3 | 12 | 13 | 10 | 12 | 13 | 2 | 2 | 4 | 9 | 9 |
| Age at onset | 3 years | 5 years | 1.5 years | 8 months | birth | 5 years | 1.5 years | birth | 8 years | 13 months | 16 months |
| Symptoms at onset | delayed walking, spastic gait | unsteadiness, spastic gait | delayed walking, unsteadiness | delayed walking, spasticity | delayed walking, toe walking | toe walking | unsteady gait, tip-toe walking | spasticity since birth | cannot run, frequent falls | delayed walking, spastic gait | spastic gait |
| Age at walking | 3 years | 1.5 years | 1.5 years | never walked | 6 years (after surgery) | 1 year | 2 years | never walked | 2 years | delayed | normal |
| Cognitive delay or mental retardation | no | no | no | yes | no | no | no | no speech | no | yes | yes |
| Lower-limb hyperreflexia and positive extensor response (Babinski) | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes | yes |
| Upper-limb hyperreflexia | no | no | no | yes | yes | yes | no | yes | yes | yes | yes |
| Dysarthria | no | no | no | no | no | no | no | no speech | no | yes, mild | yes, mild |
| Extrapyramidal signs | no | no | no | dystonia in upper limbs | no | no | no | dystonia in upper limbs | no | no | no |
| Other signs | no | no | no | no | no | no | no | no | no | maculopathy | no |
| Disability stagea (at age) | 2 (30 years) | 2 (21 years) | 2 (18 years) | 6 (11 years) | 5 (25 years) | 2 (21 years) | 4 | 4 | 2 (28 years) | 6 (16 years) | 5 |
| MRI cerebral (at age) | ND | normal (20 years) | normal (17 years) | normal (10 years) | ND | ND; CT normal | normal (5 years) | TCC, WMLs, mild cortical changes (2.5 years) | normal (28 years) | normal (14 years), then WMLs and globus pallidus hypointensities (26 years) | WMLs and globus pallidus hypointensities (26 years) |
| MRI of spine (at age) | ND | normal (20 years) | normal (17 years) | normal (10 years) | ND | normal | normal (4 years) | ND | normal (28 years) | normal | normal (19 years) |
| Nerve-conduction studies (at age) | ND | subclinical axonal neuropathy (21 years) | subclinical axonal neuropathy (18 years) | subclinical axonal neuropathy (10 years) | subclinical axonal neuropathy (25 years) | subclinical axonal neuropathy (21 years) | normal | normal | normal (28 years) | ND | ND |
None of the affected persons presented with muscle wasting, dysphagia, cerebellar signs, superficial sensory loss, or auditory loss. Muscle biopsy in one subject was unremarkable. The following abbreviations are used: CT, computed tomography; ND, not done; TCC, thin corpus callosum; and WML, white-matter lesion.
Disability scale: 1, minimal disability (slight stiffness of the legs); 2, mild disability (unable to run but full autonomy); 3, moderate disability in walking (reduced perimeter and frequent falls); 4, severe disability (unilateral assistance required for walking); 5, bilateral assistance required for walking; 6, wheelchair bound; and 7, bedridden.
Expression Profiles of CYP2U1 and DDHD1 mRNA
DDHD1 and CYP2U1 transcripts have been shown to be expressed in the brain.27–29 To identify a possible relationship between these two genes, we explored their expression profiles in multiple CNS and non-CNS mouse tissues by quantitative RT-PCR. We observed that DDHD1 and CYP2U1 mRNA levels increased during development in all tested tissues, including the cerebral cortex, but not in peripheral tissues in the case of DDHD1 (Figure S6). Moreover, we showed that their expression levels were significantly coregulated at the embryonic and adult stages in the CNS (Figure S6), but not in non-neuronal tissues, a result compatible with a concomitant need for the two proteins in the CNS.
Subcellular Localization of CYP2U1 and DDHD1 and Exploration of Mitochondrial Functions and Network Organization
We then investigated how these mutations affect cell physiology in HSP. At a subcellular level, DDHD1 fused with a FLAG tag displayed a diffuse distribution pattern almost exclusively in the cytosol (data not shown), but it has also been observed partially in microsomes and mitochondria.30 Because of its PLA1 activity, DDHD1 must transiently interact with membranes from the cytosolic pool to process its substrates. We also showed that CYP2U1 fused with enhanced GFP partially colocalized with ER and mitochondria (Figure S7), as previously reported.31
Given the partial mitochondrial localization of both proteins and the implication of this organelle in other HSPs,12,32 we investigated mitochondrial functions in lymphoblasts. At steady state in growth medium, the mean routine respiration was significantly lower in SPG49 and SPG28 cells than in controls (Figure 3A; controls versus SPG49: p = 0.053, which is very close to the limit of significance; controls versus SPG28: p = 0.011). No cell death was observed (trypan-blue staining and count), suggesting that mitochondrial alterations were not a consequence of a reduction in cell viability. As expected from the respiratory data, total and mitochondrial ATP contents were significantly reduced in SPG49 and SPG28 lymphoblasts (Figure 3B). Furthermore, a concomitant increase of the concentration of cytosolic hydrogen peroxide as measured by CM-H2DCFDA fluorescence (Figure 3C) was also observed in SPG49 and SPG28 cells when compared to controls. No major difference in mitochondrial content, measured by the comparison of the level of mitochondrial TOM20, was detected in lymphoblasts of affected cases (data not shown), suggesting that the observed reduction in mitochondrial energy fluxes results from a functional oxidative-phosphorylation-system alteration caused by mutations in SPG28/DDHD1 and SPG49/CYP2U1.
