

Abstract
Children from diabetic pregnancies have a greater incidence of Type 2 diabetes. Our objective was to determine if exposure to mild-moderate hyperglycemia, modeling managed diabetic pregnancies, affects fetal β-cell function. In sheep fetuses β-cell responsiveness was examined after two weeks of sustained hyperglycemia with 3 pulses/day, mimicking postprandial excursions, and compared to saline-infused controls (n=10). Two pulsatile hyperglycemia treatments were studied: mild (mPHG, n=5) with +15% sustained and +55% pulse; and moderate (PHG, n=10) with +20% sustained and +100% pulse. Fetal glucose-stimulated insulin secretion and glucose potentiated arginine insulin secretion were lower (P<0.05) in PHG (0.86±0.13 and 2.91±0.39 ng/ml plasma insulin) but not mPHG fetuses (1.21±0.08 and 4.25±0.56 ng/ml) compared to controls (1.58±0.25 and 4.51±0.56 ng/ml). Islet insulin content was 35% lower in PHG and 35% higher in mPHG versus controls (P<0.01). Insulin secretion and maximally stimulated insulin release were also reduced (P<0.05) in PHG islets due to lower islet insulin content. Isolated PHG islets also had 63% greater (P<0.01) ROS accumulation at 11.1 mmol/L glucose than controls (P<0.01), but oxidative damage was not detected in islet proteins. PHG fetuses showed evidence of oxidative damage to skeletal muscle proteins (P<0.05) but not insulin resistance. Our findings show that PHG induced dysregulation of islet ROS handling and decreased islet insulin content, but these outcomes are independent. The β-cell outcomes were dependent on the severity of hyperglycemia because mPHG fetuses had no distinguishable impairments in ROS handling or insulin secretion but greater insulin content.
Keywords: Gestational diabetes, GSIS, islets of Langerhans, oxidative stress, ROS
INTRODUCTION
Diabetic conditions during pregnancy have been associated with an increased incidence of impaired glucose tolerance, insulin resistance, and Type 2 diabetes in the offspring (Dabelea et al. 2000; Pettitt et al. 1985; Plagemann et al. 1997; Silverman et al. 1995; Sobngwi et al. 2003). Current clinical management guidelines allow pregnant mothers with diabetes to be mildly hyperglycemic, with fasting glucose 20-40% above normal, and to have larger postprandial excursions that are approximately twice normal fasting glucose concentrations (American Diabetes Association 2004; Gillmer et al. 1975; Parretti et al. 2001; Siegmund et al. 2008). Fetal phenotypes, such as macrosomia, are strongly associated with maternal blood glucose concentrations (Karlsson and Kjellmer 1972; Metzger et al. 2008), and postprandial glucose is more predictive of macrosomia than fasting glucose (Combs et al. 1992; de Veciana et al. 1995; Jovanovic-Peterson et al. 1991; Most and Langer 2007). Fetal glucose is dependent on maternal concentrations; therefore, fetal exposure to chronic mild hyperglycemia with postprandial pulses appears to drive the adverse outcomes in offspring of diabetic pregnancies. Although basal insulin is increased in human fetuses exposed to diabetic conditions (Metzger et al. 2008; Silverman et al. 1995), it is not known if β-cell responsiveness to glucose is affected. However, animal studies have shown that β-cell outcomes in offspring are dependent on the magnitude of hyperglycemic exposure (Aerts and van Assche 1977; Carver et al. 1996; Kervran et al. 1978).
Oxidative stress is one possible mechanism for hyperglycemia-induced β-cell dysfunction. Reactive oxygen species (ROS) such as superoxide and hydrogen peroxide (H2O2) are formed as byproducts of multiple metabolic pathways that are increased with hyperglycemia (Kaneto et al. 2005; Nakayama et al. 2005; Takahashi et al. 2004; Tsubouchi et al. 2005). In pregnant rats with severe hyperglycemia, oxidative stress increases embryological malformations and spontaneous abortions (Cederberg et al. 2001; Eriksson and Borg 1993; Kinalski et al. 1999), and similar complications have been observed in women with severe preexisting diabetes (Suhonen et al. 2000). Women with gestational diabetes also exhibit increased placental oxidative stress (Coughlan et al. 2004; Lappas et al. 2004); however, to our knowledge, fetal measures have not been obtained. Chronic oxidative stress in isolated islets and immortalized β-cell lines decreases glucose stimulated insulin secretion and lowers insulin content (Kaneto et al. 2001; Maechler et al. 1999; Noda et al. 2002; Sakai et al. 2003; Takahashi et al. 2004; Tanaka et al. 2002; Tang et al. 2007). Compared to other cell types, adult β-cells have relatively low levels of antioxidant enzymes and a limited ability to up-regulate these enzymes in response to oxidative stress (Lenzen et al. 1996; Tiedge et al. 1997), therefore making them more vulnerable to oxidative stress. It is not yet known whether fetal β-cells possess similar deficiencies and vulnerability to ROS accumulation.
Another potential mechanism linking hyperglycemia and fetal β-cell dysfunction is endoplasmic reticulum (ER) stress. Abnormal proinsulin processing and protein folding induced by hyperglycemia can result in ER stress, distended rough ER, and insulin secretion defects (Araki et al. 2003). Distension of the ER was observed in β-cells of fetal rats subjected to an experimental model of gestational diabetes (Aerts and van Assche 1977). Endoplasmic reticulum stress would reflect possible defects caused by nitric oxide, calcium storage, or cytokines (Oyadomari and Mori 2004).
We investigated insulin secretion responsiveness, islet function, islet ROS accumulation, and markers of ER stress in sheep fetuses exposed to chronic mild pulsatile hyperglycemia. The sheep model was chosen for this study, because exogenous dextrose can be chronically infused into the pregnant ewe with precision in both magnitude and pattern (Carver et al. 1996). In vivo and ex vivo insulin responsiveness can be measured in the sheep fetus (Green et al. 2011; Limesand et al. 2006), which shares similarities to the human in the progression of pancreas development (Green et al. 2010). Sheep fetuses were exposed to sustained mild to moderate hyperglycemia with three superimposed hyperglycemic pulses per day (mimicking postprandial excursions) for two weeks during late gestation. At the end of the treatment, glucose-stimulated insulin secretion (GSIS) was measured in the fetuses and their isolated pancreatic islets. Additionally, ROS accumulation in islets was measured in vitro, and markers of systemic oxidative stress and ER stress were assessed in fetal tissues. We found that pulsatile hyperglycemia lowered islet insulin content and impaired fetal GSIS and ROS regulation, though these defects occur independently and were dependent on the magnitude of pulsatile hyperglycemia.