Figure 3.
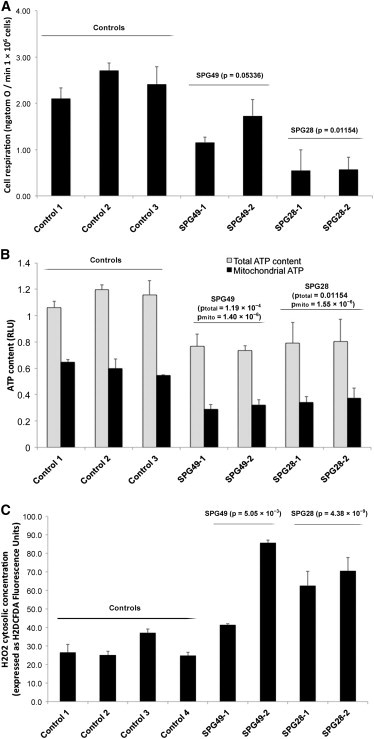
Mitochondrial Dysfunctions in Lymphoblasts from SPG28- and SPG49-Affected Individuals
(A) Mitochondrial respiration rate in SPG28-affected subjects (FSP445-V.3 [SPG28-1] and FSP445-V.1 [SPG28-2] aged 44 and 42 years, respectively, at sampling), SPG49-affected subjects (FSP719-V.3 [SPG49-1] and FSP1015-IV.3 [SPG49-2] aged 18 and 21 years, respectively, at sampling), and three control lymphoblastoid cell lines (aged 34, 37, and 40 years at sampling). The routine respiration data are shown. These measurements were taken after trypan-blue staining and a count for the exclusion of the presence of massive cell death.
(B) Total cellular (light gray bar) and mitochondrial (black bars) ATP content expressed as relative light unit (RLU) measured in SPG28, SPG49, and control lymphoblasts.
(C) H2DCFDA fluorescence gives a measure of the concentration of cytosolic hydrogen peroxide in control cells (white bar) and in CYP2U1 (light gray bar) and DDHD1 (black bars) mutant cells.
The p values comparing mutant cells to control cells are indicated above each histogram (Student’s t test). Error bars represent the SD.
Reduced oxygen consumption could be confirmed in skin fibroblasts of an SPG49-affected individual compared to controls (Figure S8A). Given that respiration originates from the activity of the respiratory chain and the consecutive buildup of an electrochemical gradient of protons, we measured those two parameters. As seen in Figure S8B, the transmembrane electric potential (Δψ) measured with TMRM was 30% lower in the SPG49 fibroblasts than in the three controls (respective p values of 0.0011, 0.0010, and 0.0033). To take into account the nonspecific fluorescence due to TMRM (data not shown), we obtained the same results with the Δψ dissipating agent FCCP (uncoupler, p-trifluoromethoxy carbonyl cyanide phenyl hydrazone). However, the catalytic activities of each of the respiratory-chain complexes assayed in vitro were not altered in the SPG49 fibroblasts or in the lymphoblasts obtained from subjects with SPG28 and SPG49 mutations (Figure S9). This spectrophotometric assay of complexes I–IV was performed in experimental conditions of a so-called “isolated enzyme” with mechanical disruption of mitochondrial membranes and the addition of excess exogenous substrates. In contrast, mitochondrial respiration, ATP synthesis, and Δψ were measured in cells with intact mitochondrial membranes, so alteration in membrane properties could be responsible for the observed bioenergetic failure in cells of affected individuals. Interestingly, we observed structural abnormalities of the mitochondrial membrane, given that the mitochondrial network presented with unusual signs of tubule self-fusions in the SPG49 fibroblasts. These abnormalities included the presence of numerous (1) “donut-like” vesicles 800 nm in diameter within the mitochondrial tubules and (2) circular subnetworks 5 μm in diameter (Figure 4). These features were observed with either outer-membrane TOM20 labeling (Figure 4) or mitochondrial-matrix dyes such as mitotracker (data not shown) or mito-GFP (Figure S10). All together, our findings indicate mitochondrial-membrane-organization alteration associated with a reduction of energy production. Previous studies revealed that such remodeling of mitochondrial tubules could influence the internal diffusion of energy metabolites, the sequestration and conduction of the electric membrane potential (ΔΨ), the stability of the respirasome, the efficiency of oxidative phosphorylation, the diffusion of proteins, or the delivery of newly synthesized ATP to various cellular areas.33
Figure 4.
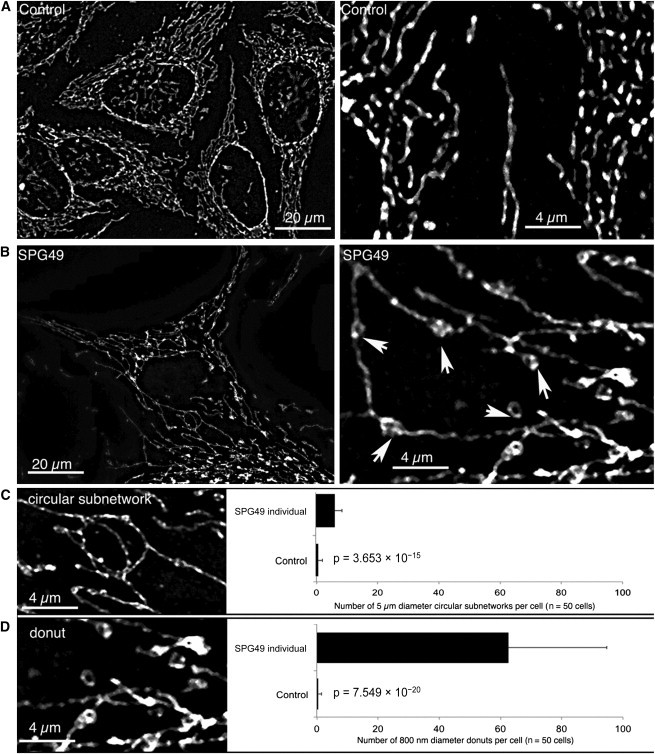
Abnormal Structures in the Mitochondrial Network of SPG49-Affected Skin Fibroblasts
Mitochondrial-network morphology was analyzed by confocal microscopy of skin fibroblasts obtained from control individuals (A) and from individual HSP1363-IV.1 (B). The images on the right are high magnifications (5× zoomed in) of the images on the left. The mitochondrial network was labeled by immunocytochemistry with specific antibodies directed against TOM20, an outer-membrane mitochondrial protein, and fluorescence confocal imaging on a Zeiss Vivatome microscope followed. Three-dimensional stacks were obtained, and the projection images are shown here. We observed two types of abnormal structures on the mitochondrial network of SPG49 cells: small “donut-like” vesicles (800 nm in diameter) suggestive of self-fused mitochondrial filaments (D) and larger circular subnetworks (5 μM in diameter) (C). Counting of these two types of abnormal structures was performed on 50 control cells and 50 cells from the tested subject (right panels). Mean values and p values (Student’s t test) are shown, and error bars represent the SD.
Discussion
We have described a clinicogenetic entity, SPG49, associated with a wide range of phenotypes from pure to complex forms causing a thin corpus callosum and mental impairment, as in SPG11 and SPG15,34,35 or basal-ganglia calcification (see GeneReviews in Web Resources). Second, the identification of the causative mutations in SPG28- and SPG49-affected families highlights lipid metabolism as a critical pathway in HSP given that CYP2U1 and DDHD1 encode fatty-acid- and/or phospholipid-metabolizing enzymes.
DDHD1 was previously identified as a PA-preferring phospholipase A1 (PA-PLA1) but is also known to serve as a substrate for phosphatidylinositol to form 2-arachidonoyl lysophosphatidylinositol.27,36 DDHD1 is ubiquitously expressed in human tissues such as the brain and testis, but the physiological role of this enzyme has not been fully established. DDHD1 orthologs, p125 and DDHD2 (KIAA0725p), are involved in the maintenance of the ER and/or Golgi structures.37,38 Therefore, DDHD1 might also be involved in similar functions in the maintenance of organelle membranes and intracellular trafficking.
CYP2U1 is one of the oldest identifiable vertebrate cytochrome P450 proteins implicated in ω- and ω-1 fatty-acid (C16–C22) hydroxylation.28 In vitro, CYP2U1 is able to catalyze the hydroxylation of arachidonic acid and related long-chain fatty acids such as eicosapentaenoic (EPA) and docosahexaenoic (DHA) acids. Two known metabolites, 19- and 20-hydroxyeicosatetraenoic (HETE) acids, are local mediators of signal transduction.28,39–41
These two enzymes most likely act in the same pathway related to phospholipid degradation and fatty-acid metabolism, which is in agreement with (1) the coregulation of the expression of DDHD1 and CYP2U1, specifically in the CNS, (2) the identification of mutations in both genes in affected subjects with relatively similar clinical presentations, and (3) similar consequences of these mutations for mitochondrial physiology. In addition, other enzymes involving the metabolism of fatty acids and phospholipids have been implicated in neurodegeneration.8,42–44 PA and phosphatidylinositol, as well as the bioactive lipids resulting from their metabolism, modulate membrane properties and play a role in signal-transduction pathways.45,46 Alterations of this pathway (Figure 5) in individuals with HSP could thus have various consequences leading to the observed phenotypes.
Figure 5.
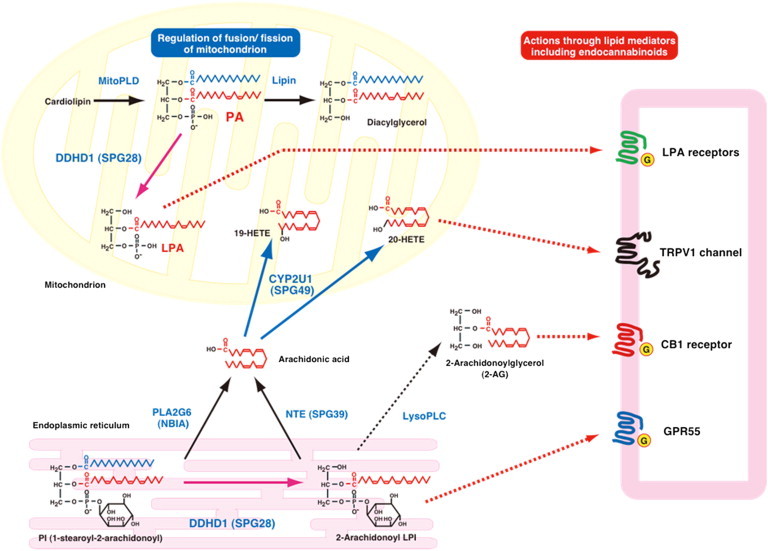
Schematic Representation of the Metabolic Connections between DDHD1 and CYP2U1 Enzymatic Activities
The content of PA on the surface of mitochondria is known to regulate mitochondrial fusion; PA is generated by mitochondrial phospholipase D (MitoPLD) and is further degraded by lipin PA phosphatase. DDHD1 was previously identified as PA-phospholipase A1 (PLA1). The action of DDHD1 might regulate the content of PA on the surface of mitochondria and then be involved in mitochondrial fusion. The DDHD1 mutation causes reduced PA-PLA1 activity, and the resultant increased PA content on the surface of mitochondria might cause the impairment of mitochondrial fusion and lead to the dysfunction of mitochondria. In contrast, phosphatidylinositol (PI) serves as a substrate of DDHD1 to form 2-arachidonoyl lysophosphatidylinositol (LPI). 2-arachidonoyl LPI is known to act on GPR55, which is assumed to be a cannabinoid receptor. 2-arachidonoyl LPI might be hydrolyzed by lysophospholipase C into 2-arachidonoylglycerol, which is an endogenous agonist for cannabinoid receptors CB1 and CB2. Arachidonic acid can be released from PI through phospholipase A2, which includes PLA2G6 (iPLA2 and PNPLA9), and can also be generated from 2-arachidonoyl LPI by neuropathy target esterase (NTE, PNPLA6) given that this enzyme exhibits high lysophospholipase activity. Arachidonic acid is converted to various eicosanoids through the cyclooxygenase and lipoxygenase pathways. In addition, arachidonic acid is known to be the preferred substrate of CYP2U1 to form 19- or 20-HETE acids. Among these, 20-HETE acid is reported as a potent activator of the TRPV1 cation channel, which is a receptor of endocannabinoids, including anandamide and N-arachidonoyl dopamine. CYP2U1 can also metabolize esterified forms of arachidonic acid (EPA and DHA). These common arachidonic-acid metabolites can have effects on the endocannabinoid system through the CB1, GPR55, and TRPV1 receptors.