MATERIALS AND METHODS
Animal preparation
Pregnant Columbia-Rambouillet ewes carrying singletons were purchased from Nebeker Ranch (Lancaster, CA, USA) and managed in compliance with the Institutional Animal Care and Use Committee of the University of Arizona. All animal experiments were conducted at the William J. Parker Agricultural Research Complex, Tucson, AZ, USA, which is accredited by the National Institutes of Health, the United States Department of Agriculture, and the American Association for the Accreditation of Laboratory Animal Care. Animal rooms were maintained at 22±1 C with a 14-h light/10-h dark cycle. Except as noted below, food and water were available ad libitum, and food intake was recorded daily. Food was withheld for 24 hours and water for 18 hours prior to surgery. At ~119 dGA, fetuses were surgically instrumented with indwelling polyvinyl catheters as described previously (Limesand and Hay 2003; Limesand et al. 2007). Fetal catheters for blood sampling were placed in the abdominal aorta via hind limb pedal arteries and infusion catheters were placed in the femoral veins via the saphenous veins. Maternal catheters were placed in the femoral artery for arterial sampling and the femoral vein for infusions. All catheters were tunneled subcutaneously to the ewe’s flank, exteriorized through a skin incision, and kept in a plastic mesh pouch sutured to the ewe’s skin. Ewes were given 4-5 days to recover from surgery before treatment.
Study design
Animals were randomly assigned to one of three treatment groups: control (n=10), mild pulsatile hyperglycemia (mPHG, n=5), and pulsatile hyperglycemia (PHG, n=10). Treatments were initiated at 124±0.5 dGA with maternal intravenous infusions of 50% dextrose (wt/vol in H2O) and were maintained for 14 days. In the mPHG and PHG animals, dextrose was infused to raise maternal plasma glucose concentrations 15% and 20% above euglycemia (determined in control fetuses), respectively. Arterial plasma glucose concentrations were measured at least twice daily, and dextrose infusion rates were adjusted to maintain the specified treatment conditions. In addition, mPHG and PHG ewes received 45 min boluses of dextrose at 0800, 1400, and 2000 hours each day at a rate sufficient to achieve peak plasma glucose concentrations 55% and 100% above euglycemic values (compared to controls), respectively. The magnitude and pattern of the hyperglycemia treatments were specifically chosen to represent pregnant women with well-controlled diabetes (mPHG) and marginal glucose control (PHG), according to the current clinical guidelines (American Diabetes Association 2004; Carpenter and Coustan 1982; Gilmartin et al. 2008). Plasma glucose concentrations during boluses were checked at least every other day and bolus infusion rates adjusted when needed. Control ewes were randomly paired to PHG ewes and received saline infusions of equal volumes. Maternal and fetal arterial blood samples were collected daily between 0700 and 0800, and basal plasma glucose, lactate, and insulin, and blood gas and pH levels were measured. Seven maternal and fetal plasma samples (time 0 to 120 min) were collected for the 1400 bolus on day 9 of treatment to quantify the fetal plasma insulin response to the boluses.
Glucose stimulated insulin secretion and glucose potentiated arginine-stimulated insulin secretion studies
On d 13 or 14 of treatment, while continuing the chronic sustained maternal dextrose infusions and in place of the 0800 bolus, GSIS was measured in nine control, five mPHG, and ten PHG fetuses with a square-wave hyperglycemic clamp as previously reported (Green et al. 2011; Limesand et al. 2006). Briefly, a continuous transfusion of maternal arterial blood into the fetus (5 ml/h) was started 45 min prior to baseline sampling and maintained for the duration of the study to compensate for blood collection. All sample times are presented relative to the start of the fetal glucose bolus and continuous glucose infusion at time 0. Basal period plasma glucose and insulin concentrations were determined at −21, −13, and −5 min. The hyperglycemic clamp was initiated with a dextrose bolus of 1.19±0.04 mmol/kg estimated fetal weight to the fetus followed by a constant infusion of 33% dextrose in saline to increase and maintain fetal arterial plasma glucose concentration at 2.4 mmol/l, which produces a near-maximal GSIS response in singleton sheep fetuses (Green et al. 2011; Limesand et al. 2006). At the onset of the glucose infusion, fetal arterial samples were collected every 5-10 min for the initial 30 min to establish the hyperglycemic steady state, after which fetal samples were collected at 45, 53, and 61 min (hyperglycemic period). At basal (−21 – −5 min) and hyperglycemic (45 – 61 min) periods, blood gas and pH and plasma glucose, lactate, and insulin concentrations were measured. Following the 61 min hyperglycemic sample, a glucose potentiated arginine-stimulated insulin secretion (GPAIS) test was conducted by injecting a bolus of arginine (0.5 mmol/kg estimated fetal weight mixed with 1 ml of 2 mol/L sodium acetate and 4 ml saline) over 4 min into the fetal circulation and collecting plasma samples at 5, 15, and 30 min for subsequent measurement of insulin concentrations.
Biochemical Analyses
Blood gases, pH, and oximetry parameters were measured in whole blood collected in heparin-lined syringes (Elkins-Sinn, Inc., Cherry Hill, NJ) with an ABL 720 (Radiometer, Copenhagen, Denmark). Sample values were temperature corrected at 39.1 C. Whole blood collected in EDTA-lined syringes (Sigma-Aldrich, St. Louis, MO) was centrifuged (13,000×g) for 2 min at 4 C, and the plasma was aspirated from the red blood cells. Plasma glucose and lactate concentrations were measured with a YSI Model 2700 SELECT Biochemistry Analyzer (Yellow Springs Instruments, Yellow Springs, OH). The remaining plasma was stored at −80 C until insulin was measured with an ovine insulin ELISA (ALPCO Diagnostics, Windham, NH; intra-assay and inter-assay coefficients of variation were 5.6% and 2.9% respectively). In the control and PHG fetuses, remaining plasma from the −21 and −13 min samples were pooled, and norepinephrine concentrations were analyzed by Noradrenaline ELISA (Labor Diagnostika Nord GmbH & Co.KG, Germany; intra-assay and inter-assay coefficients of variation were 20% and 22%, respectively).
Necropsy and islet isolation
Within 24 hours of the GSIS studies, while remaining under treatment conditions, ewes and fetuses were killed with an intravenous overdose of sodium pentobarbital (86 mg/kg) and phenytoin sodium (11 mg/kg; Euthasol, Virbac Animal Health, Fort Worth, TX, USA). The fetus was blotted dried and weighed. The fetal pancreas was perfused and digested with Liberase BlendZyme III (0.175 mg/ml; Roche, Indianapolis, IN) in Krebs Ringer buffer (KRB; 118 mmol/L NaCl, 4.8 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L MgSO4, 1.2 mmol/L KH2PO4, 25 mmol/L NaHCO3, pH 7.3) and then individual islets were purified as described previously (Leos et al. 2010; Limesand et al. 2006; Rozance et al. 2006). After removal of the pancreas, brain, liver, skeletal muscle, spleen, kidney, lung, heart and perirenal adipose tissues were dissected and weighed. Liver (left lobe) and semitendinosus muscle samples were snap frozen in liquid nitrogen and stored at −80°C for RNA and protein extraction.
Islet insulin secretion and glucose oxidation
Isolated islets were cultured overnight at 37 C in 95% O2/5% CO2 in RPMI-1640 medium (Sigma-Aldrich) supplemented with 2% FBS, 2.8 mmol/L glucose, and penicillin-streptomycin (50 U and 50 μg; Sigma-Aldrich). One day after isolation, in vitro experiments on islets were performed, and an aliquot of hand-picked islets was frozen and stored at −80 C for RNA and protein extraction.
In seven control, five mPHG, and eight PHG fetuses, insulin secretion was measured in static islet incubations (Limesand et al. 2006). Fetal islets were washed twice in KRB with 0.5% BSA and a third time in KRB/BSA supplemented with 10 μmol/L Forskolin (Sigma-Aldrich) equilibrated to 37 C and 95% O2/5% CO2. Ten islets were hand-picked (n=3-4 replicates/condition) and incubated at 37 C for one hour in KRB/BSA/Forskolin media with the following conditions: no glucose, 1.1 mmol/L glucose (non-stimulatory concentration), 11.1 mmol/L glucose (maximal glucose stimulatory concentration for fetal sheep islets (Limesand et al. 2006)), or 1.1 mmol/L glucose plus 30 mmol/L KCl. Negative control islet incubations in 11.1 mmol/L glucose on ice were included to evaluate cellular integrity. Following the incubation, islets were pelleted by centrifugation (3 min at 800 × g) at 4 C. The media was removed and frozen, and islet insulin was extracted with acid-ethanol (1 mol/L HCl/70% ethanol). Insulin concentrations were measured with the ovine insulin ELISA. The data were analyzed as ng insulin release per islet.
Rates of glucose oxidation were measured by the formation of 14CO2 as previously described (Limesand et al. 2006) for islets from five control and five PHG fetuses; mPHG islets were not assessed. Twenty-five islets were hand-picked into a 1 ml cryotube affixed inside a scintillation vial and sealed. The islets were incubated for 2 h at 37 C in RPMI-1640 with 1% fetal bovine serum containing 1.1 or 11.1 mmol/L D-glucose and [U-14C]D-glucose (8 or 16 μCi/ml, respectively; PerkinElmer, Boston, MA).
Islet ROS measurements
Following isolation, islets were incubated overnight on glass cover slips pre-coated with human fibronectin (10 mg/L in PBS; BD Biosciences, Bedford, MA). Cover slips with adhered islets were transferred to a temperature-controlled chamber that was mounted on the stage of an Olympus (Center Valley, PA) IX-70 microscope. Islets were preloaded for 20 min at 37 C in HBSS containing 1.1 mmol/L glucose and 2 μmol/L of the ROS-sensitive probe CM-H2DCFDA (Invitrogen, Carlsbad, CA). The media was then replaced with fresh 1.1 mmol/L glucose in HBSS (37 C). Fluorescent images of a single islet were captured every minute with 100 msec exposures using a Photometrics (Tucson, AZ) Coolsnap camera under the following sequence of conditions for 15 min each: 1.1 mmol/L glucose; 11.1 mmol/L glucose; and 11.1 mmol/L glucose + 9 mmol/L H2O2 to evaluate the dynamic range of the probe. Preliminary experiments with both control and PHG islets in 1.1 mmol/L glucose showed a slow constant increase in fluorescence intensity for 60 min, indicating a normal basal rate of H2O2 production regardless of fetal treatment. The rate of fluorescence increase at 1.1 mmol/L glucose was used normalize rates during the hyperglycemic and hydrogen peroxide conditions in order to account for inter-islet variation in CM-H2DCFDA loading. ROS measurements were obtained from two to five islets (5-10 ROI/islet) per fetal sheep and determined in eight control, three mPHG, and six PHG fetuses.
Tissue preparation
Total RNA was extracted from control and PHG liver and skeletal muscle tissues with Tri Reagent (Molecular Research Center, Cincinnati, OH) and cleaned up using a QIAGEN Mini RNeasy column (QIAGEN, Valencia, CA). Liver and skeletal muscle was homogenized in cold lysis buffer containing: 1% Nonidet P-40, 150 mmol/L NaCl, 1 mM EDTA, 1 mmol/L Na3VO4, 1mmol/L NaF, 50 mmol/L Tris, pH 7.4, 0.5 mmol/L phenylmethylsulfonyl fluoride (PMSF), 1 mM DTT, 0.4 ng/ml aprotinin, and 6.3 μg/ml leupeptin. Protein lysates were centrifuged at 13,000 × g at 4 C for 10 min, and the supernatant was frozen and stored at −80 C. Total RNA and protein were extracted from purified islets of Langerhans using the Allprep DNA/RNA/Protein Mini Kit (QIAGEN). RNA concentrations were determined by measuring absorbance at 260 and 280 nm (NanoDrop ND-1000 Spectrophotometer, Wilmington, DE), and RNA integrity was confirmed with an Experion Automated Electrophoresis System (BioRad Laboratories, Hercules, CA). Protein concentrations were determined with the BCA protein assay (Thermo Fisher Scientific Inc; Rockford, IL).
Tissue protein oxidative damage
Protein carbonyl incorporation was measured in skeletal muscle and liver from 8 PHG and 10 control fetuses and in islet protein lysates from 6 PHG and 6 control fetuses using the OxyBlot Protein Oxidation Detection Kit (Millipore, Billerica, MA) following the manufacturer’s instructions except as noted here. Five μg of islet protein and 7.5 μg of liver and skeletal muscle protein lysates were derivatized to 2,4-dinitrophenylhydrazone (DNP) by reaction with 2,4-dinitrophenylhydrazine or were treated with a negative solution for a negative control. Proteins were separated on 10% SDS-PAGE, transferred to a polyvinylidene fluoride membrane (BioRad), and blocked in the provided blocking/dilution buffer at room temperature for 1 h. Immunoblot detection was achieved by overnight incubation at room temperature with the provided rabbit anti-DNP antibody. The primary antibody was detected with anti-rabbit immunoglobulin G horseradish peroxidase conjugated secondary antibody (1:10,000; BioRad) for 1 h at room temperature and detected using SuperSignal West Pico (Thermo Fisher Scientific) exposed to Kodak x-ray film. Because the density of individual bands did not change independently, the sum of the density of all DNP bands in each lane was quantified with ImageJ Software v. 1.41 (National Institutes of Health, Bethesda, MD). Multiple exposure times were analyzed to confirm that the films quantified were not saturated. Protein from each fetus was analyzed in triplicate, and paired control and PHG fetuses were analyzed on the same gel. In addition, protein from one select control fetus was included in triplicate on every gel, and the band density for all other lanes was normalized to the mean for the control protein.