First, phospholipids and fatty acids can serve as precursors of a wide variety of bioactive lipid messengers45,46 that exhibit hormone- or neurotransmitter-like activity through membrane receptors. Previously, one of the authors found that the major molecular species of phosphatidylinositol (PI), 1-stearoyl-2-arachidonoyl PI, was able to serve as substrate of DDHD1 to generate 2-arachidonoyl lysophosphatidylinositol, a potent agonist of GPR55,47,48 considered a cannabinoid receptor,49 like CB1 or CB2.30,50 Arachinonic acid is metabolized to various eicosanoids through cyclooxygenase and lipoxygenase pathways. CYP2U1 is known to convert arachidonic acid into 19- and 20-HETE acids, which are known to regulate ion channels and neurotransmitter release.28,51,52 In particular, 20-HETE acid has recently been reported as a potent activator of the transient receptor potential vanilloid 1 (TRPV1) channel. TRPV1 colocalizes with cannabinoid receptors CB1 and CB2 in brain and sensory neurons and seems to be gated by endocannabinoids, such as anandamide and N-arachidonoyl dopamine.51,52 Although the bioactive lipids synthesized by DDHD1 and CYP2U1 have not been shown to act directly on mitochondria, their action on receptors might mediate their effect, as has been demonstrated for the CB1 cannabinoid receptors, which can regulate mitochondrial respiration and energy production.53
Second, mitochondrial-membrane lipid composition is critical for maintaining proper bioenergetic functions,23,54 and the alteration of phospholipid metabolism has already been shown to impact mitochondrial functions and trigger secondary cellular dysfunction.54–56 Indeed, maintenance of membrane composition, particularly of the mitochondrial membrane, should be critical for the functions of the long axons of the corticospinal tracts. The shape of the mitochondrion is also known to be linked to mitochondrial functions, including respiration, and the dysregulation of the mitochondria dynamics causes mitochondrial dysfunction.57 PA, a fusogenic phospholipid at the mitochondrion surface, is postulated to regulate the fusion of mitochondria.56 Overexpression of mitochondria phospholipase D (MitoPLD) causes continuous giant perinuclear mitochondria, but the reduced function of MitoPLD causes noncontiguous mitochondrial fragments. In the present study, the mutation of DDHD1 (SPG28) was shown to cause mitochondrial dysfunction, including reduced respiration and ATP production. It is tempting to postulate that the associated mitochondrial bioenergetic dysfunction might result from the accumulation of PA in mitochondria given that DDHD1 exhibits PA-degrading activity (PA-PLA1). SPG28 fibroblasts were not available for exploring this finding, but the abnormal mitochondrial organization in SPG49 fibroblasts suggests that the pathway implicated in SPG28 and SPG49 has a role in this process. Mitochondrial abnormalities could also result from other, unknown, mechanisms because the link between altered mitochondrial morphogenesis and neuronal impairment has already been shown in other diseases, including other forms of HSP.12,32,58–63 Furthermore, because these results on mitochondrial-network disorganization were derived from results from one single fibroblast culture, additional individuals with CYP2U1 and DDHD1 mutations are required for making firm conclusions.
Lastly, other mechanisms could also contribute to the disease. Indeed, in numerous conditions of mitochondrial respiratory impairment, increased reactive oxygen species (ROS) were observed, as in SPG28 and SPG49 cells. Such ROS overproduction could play a role in neuronal degeneration, as previously suggested for HSP pathophysiology.64
In conclusion, our identification of causative mutations in two genes demonstrates the importance of combining systematic gene mapping with large-scale sequencing for elucidating the molecular basis of HSP. Unraveling the role of different proteins involved in the same biological pathway might pave the way for common therapeutic possibilities for individuals with different gene mutations. Our results suggest that the membrane itself or membrane-derived mediators are very important for the neuronal functions in the corticospinal tracts.
The wide expression of DDHD1 and CYP2U1 suggests that it should be possible to explore metabolic dysregulation in peripheral tissues, as in SPG5,65 and develop biomarkers for potential treatment outcome. In particular, the probable implication of mitochondria in the pathophysiology of SPG28 and SPG49 could allow the development of innovative therapeutic strategies focused on organelle dynamics and bioenergetics, as well as ROS scavenging.