PCR and quantitative real-time PCR (qPCR)
Synthetic oligonucleotide primers were designed against sequences for genes of interest (GenBank accession numbers listed in parentheses and are for ovine sequences unless otherwise noted): antioxidants SOD-1 (FJ546075), SOD-2 (GQ221055.1), GPx-1 (JF728302), catalase (GQ421282) and uncoupling protein 2 (UCP2; bovine sequence NM_001033611); insulin (U00659) and insulin transcription factors pancreatic and duodenal homeobox 1 (PDX-1; JF728303) and V-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA; bovine sequence NM_001105637.1); endoplasmic reticulum stress response genes glucose regulatory protein-78 (GRP78; DQ029323), and DNA-damage inducible transcript-3 (DDIT-3; AY943948); and the reference gene ribosomal protein S15 (S15; AY949774) were designed with the aid of Primer-BLAST (NCBI, Bethesda, MD) software and purchased from Eurofins MWG Operon (Huntsville, AL) (primer sequences are available upon request). PCR products for ovine genes were amplified from fetal ovine mRNA by reverse transcription-PCR using Superscript III reverse transcriptase and Taq DNA polymerase (QIAGEN) according to the manufacturer’s instructions. Correct PCR products were verified by confirming product size after separation in a 1% agarose DNA gel. PCR products were then inserted into the TOPO TA cloning expression vector pCRII (Invitrogen, Carlsbad, CA) and transformed into One Shot Mach T1 Phage-Resistant Chemically Competent E. coli (Invitrogen). Plasmids were prepared for nucleotide sequencing with a QIAprep Spin Miniprep Kit (QIAGEN) and sequenced at the University of Arizona DNA Sequencing Service.
One μg of RNA extracted from islets from nine control and five PHG fetuses was used to generate cDNA with SuperScript III Reverse Transcriptase (Invitrogen). The relative mRNA expression for each gene of interest was determined by qPCR using SYBR Green (QIAGEN) in an iQ5 Real-Time PCR Detection System (BioRad) as reported previously (Chen et al. 2010). After initial denaturation at 95°C for 15 min, all reactions went through 40 cycles of 96°C (30 sec), annealing temperature of 60-62°C (30 sec), and 72°C (10 sec) at which point the fluorescence was measured. Melt curve analysis was performed at the end of the amplification to confirm product homogeneity. PCR efficiency was determined with islet cDNA (80-100%) and was linear over six orders of magnitude. RNA samples were run in triplicate for each qPCR reaction. The results were normalized to the reference gene S15 for each qPCR, and the average ΔCT (cycle threshold) was analyzed by the comparative ΔCT method (CT gene of interest - CT reference gene) (Schmittgen and Livak 2008).
Western immunoblot analyses
Western immunoblot analyses for superoxide dismutase 1 (SOD-1) and 2 (SOD-2), glutathione peroxidase 1/2 (GPx-1/2), insulin receptor-β (Rβ), β tubulin, and ribosomal protein S6 were conducted as previously described (Chen et al. 2010) on liver and skeletal muscle protein preparations. Protein samples from each fetus were run in duplicate, and 30 μg of protein were loaded per lane. Paired control and PHG fetuses were analyzed on the same gel, and protein from one control fetus was included in duplicate on every gel for the purposes of normalizing the band density from all other lanes on the gel. Ribosomal protein S6 was detected on a separate but identical gel loaded concurrently to test for equality of protein loading between animals. Immunoblot detection was accomplished with the following polyclonal antibodies purchased from Santa Cruz Biotechnology (Santa Cruz, CA) unless otherwise noted: rabbit anti-SOD-1 (FL-154, sc-11407, diluted 1:1000); rabbit anti-SOD-2 (FL-222, sc-30080, diluted 1:1000); rabbit anti-GPx-1/2 (H-151, sc-30147, diluted 1:1000); rabbit anti-insulin Rβ (C-19, sc-711, diluted 1:200); rabbit anti-ribosomal protein S6 (FL-249, sc-20085, diluted 1:200); and rabbit anti-β-tubulin (Thermo Scientific (Waltham, MA), RB-9249-PO, diluted 1:1000).
Statistics
All data are expressed as mean ± SEM, and P-values less than 0.05 were considered significant. Data for feed and water intakes, maternal and fetal daily plasma values, organ weights, and qPCR were analyzed by one-way ANOVA and means compared with a Tukey’s HSD test. Data from the fetal GSIS and islet insulin secretion experiments (including insulin contents) were subjected to an ANOVA with sheep as a random effect and treatment and period or treatment and media condition, respectively, as fixed effects. Differences were determined with an LSD test. In the GSIS, insulin responsiveness data had unequal variances, so a Welch’s ANOVA was also run but gave the same results. Analysis for GPAIS was an ANOVA that accounted for the sheep as a random effect and draw and treatment as fixed effects (SAS GLM procedure). ROS data were analyzed by ANOVA with sheep as a random effect. Protein carbonyl data and other Western immunoblot data were analyzed by paired t-test to determine differences between protein expression in paired control and PHG fetuses separated in the same gel. Statistical analyses were completed in JMP 8 and SAS 9.2 (SAS; Cary, NC).
RESULTS
Maternal body weight and feed intake
Before treatment, maternal body weight was not different between control (44.2±1.7 kg), PHG (41.1±2.7 kg), and mPHG ewes (49.9±3.0 kg). During the treatment period, feed intake was lower (P<0.01) in PHG ewes (1.28±0.06 kg/d) compared to mPHG (1.78±0.08 kg/d) and control ewes (1.84±0.07 kg/d). However, total daily energy intake (feed + dextrose infusion) was not different between mPHG (2.88±0.13 Mcal/d) and PHG ewes (2.61±0.10 Mcal/d), and both were greater than that of control ewes (2.26±0.09 Mcal/d; P<0.01). In all treatments, energy intake was in excess of the requirements for maintenance and gestation (National Research Council 2007). Ewe body weight was included as a covariate in the analyses of feed and energy intake; the effect of body weight was significant for both parameters (P<0.01), but no interaction was found between body weight and treatment.
Fetal body and organ weights
At necropsy, average fetal body weights were similar between control (3.9±0.2 kg), mPHG (4.3±0.2 kg), and PHG fetuses (3.6±0.1kg). Fetal body weight was included as a covariate for all analyses of fetal organ weights, and no treatment effect was found for individual fetal organ weights (data not shown).
Maternal and fetal plasma values during treatment
Prior to treatment, maternal and fetal plasma glucose and fetal plasma insulin concentrations were not different between treatment groups (Table 1). Per our experimental design, the magnitude and pattern of hyperglycemia was tightly controlled during the treatment period (Table 1, Figure 1). The inter-animal coefficients of variation in control, mPHG, and PHG sheep, respectively, were 3.2%, 4.0%, and 3.3% for maternal plasma glucose and 20.7%, 13.6%, and 13.2% for fetal plasma glucose. The within-animal coefficients of variation for daily samples did not differ between treatments for either maternal (mean 5.4±0.4%; P=0.50) or fetal plasma glucose concentrations (mean 8.7±0.7%; P=0.26). Basal maternal plasma glucose was 15±2% higher in mPHG ewes and 20±2% higher in PHG ewes compared to euglycemic controls (P<0.01; Table 1, Figure 1). Fetal plasma glucose was 16±7% and 21±5% higher in the mPHG and PHG fetuses, respectively (P<0.05). Fetal insulin concentrations were not different between groups during the treatment period (Table 1).
Table 1.
Ovine maternal plasma glucose and lactate and fetal plasma glucose, lactate, and insulin prior to and during the treatment period.
| Control | mPHG | PHG | |
|---|---|---|---|
| N | 10 | 5 | 10 |
| Pre-treatment | |||
| Maternal glucose (mmol/L) | 3.83±0.06 | 4.07±0.07 | 3.78±0.09 |
| Fetal glucose (mmol/L) | 1.19±0.07 | 1.15±0.07 | 1.11±0.05 |
| Fetal insulin (ng/ml) | 0.34±0.04 | 0.53±0.07 | 0.36±0.05 |
| Treatment | |||
| Maternal glucose (mmol/L) | 3.66±0.04b | 4.22±0.06a | 4.39±0.06a |
| Fetal glucose (mmol/L) | 1.07±0.07b | 1.23±0.07ab | 1.29±0.06a |
| Fetal insulin (ng/ml) | 0.39±0.05 | 0.53±0.08 | 0.38±0.05 |
Values are means ± SEM. Pre-treatment and treatment values are the means from samples taken daily prior to the 0800 bolus. Values not sharing the same letter within each row are different, P<0.05.
Figure 1. Maternal (A) and Fetal (B) Plasma Glucose Concentrations.

Sustained mild (mPHG) or moderate (PHG) hyperglycemia was initiated on day 0. Plasma was sampled prior to initiating treatment on day 0 and prior to the 0800 dextrose pulse daily thereafter. Panel C shows maternal and fetal plasma glucose concentrations during the 45-min dextrose pulse administered at 1400 on day 9 of treatment. The two-hour sampling period illustrates the magnitude and duration of hyperglycemia resulting from one of the dextrose pulses, which were given 3x/day throughout the treatment period.
At the peak of the boluses, plasma glucose concentrations were 1.56 fold higher than euglycemic control concentrations in mPHG ewes (5.72±0.09 mmol/L) and fetuses (1.67±0.10 mmol/L), 2.03 fold higher in PHG ewes (7.44±0.03 mmol/L), and 2.01 fold higher in PHG fetuses (2.16±0.10 mmol/L, P<0.01; Figure 1C). Plasma insulin concentrations were increased 1.40-fold (0.74±0.11 ng/ml; P<0.05) in mPHG and 1.58-fold (1.60±0.07 ng/ml; P<0.01) in PHG compared to basal values (P<0.05). The peak bolus glucose concentrations had inter-animal coefficients of variation of 3.5% and 7.0% for maternal glucose and 13.9% and 12.5% for fetal glucose in mPHG and PHG animals, respectively. The average within-animal coefficients of variation (determined from at least six boluses on different days) were 6.4±0.5% for ewes and 8.7±0.7% for fetuses and were not different between treatments.
Treatment infusion rates
The amount of dextrose required to maintain basal maternal glucose at target concentrations increased during the treatment in the mPHG and PHG ewes (Figure 2). The constant infusion rates on d 14 of treatment were 3.6-fold and 2.4-fold higher than d 1 in the mPHG and PHG groups, respectively, and the slopes of the infusion rates over time were positive for both treatments (P<0.01; Figure 2). The bolus infusion rates also increased 1.3-fold (positive slope, P<0.01) and 1.1-fold (slope P=0.12) during the treatments, averaging 0.17±0.01 g•kg BW−1•h−1 and 0.31±0.02 g•kg BW−1•h−1 in the mPHG and PHG groups, respectively.
Figure 2. Dextrose Infusion Rates Increase during mPHG and PHG Treatment Periods.

Rates of dextrose infusion required to chronically maintain maternal plasma glucose 15% (mPHG) and 20% (PHG) above euglycemia increased during the course of the 14-day treatment.
Hematological, lactate, and norepinephrine values during fetal GSIS
Blood gases and blood pH were measured to evaluate physiological responses associated with hyperglycemia during the treatment and GSIS study. The fetal partial pressure of CO2 was greater in PHG fetuses (51.50±0.18 mmHg; P<0.01 for treatment effect) compared to controls (49.30±0.21 mmHg), and mPHG fetuses (49.99±0.23 mmHg) were not different from either control or PHG treatments. The pCO2 increased in the hyperglycemic period (50.80±0.17 mmHg, P<0.01 for period) versus basal (49.72±0.17 mmHg), and there was no treatment by period interaction. Fetal blood oxygen content was not different between treatments but declined (P<0.01) during the GSIS hyperglycemic clamp (3.3±0.5 mmol/L) compared to the basal period (3.7±0.10 mmol/L). Average fetal arterial pO2 was 20.8±0.9 mmHg at basal and was unaffected by treatment or period. Fetal blood pH was also not affected by treatment, but pH was decreased (P<0.01) in the hyperglycemic period (7.35 ±0.003) compared to basal period (7.37±0.004). Maternal hematological parameters were not different between treatments (data not shown).
Differences within treatment and period were found for fetal plasma lactate concentrations, but no treatment × period interaction was observed. Plasma lactate concentrations were greater (P<0.01) in PHG fetuses (2.63±0.03 mmol/L) than mPHG fetuses (2.22±0.04 mmol/L) and both were greater (P<0.01) than control fetuses (1.88±0.03 mmol/L). Plasma lactate concentrations increased (P<0.01) during the hyperglycemic clamp (2.48±0.03 mmol/L) from a basal period (1.99±0.03 mmol/L). Maternal plasma lactate concentrations were lower (P<0.05) in PHG ewes (0.45±0.01 mmol/L) than mPHG ewes (0.49±0.01 mmol/L) and were lower in both treatments than in controls (0.82±0.01 mmol/L; P<0.01). The fetal hyperglycemic clamp did not alter maternal plasma lactate concentrations.