Acknowledgments
We are grateful to the affected family members and their relatives who participated in this study. We thank S. Rivaud-Pechoux and C. Gautier for their advice and D. Zelenika, E. Mundwiller, L. Orlando, D. Bouteiller, A. Rastetter, A. Méneret, the DNA and Cell Bank of the Centre de Recherche de l’Institut du Cerveau et de la Moelle Épinière, and the Plateforme d’Imagerie de la Pitié-Salpêtrière for their contribution. We also thank J.-P. Azulay, A. Lossos, and A. Cherif, who referred some of the affected individuals. This work was supported by the Association Strumpell-Lorrain (to the Spastic Paraplegia and Ataxia Network and C.G.), the Association contre les Maladies Mitochondriales (to C.G. and R.R.), the Agence Nationale de la Recherche (to A.D., G.S., and C.G.), the Association Française contre les Myopathies (to C.G. and G.S.), the European Union E-Rare program (to A.Br.), the University of Tübingen (to R.S.), the Conseil Régional d’Aquitaine (to C.G.), and the Verum Foundation (to A.Br.). M.A.M.S. was supported by the College of Medicine Research Center (project 07-581) at King Saud University, Saudi Arabia. M.Al.B. and I.Al.A. were supported by the King Abdullah International Medical Research Center, Riyadh, Saudi Arabia. M.N. and C.T. were recipients of fellowships from the Neuroscience Research Pole in Ile de France and the French Ministry of Research, respectively. F.M.S. was supported by Fondazione Telethon project GGP10121A. This study also received funding from the program “Investissements d’avenir” ANR-10-IAIHU-06 (to the Institut du Cerveau et de la Moelle Épinière).
Contributor Information
Alexis Brice, Email: alexis.brice@upmc.fr.
Giovanni Stevanin, Email: giovanni.stevanin@upmc.fr.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
GeneReviews, Sobrido, M.J., Hopfer, S., and Geschwind, D.H. (2004). Familial Idiopathic Basal Ganglia Calcification, http://www.ncbi.nlm.nih.gov/books/NBK1421
MAP-O-MAT, http://compgen.rutgers.edu/mapomat/
Mutation Taster, http://www.mutationtaster.org/
Online Mendelian Ihneritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
Splice Site Prediction by Neural Network, http://www.fruitfly.org/seq_tools/splice.html
References
- 1.Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. doi: 10.1016/s0140-6736(83)92879-9. [DOI] [PubMed] [Google Scholar]
- 2.Tallaksen C.M., Dürr A., Brice A. Recent advances in hereditary spastic paraplegia. Curr. Opin. Neurol. 2001;14:457–463. doi: 10.1097/00019052-200108000-00005. [DOI] [PubMed] [Google Scholar]
- 3.Fink J.K. Advances in the hereditary spastic paraplegias. Exp. Neurol. 2003;184(Suppl 1):S106–S110. doi: 10.1016/j.expneurol.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Schüle R., Schöls L. Genetics of hereditary spastic paraplegias. Semin. Neurol. 2011;31:484–493. doi: 10.1055/s-0031-1299787. [DOI] [PubMed] [Google Scholar]
- 5.Finsterer J., Löscher W., Quasthoff S., Wanschitz J., Auer-Grumbach M., Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 2012;318:1–18. doi: 10.1016/j.jns.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 6.Salinas S., Proukakis C., Crosby A., Warner T.T. Hereditary spastic paraplegia: Clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- 7.Stevanin G., Ruberg M., Brice A. Recent advances in the genetics of spastic paraplegias. Curr. Neurol. Neurosci. Rep. 2008;8:198–210. doi: 10.1007/s11910-008-0032-z. [DOI] [PubMed] [Google Scholar]
- 8.Blackstone C., O’Kane C.J., Reid E. Hereditary spastic paraplegias: Membrane traffic and the motor pathway. Nat. Rev. Neurosci. 2011;12:31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coutinho P., Barros J., Zemmouri R., Guimarães J., Alves C., Chorão R., Lourenço E., Ribeiro P., Loureiro J.L., Santos J.V. Clinical heterogeneity of autosomal recessive spastic paraplegias: Analysis of 106 patients in 46 families. Arch. Neurol. 1999;56:943–949. doi: 10.1001/archneur.56.8.943. [DOI] [PubMed] [Google Scholar]
- 10.Boukhris A., Stevanin G., Feki I., Denora P., Elleuch N., Miladi M.I., Goizet C., Truchetto J., Belal S., Brice A., Mhiri C. Tunisian hereditary spastic paraplegias: Clinical variability supported by genetic heterogeneity. Clin. Genet. 2009;75:527–536. doi: 10.1111/j.1399-0004.2009.01176.x. [DOI] [PubMed] [Google Scholar]
- 11.Bouslam N., Benomar A., Azzedine H., Bouhouche A., Namekawa M., Klebe S., Charon C., Durr A., Ruberg M., Brice A. Mapping of a new form of pure autosomal recessive spastic paraplegia (SPG28) Ann. Neurol. 2005;57:567–571. doi: 10.1002/ana.20416. [DOI] [PubMed] [Google Scholar]
- 12.Casari G., De Fusco M., Ciarmatori S., Zeviani M., Mora M., Fernandez P., De Michele G., Filla A., Cocozza S., Marconi R. Spastic paraplegia and OXPHOS impairment caused by mutations in paraplegin, a nuclear-encoded mitochondrial metalloprotease. Cell. 1998;93:973–983. doi: 10.1016/s0092-8674(00)81203-9. [DOI] [PubMed] [Google Scholar]