In the sheep fetus, norepinephrine in response to fetal stress has been shown to increase oxygen tension and lactate concentrations and decrease insulin secretion (Bassett and Hanson 2000; Jackson et al. 2000). To confirm that adrenergic stimulation was not responsible for the hematological and metabolic responses described above, we measured fetal plasma norepinephine concentrations during the basal period of the GSIS study(Jackson et al. 2000). Fetal norepinephrine concentrations were not different between control (339±78 pg/ml) and PHG fetuses (402±182 pg/ml; P=0.75).
Fetal GSIS and GPAIS
In the GSIS basal period fetal plasma glucose concentrations were not different from the treatment values reported above (Table 1 and Figure 3). During the square-wave hyperglycemic clamp, glucose concentrations were increased to 2.3±0.2 mmol/L, which was not different between treatments (Figures 3A and 3B). Basal plasma insulin concentrations were not different between treatments and were 0.44±0.08 ng/ml in control, 0.50±0.06 ng/ml in mPHG, and 0.42±0.07 ng/ml in PHG fetuses. At the hyperglycemic steady state, plasma insulin concentrations were lower (P<0.05) in PHG fetuses (0.86±0.13 ng/ml) compared to control fetuses (1.58±0.25 ng/ml). Hyperglycemic insulin concentrations in the mPHG fetuses (1.21±0.08 ng/ml) were not different from the control (P<0.08) or PHG (P<0.09) treatments. GSIS responsiveness was calculated as the difference between hyperglycemic and basal steady state plasma insulin concentrations. PHG fetuses had lower (P<0.05) GSIS responsiveness (0.45±0.07 ng/ml) compared to controls (1.14±0.25 ng/ml), while the mPHG fetuses were intermediate (0.71±0.12 ng/ml) and not different from either control or PHG fetuses (Figures 3C and 3D). The fetal plasma insulin to glucose ratio did not differ between treatments during the basal period, but during the hyperglycemic clamp it was lower in PHG fetuses (0.35±0.05; P<0.05) than in controls (0.68±0.11); mPHG fetuses (0.52±0.03) were intermediate and not different from the other treatments (Figure 3E).
Figure 3. Impaired Glucose Stimulated Insulin Secretion in PHG Fetuses.

Graphs A and C show mean plasma glucose and insulin concentrations during basal (−21 to 0 minutes) and hyperglycemic (after time 0) periods. The bar graphs show the changes in glucose (B) and insulin (D) between the basal and hyperglycemic (45-61 minutes) steady state periods, which were calculated as the difference between hyperglycemia and basal mean concentrations. The elevation in glucose was similar between treatments, but PHG fetuses had a lower insulin response than control fetuses (P<0.05). Plasma insulin/glucose ratios (panel E) are shown for basal and hyperglycemic steady state periods. Bars not sharing the same letter are different, P<0.05.
Insulin concentrations following the arginine bolus reached maximum values after five minutes in all treatments (Figure 4). Insulin concentrations during the GPAIS were lower in PHG fetuses compared with control and mPHG fetuses at both 5 min (P<0.01) and 15 min (P<0.05) following administration of arginine. The net incremental insulin area under the curve (AUC), when calculated from basal insulin concentrations, was lower in PHG fetuses (P≤0.05) than in control fetuses (Figure 4B), reflecting impairment in the combined ability of glucose and arginine to enhance insulin secretion. In contrast, when net incremental AUC was calculated from the hyperglycemic steady state insulin concentrations, the AUC values were not different between groups (P=0.40; not shown).
Figure 4. Impaired GPAIS in PHG Fetuses.

The follow-on arginine bolus administered at ~65 min into the square-wave hyperglycemic clamp stimulated insulin secretion in all treatment groups and is presented relative to the start of the arginine infusion in panel A. An interaction between treatment and sample time (P<0.05) was found, and insulin concentrations were lower (P<0.05) in PHG fetuses at 5 and 15 minutes, indicated by the asterisks. In panel B, GPAIS area under the curve (ng/mL X minute), calculated using basal steady state period insulin concentration as baseline, is reduced in PHG fetuses (* = P≤0.05).
Isolated islet insulin contents, insulin secretion, and glucose oxidation
Islet insulin contents varied with treatment (P<0.05; Figure 5A); mPHG islets had greater insulin contents and PHG islets had lower insulin contents than control islets (P<0.05). Mean insulin release across the islet static incubations was reduced (P<0.05) in PHG islets (2.1±0.3 ng/islet) compared to control (3.2±0.4 ng/islet) and mPHG islets (3.9±0.4 ng/islet), but control and mPHG means were not different. Among all treatments, insulin release was increased (P≤0.05) in media conditions containing 11.1 mmol/L glucose (2.7±0.4 ng/islet) and 30 mmol/L KCl (8.8±0.4 ng/islet) compared to incubation conditions with 0 mmol/L and 1.1 mmol/L glucose (1.2±0.4 and 1.4±0.4 ng/islet; Figure 5B). The only difference between treatment groups within an incubation condition was found in the 30 mmol/L KCl condition; insulin release in PHG islets was less (P<0.05) than control and mPHG islets. Islets incubated on ice in 11.1 mmol/L glucose released equivalent amounts of insulin (1.4±0.4 ng/islet) as islets in no glucose, demonstrating that insulin in the media was due to secretion and not cellular breakdown. Fractional insulin secretion, calculated as the percentage of total islet insulin released into the media, did not vary by treatment (data not shown).
Figure 5. Decreased Insulin Content and Release in PHG Islets.

In panel A, mean insulin content (ng/islet) is presented for islets isolated from control (n=7), mPHG (n=5), and PHG (n=8) fetuses (indicated on the x-axis). In panel B, insulin release (ng/islet) is presented for static incubations in KRB/BSA media containing 0 mmol/L glucose, 1.1 mmol/L glucose, 11.1 mmol/L glucose, 30 mmol/L KCl and 1.1 mmol/L glucose, or 11.1 mmol/L glucose incubated on ice (indicated on the abscissa). Insulin release was increased in 11.1 mmol/L glucose and 30 mmol/L KCl compared to incubation with 0 and 1.1 mmol/L glucose, and is indicated by the horizontal bars with an asterisk (P<0.05). In the PHG islets, KCl stimulated insulin release was less than control and mPHG islets, which is indicated with the number symbol (P<0.05).
The rate of glucose oxidation was measured in control and PHG islets. Stimulatory glucose concentrations (11.1 mmol/L) increased the rate of glucose oxidative phosphorylation in isolated islets two-fold (8.1±1.2 pmol/islet/hr) compared to basal rates measured in 1.1 mmol/L glucose (4.1±0.6 pmol/islet/hr; P<0.01). No differences between treatments were found.
Islet reactive oxygen species accumulation
Fluorescence imaging of single isolated islets preloaded with CM-H2DCFDA showed no difference between treatments in the rate of islet ROS accumulation at basal glucose concentrations (1.1 mmol/l); basal rates were 103±17 a.u./min for control islets, 118±37 a.u./min for mPHG islets, and 94±33 a.u./min for PHG islets. Experimental intra-islet variation due to CM-H2DCFDA loading was normalized to the basal rate in each individual islet to compare rates of ROS accumulation in the presence of stimulatory glucose and H2O2. Elevation of glucose from 1.1 mmol/L to 11.1 mmol/L lead to a 63% increase in the ROS accumulation rate in PHG islets, but there were no changes in control or mPHG islets. The relative glucose-stimulated (11.1 mmol/L) rate of ROS accumulation was greater in PHG islets compared with control and mPHG islets (P<0.05; Figure 6). The subsequent addition of 9 mmol/L H2O2 further increased the rate of ROS accumulation similarly in all treatments (Figure 6), showing that the dynamic range of the probe was not exceeded during the experiment. These data indicate that isolated PHG islets have a greater rate of ROS accumulation under stimulatory glucose conditions.
Figure 6. Greater Glucose Stimulated ROS Accumulation in PHG Islets.
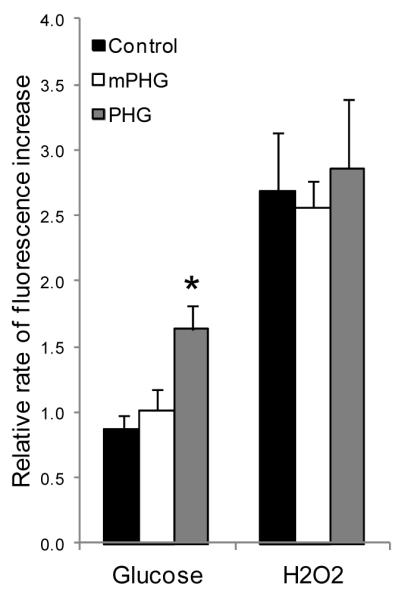
In isolated fetal sheep islets, the fluorescence intensities from oxidation of the CM-H2DCFDA probe were determined in 1.1 mmol/L glucose, 11.1 mmol/L glucose, and 9 mmol/L H2O2 every minute for 15 minutes in each condition. Rates of ROS accumulation in stimulatory glucose and H2O2 were normalized to the basal rate (1.1 mmol/L glucose) for each islet, and the bars represent the mean ± SEM for 2-5 islets from each of eight control, five mPHG, and six PHG fetuses. The asterisk indicates a significant difference (P<0.05) between control and PHG treatments in ROS accumulation at 11.1 mmol/L glucose.
Islet gene expression for antioxidant enzymes, insulin expression, and ER stress
In isolated islets, relative mRNA expression levels for SOD-1, SOD-2, catalase, and GPx-1 were similar between treatments (Figure 7), indicating that the greater glucose-stimulated ROS accumulation in PHG islets was caused by increased production of ROS rather than a change in enzymatic ROS clearance. In addition, mRNA expression levels for insulin, PDX-1, MafA, UCP2, and endoplasmic reticulum stress response genes (GRP78 and DDIT-3) were not different between control and PHG islets (Figure 7). Islets from mPHG fetuses were not evaluated.
Figure 7. Islet Gene Expression for Antioxidant Enzymes, Insulin, and ER Stress.

The mRNA expression normalized to ribosomal protein s15 is shown for enzymatic antioxidant defense (SOD-1, SOD-2, catalase, and GPX-1); insulin transcription (insulin, PDX-1, and MafA); and endoplasmic reticulum stress (GRP78 and DDIT3) in islets isolated from control and PHG fetuses. Values shown are calculated by the comparative ΔCT method (CT gene of interest - CT reference gene). Gene expression was not different between control and PHG islets.
Tissue protein oxidative stress, antioxidant enzymes, and insulin receptor
We evaluated whether chronic PHG caused islet or systemic oxidative stress by measuring carbonyl incorporation of proteins, which is an indicator of protein oxidative damage. Skeletal muscle from PHG fetuses had 46% greater intensities for carbonyl moieties compared to control fetuses (P<0.05). However, there was no difference between PHG and control fetuses in carbonyl incorporation in liver or islet proteins. Protein concentrations of SOD1, SOD2, and GPx-1/2 in skeletal muscle and liver measured by Western blot were not different between control and PHG treatments (Figure 8A-D). Tissues from mPHG fetuses were not evaluated.
Figure 8. Antioxidant Enzyme and Insulin Receptor Concentrations in Liver and Skeletal Muscle.

Representative Western immunoblots show protein concentrations of SOD-1, SOD-2, GPx-1/2, and the reference protein ribosomal protein S6, in skeletal muscle (A and B) and liver (C and D) and of insulin receptor β and the reference protein β-tubulin in skeletal muscle (SM) and liver (E and D). Bar graphs show quantification of band density by analysis in ImageJ. Antioxidant enzyme and insulin receptor β protein concentrations were not different between control and PHG treatments.
Insulin receptor concentrations were measured in control and PHG insulin-sensitive tissues to evaluate whether receptor-mediated insulin clearance was enhanced by PHG exposure. No differences were found for insulin receptor concentrations in liver or skeletal muscle protein extracts (Figure 8E and F).
DISCUSSION
The objective of this study was to determine if two weeks of PHG exposure, similar to conditions in managed diabetic pregnancies, would impact fetal β-cell responsiveness. The major finding was that PHG treatment attenuated fetal insulin secretion in response to glucose and glucose-potentiated arginine. The GPAIS study showed that the readily releasable pool of insulin was reduced in PHG fetuses compared to controls. Islets isolated from PHG fetuses also had reduced insulin content and insulin release. Moreover, maximal insulin release induced by depolarizing the islets with potassium chloride was lower in PHG islets. These data, together with the fetal GPAIS results, indicate that insulin content was a limiting factor for PHG islets because both measures are dependent on β-cell insulin content or β-cell mass (Robertson 2007; Seaquist and Robertson 1992). Fractional islet insulin release and islet glucose metabolism were not affected by PHG treatment, further supporting the hypothesis that reduced β-cell insulin content, rather than a defect in stimulus-secretion coupling, was the major factor explaining attenuated fetal GSIS. The second major finding of this study was that glucose-stimulated ROS accumulation was greater in the PHG islets, but there was no evidence for islet oxidative stress or ER stress. Therefore, oxidative stress does not appear to initiate the decline in insulin content; however, if persistent, it will cause islet oxidative damage and β-cell failure, as shown for adult models of hyperglycemia-induced diabetes (Kaneto et al. 1999; Tanaka et al. 1999; Tang et al. 2007). The major outcomes from PHG exposure were reduced fetal insulin secretion due to less insulin content and impaired islet ROS handling, but these two deficiencies do not appear to have a causal relationship.