- 13.Goizet C., Boukhris A., Durr A., Beetz C., Truchetto J., Tesson C., Tsaousidou M., Forlani S., Guyant-Maréchal L., Fontaine B. CYP7B1 mutations in pure and complex forms of hereditary spastic paraplegia type 5. Brain. 2009;132:1589–1600. doi: 10.1093/brain/awp073. [DOI] [PubMed] [Google Scholar]
- 14.Klebe S., Lossos A., Azzedine H., Mundwiller E., Sheffer R., Gaussen M., Marelli C., Nawara M., Carpentier W., Meyer V. KIF1A missense mutations in SPG30, an autosomal recessive spastic paraplegia: Distinct phenotypes according to the nature of the mutations. Eur. J. Hum. Genet. 2012;20:645–649. doi: 10.1038/ejhg.2011.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsaousidou M.K., Ouahchi K., Warner T.T., Yang Y., Simpson M.A., Laing N.G., Wilkinson P.A., Madrid R.E., Patel H., Hentati F. Sequence alterations within CYP7B1 implicate defective cholesterol homeostasis in motor-neuron degeneration. Am. J. Hum. Genet. 2008;82:510–515. doi: 10.1016/j.ajhg.2007.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stevanin G., Azzedine H., Denora P., Boukhris A., Tazir M., Lossos A., Rosa A.L., Lerer I., Hamri A., Alegria P., SPATAX consortium Mutations in SPG11 are frequent in autosomal recessive spastic paraplegia with thin corpus callosum, cognitive decline and lower motor neuron degeneration. Brain. 2008;131:772–784. doi: 10.1093/brain/awm293. [DOI] [PubMed] [Google Scholar]
- 17.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 18.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 20.Murmu R.P., Martin E., Rastetter A., Esteves T., Muriel M.-P., El Hachimi K.H., Denora P.S., Dauphin A., Fernandez J.C., Duyckaerts C. Cellular distribution and subcellular localization of spatacsin and spastizin, two proteins involved in hereditary spastic paraplegia. Mol. Cell. Neurosci. 2011;47:191–202. doi: 10.1016/j.mcn.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 21.Jose C., Hébert-Chatelain E., Bellance N., Larendra A., Su M., Nouette-Gaulain K., Rossignol R. AICAR inhibits cancer cell growth and triggers cell-type distinct effects on OXPHOS biogenesis, oxidative stress and Akt activation. Biochim. Biophys. Acta. 2011;1807:707–718. doi: 10.1016/j.bbabio.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Benard G., Faustin B., Passerieux E., Galinier A., Rocher C., Bellance N., Delage J.-P., Casteilla L., Letellier T., Rossignol R. Physiological diversity of mitochondrial oxidative phosphorylation. Am. J. Physiol. Cell Physiol. 2006;291:C1172–C1182. doi: 10.1152/ajpcell.00195.2006. [DOI] [PubMed] [Google Scholar]
- 23.Benard G., Bellance N., James D., Parrone P., Fernandez H., Letellier T., Rossignol R. Mitochondrial bioenergetics and structural network organization. J. Cell Sci. 2007;120:838–848. doi: 10.1242/jcs.03381. [DOI] [PubMed] [Google Scholar]
- 24.Li Q., Lau A., Morris T.J., Guo L., Fordyce C.B., Stanley E.F. A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J. Neurosci. 2004;24:4070–4081. doi: 10.1523/JNEUROSCI.0346-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown M., Adyshev D., Bindokas V., Moitra J., Garcia J.G.N., Dudek S.M. Quantitative distribution and colocalization of non-muscle myosin light chain kinase isoforms and cortactin in human lung endothelium. Microvasc. Res. 2010;80:75–88. doi: 10.1016/j.mvr.2009.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Darios F., Lambeng N., Troadec J.-D., Michel P.P., Ruberg M. Ceramide increases mitochondrial free calcium levels via caspase 8 and Bid: role in initiation of cell death. J. Neurochem. 2003;84:643–654. doi: 10.1046/j.1471-4159.2003.01590.x. [DOI] [PubMed] [Google Scholar]
- 27.Higgs H.N., Han M.H., Johnson G.E., Glomset J.A. Cloning of a phosphatidic acid-preferring phospholipase A1 from bovine testis. J. Biol. Chem. 1998;273:5468–5477. doi: 10.1074/jbc.273.10.5468. [DOI] [PubMed] [Google Scholar]
- 28.Chuang S.S., Helvig C., Taimi M., Ramshaw H.A., Collop A.H., Amad M., White J.A., Petkovich M., Jones G., Korczak B. CYP2U1, a novel human thymus- and brain-specific cytochrome P450, catalyzes omega- and (omega-1)-hydroxylation of fatty acids. J. Biol. Chem. 2004;279:6305–6314. doi: 10.1074/jbc.M311830200. [DOI] [PubMed] [Google Scholar]
- 29.Karlgren M., Backlund M., Johansson I., Oscarson M., Ingelman-Sundberg M. Characterization and tissue distribution of a novel human cytochrome P450-CYP2U1. Biochem. Biophys. Res. Commun. 2004;315:679–685. doi: 10.1016/j.bbrc.2004.01.110. [DOI] [PubMed] [Google Scholar]
- 30.Yamashita A., Kumazawa T., Koga H., Suzuki N., Oka S., Sugiura T. Generation of lysophosphatidylinositol by DDHD domain containing 1 (DDHD1): Possible involvement of phospholipase D/phosphatidic acid in the activation of DDHD1. Biochim. Biophys. Acta. 2010;1801:711–720. doi: 10.1016/j.bbalip.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 31.Dutheil F., Dauchy S., Diry M., Sazdovitch V., Cloarec O., Mellottée L., Bièche I., Ingelman-Sundberg M., Flinois J.-P., de Waziers I. Xenobiotic-metabolizing enzymes and transporters in the normal human brain: Regional and cellular mapping as a basis for putative roles in cerebral function. Drug Metab. Dispos. 2009;37:1528–1538. doi: 10.1124/dmd.109.027011. [DOI] [PubMed] [Google Scholar]
- 32.Rugarli E.I., Langer T. Mitochondrial quality control: A matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benard G., Rossignol R. Mitochondrial fluidity matters. Focus on “Inherited complex I deficiency is associated with faster protein diffusion in the matrix of moving mitochondria”. Am. J. Physiol. Cell Physiol. 2008;294:C1123. doi: 10.1152/ajpcell.00143.2008. [DOI] [PubMed] [Google Scholar]
- 34.Hanein S., Martin E., Boukhris A., Byrne P., Goizet C., Hamri A., Benomar A., Lossos A., Denora P., Fernandez J. Identification of the SPG15 gene, encoding spastizin, as a frequent cause of complicated autosomal-recessive spastic paraplegia, including Kjellin syndrome. Am. J. Hum. Genet. 2008;82:992–1002. doi: 10.1016/j.ajhg.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevanin G., Santorelli F.M., Azzedine H., Coutinho P., Chomilier J., Denora P.S., Martin E., Ouvrard-Hernandez A.-M., Tessa A., Bouslam N. Mutations in SPG11, encoding spatacsin, are a major cause of spastic paraplegia with thin corpus callosum. Nat. Genet. 2007;39:366–372. doi: 10.1038/ng1980. [DOI] [PubMed] [Google Scholar]
- 36.Higgs H.N., Glomset J.A. Identification of a phosphatidic acid-preferring phospholipase A1 from bovine brain and testis. Proc. Natl. Acad. Sci. USA. 1994;91:9574–9578. doi: 10.1073/pnas.91.20.9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tani K., Mizoguchi T., Iwamatsu A., Hatsuzawa K., Tagaya M. p125 is a novel mammalian Sec23p-interacting protein with structural similarity to phospholipid-modifying proteins. J. Biol. Chem. 1999;274:20505–20512. doi: 10.1074/jbc.274.29.20505. [DOI] [PubMed] [Google Scholar]
- 38.Nakajima K., Sonoda H., Mizoguchi T., Aoki J., Arai H., Nagahama M., Tagaya M., Tani K. A novel phospholipase A1 with sequence homology to a mammalian Sec23p-interacting protein, p125. J. Biol. Chem. 2002;277:11329–11335. doi: 10.1074/jbc.M111092200. [DOI] [PubMed] [Google Scholar]
- 39.Gebremedhin D., Lange A.R., Narayanan J., Aebly M.R., Jacobs E.R., Harder D.R. Cat cerebral arterial smooth muscle cells express cytochrome P450 4A2 enzyme and produce the vasoconstrictor 20-HETE which enhances L-type Ca2+ current. J. Physiol. 1998;507:771–781. doi: 10.1111/j.1469-7793.1998.771bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carroll M.A., McGiff J.C. A new class of lipid mediators: Cytochrome P450 arachidonate metabolites. Thorax. 2000;55(Suppl 2):S13–S16. doi: 10.1136/thorax.55.suppl_2.S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imig J.D., Pham B.T., LeBlanc E.A., Reddy K.M., Falck J.R., Inscho E.W. Cytochrome P450 and cyclooxygenase metabolites contribute to the endothelin-1 afferent arteriolar vasoconstrictor and calcium responses. Hypertension. 2000;35:307–312. doi: 10.1161/01.hyp.35.1.307. [DOI] [PubMed] [Google Scholar]
- 42.Chang P.-A., Wu Y.-J. Neuropathy target esterase: An essential enzyme for neural development and axonal maintenance. Int. J. Biochem. Cell Biol. 2010;42:573–575. doi: 10.1016/j.biocel.2009.12.007. [DOI] [PubMed] [Google Scholar]
- 43.Aldahmesh M.A., Mohamed J.Y., Alkuraya H.S., Verma I.C., Puri R.D., Alaiya A.A., Rizzo W.B., Alkuraya F.S. Recessive mutations in ELOVL4 cause ichthyosis, intellectual disability, and spastic quadriplegia. Am. J. Hum. Genet. 2011;89:745–750. doi: 10.1016/j.ajhg.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gregory A., Hayflick S.J. Genetics of neurodegeneration with brain iron accumulation. Curr. Neurol. Neurosci. Rep. 2011;11:254–261. doi: 10.1007/s11910-011-0181-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schaloske R.H., Dennis E.A. The phospholipase A2 superfamily and its group numbering system. Biochim. Biophys. Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 46.Kienesberger P.C., Oberer M., Lass A., Zechner R. Mammalian patatin domain containing proteins: A family with diverse lipolytic activities involved in multiple biological functions. J. Lipid Res. 2009;50(Suppl):S63–S68. doi: 10.1194/jlr.R800082-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oka S., Nakajima K., Yamashita A., Kishimoto S., Sugiura T. Identification of GPR55 as a lysophosphatidylinositol receptor. Biochem. Biophys. Res. Commun. 2007;362:928–934. doi: 10.1016/j.bbrc.2007.08.078. [DOI] [PubMed] [Google Scholar]
- 48.Oka S., Toshida T., Maruyama K., Nakajima K., Yamashita A., Sugiura T. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: A possible natural ligand for GPR55. J. Biochem. 2009;145:13–20. doi: 10.1093/jb/mvn136. [DOI] [PubMed] [Google Scholar]
- 49.Oka S., Ota R., Shima M., Yamashita A., Sugiura T. GPR35 is a novel lysophosphatidic acid receptor. Biochem. Biophys. Res. Commun. 2010;395:232–237. doi: 10.1016/j.bbrc.2010.03.169. [DOI] [PubMed] [Google Scholar]
- 50.Anavi-Goffer S., Baillie G., Irving A.J., Gertsch J., Greig I.R., Pertwee R.G., Ross R.A. Modulation of L-α-lysophosphatidylinositol/GPR55 mitogen-activated protein kinase (MAPK) signaling by cannabinoids. J. Biol. Chem. 2012;287:91–104. doi: 10.1074/jbc.M111.296020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rimmerman N., Bradshaw H.B., Basnet A., Tan B., Widlanski T.S., Walker J.M. Microsomal omega-hydroxylated metabolites of N-arachidonoyl dopamine are active at recombinant human TRPV1 receptors. Prostaglandins Other Lipid Mediat. 2009;88:10–17. doi: 10.1016/j.prostaglandins.2008.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wen H., Östman J., Bubb K.J., Panayiotou C., Priestley J.V., Baker M.D., Ahluwalia A. 20-Hydroxyeicosatetraenoic acid (20-HETE) is a novel activator of transient receptor potential vanilloid 1 (TRPV1) channel. J. Biol. Chem. 2012;287:13868–13876. doi: 10.1074/jbc.M111.334896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bénard G., Massa F., Puente N., Lourenço J., Bellocchio L., Soria-Gómez E., Matias I., Delamarre A., Metna-Laurent M., Cannich A. Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat. Neurosci. 2012;15:558–564. doi: 10.1038/nn.3053. [DOI] [PubMed] [Google Scholar]
- 54.Steenbergen R., Nanowski T.S., Beigneux A., Kulinski A., Young S.G., Vance J.E. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 2005;280:40032–40040. doi: 10.1074/jbc.M506510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brandner K., Mick D.U., Frazier A.E., Taylor R.D., Meisinger C., Rehling P. Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: implications for Barth Syndrome. Mol. Biol. Cell. 2005;16:5202–5214. doi: 10.1091/mbc.E05-03-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Choi S.-Y., Huang P., Jenkins G.M., Chan D.C., Schiller J., Frohman M.A. A common lipid links Mfn-mediated mitochondrial fusion and SNARE-regulated exocytosis. Nat. Cell Biol. 2006;8:1255–1262. doi: 10.1038/ncb1487. [DOI] [PubMed] [Google Scholar]
- 57.Benard G., Karbowski M. Mitochondrial fusion and division: Regulation and role in cell viability. Semin. Cell Dev. Biol. 2009;20:365–374. doi: 10.1016/j.semcdb.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Niemann A., Wagner K.M., Ruegg M., Suter U. GDAP1 mutations differ in their effects on mitochondrial dynamics and apoptosis depending on the mode of inheritance. Neurobiol. Dis. 2009;36:509–520. doi: 10.1016/j.nbd.2009.09.011. [DOI] [PubMed] [Google Scholar]
- 59.Chang C.-R., Manlandro C.M., Arnoult D., Stadler J., Posey A.E., Hill R.B., Blackstone C. A lethal de novo mutation in the middle domain of the dynamin-related GTPase Drp1 impairs higher order assembly and mitochondrial division. J. Biol. Chem. 2010;285:32494–32503. doi: 10.1074/jbc.M110.142430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goizet C., Depienne C., Benard G., Boukhris A., Mundwiller E., Solé G., Coupry I., Pilliod J., Martin-Négrier M.-L., Fedirko E. REEP1 mutations in SPG31: frequency, mutational spectrum, and potential association with mitochondrial morpho-functional dysfunction. Hum. Mutat. 2011;32:1118–1127. doi: 10.1002/humu.21542. [DOI] [PubMed] [Google Scholar]
- 61.Chevrollier A., Cassereau J., Ferré M., Alban J., Desquiret-Dumas V., Gueguen N., Amati-Bonneau P., Procaccio V., Bonneau D., Reynier P. Standardized mitochondrial analysis gives new insights into mitochondrial dynamics and OPA1 function. Int. J. Biochem. Cell Biol. 2012;44:980–988. doi: 10.1016/j.biocel.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 62.Girard M., Larivière R., Parfitt D.A., Deane E.C., Gaudet R., Nossova N., Blondeau F., Prenosil G., Vermeulen E.G.M., Duchen M.R. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS) Proc. Natl. Acad. Sci. USA. 2012;109:1661–1666. doi: 10.1073/pnas.1113166109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Misko A.L., Sasaki Y., Tuck E., Milbrandt J., Baloh R.H. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 2012;32:4145–4155. doi: 10.1523/JNEUROSCI.6338-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gücüyener K., Pinarli F.G., Erbaş D., Hasanoğlu A., Serdaroğlu A., Topaloğlu H. Is oxidative damage in operation in patients with hereditary spastic paraparesis? Brain Dev. 2010;32:130–136. doi: 10.1016/j.braindev.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 65.Schüle R., Siddique T., Deng H.-X., Yang Y., Donkervoort S., Hansson M., Madrid R.E., Siddique N., Schöls L., Björkhem I. Marked accumulation of 27-hydroxycholesterol in SPG5 patients with hereditary spastic paresis. J. Lipid Res. 2010;51:819–823. doi: 10.1194/jlr.M002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.