The results of this study show that islet dysfunction is dependent on the magnitude of pulsatile hyperglycemia. Strikingly, GSIS responsiveness was intermediate in mPHG fetuses, and the mPHG response to the GPAIS study was not different from controls but greater than PHG fetuses. The mPHG islet insulin content was also greater than controls and, coupled with normal fetal GSIS, indicates insulin stimulus-secretion may be reduced. This is also supported in the islet experiments, because no enhancement of islet insulin secretion was observed even with elevated insulin content. Together, these findings indicate that within a narrow range of exposure to mild or moderate pulsatile hyperglycemia, fetal β-cells exhibit impaired insulin stimulus-secretion with a compensatory increase in insulin contents (mPHG) or lower insulin contents (PHG), depending on the magnitude of hyperglycemia.
Insulin secretion responsiveness varies in human neonates born to diabetic mothers, and this variation is probably a result of differences in the duration and magnitude of exposure to hyperglycemia, as in our study and other animal studies described below. One human study shows no difference in infants’ insulin responses to a glucose infusion (King et al. 1989). Others show higher insulin concentrations in infants from diabetic mothers (Obenshain et al. 1970; Pildes et al. 1969; Pribylova and Kozlova 1979), similar to the findings from our mPHG group, which had increased islet insulin content. Another study that directly measured insulin responsiveness to an intravenous glucose infusion in 2-hour old infants of normal and diabetic mothers found that infants of diabetic mothers had a higher first phase but lower second phase insulin response (Isles et al. 1968), indicating β-cell dysfunction. Regardless, children of diabetic pregnancies exhibit an increased incidence of glucose intolerance (Pettitt et al. 1985; Silverman et al. 1995), and by the time these offspring reach their early twenties, they have compromised acute or early insulin secretion compared to control subjects (Gautier et al. 2001; Sobngwi et al. 2003). Together, these data support the notion that insulin secretion is programmed in utero, and our findings suggest that there is a narrow range of hyperglycemia within which either insulin secretion or production can be impaired. It should also be noted that basal glucose concentrations were not significantly different between the mPHG and PHG treatments in our study, but the pulsatile excursions were greater in the PHG treatment. Thus, differences in outcomes between the mPHG and PHG fetuses appear to be driven by the pulsatile excursions rather than chronic sustained hyperglycemia, an observation that has also been made in human studies (Combs et al. 1992; de Veciana et al. 1995; Jovanovic-Peterson et al. 1991; Most and Langer 2007).
Other studies in pregnant sheep have evaluated effects of hyperglycemia on fetal insulin secretion. Carver et al. (Carver et al. 1996) tested 10 days of chronic sustained hyperglycemia (+35%) and a pulsatile treatment (+17% sustained, +60% pulses 3x/d) similar to our mPHG group. They found that the magnitude and pattern of hyperglycemia affected β-cell function differently, because insulin secretion was suppressed with chronic constant hyperglycemia and enhanced in the pulsatile hyperglycemia treatment (Carver et al. 1995; Carver et al. 1996). In our study, insulin secretion was suppressed after 14 days of PHG treatment (+20% sustained, +100% pulses 3x/d) but not mPHG treatment. We do not believe this difference was due to duration of treatment, at least for the PHG fetuses, because comparison of the insulin/glucose ratio during one of the boluses on day 9 to the hyperglycemic clamp on day 14, which reached similar glucose concentrations, showed no difference between days 9 (0.32±0.03) and 14 (0.31±0.03). This indicates that the lower insulin secretion responsiveness in PHG fetuses had occurred by 9 days. Therefore, PHG might exceed a threshold of severity of hyperglycemia, resulting in lower insulin secretion as found in fetuses with 35% chronic sustained hyperglycemia (Carver et al. 1996). We found greater insulin content in islets isolated from mPHG fetuses compared to control fetuses, while PHG fetuses had less islet insulin content (Figure 5). These data also reflect a β-cell response that is dependent on the magnitude of hyperglycemic exposure. In the milder PHG treatments by Carver and colleagues, duration could still be a factor and the increased insulin production or β-cell mass, which appears to occur in mPHG islets, could occur initially in PHG islets. However, with continued hyperglycemic exposure, the β-cells become exhausted and insulin content decreases.
Rodent models of diabetic pregnancies generated by streptozotocin injections or direct glucose infusions have outcomes in offspring that are also dependent upon the severity of hyperglycemia. Severe hyperglycemia (usually 3-4x normal) in rat fetuses leads to β-cell degranulation and exhaustion, hypoinsulinemia, and impaired insulin secretion (Aerts and van Assche 1977; Kervran et al. 1978). Postnatally, offspring have hypertrophy of the endocrine pancreas with an excess of small islets (Aerts et al. 1997) and are hypoglycemic and insulin resistant (Aerts and Van Assche 1981; Holemans et al. 1991). In contrast, rat fetuses exposed to moderate hyperglycemia (1.3-2x normal) have β-cell hyperplasia, increased insulin synthesis, and enhanced glucose stimulated insulin secretion (Aerts et al. 1997; Kervran et al. 1978). However, withdrawal of hyperglycemia after birth results in impaired GSIS with normal pancreatic endocrine mass and cell distribution at weaning (Aerts et al. 1990; Aerts et al. 1997; Bihoreau et al. 1986; Boloker et al. 2002; Gauguier et al. 1991; Han et al. 2007; Van Assche et al. 2001). The work in rodent models further highlights differential outcomes resulting from moderate or severe hyperglycemia, similar to those found in our mPHG and PHG treatments, and it exemplifies the need for fetal evaluation of relevant hyperglycemic paradigms.
PHG islets demonstrated increased glucose-stimulated ROS accumulation compared to control or mPHG islets. Antioxidant enzyme expression was not different between control and PHG islets, indicating that the increased ROS accumulation was due to greater ROS production rather than impaired ROS clearance. It is worth noting that preliminary experiments in our laboratory indicate that overnight culture of fetal sheep islets, as was used in our study, induced a six-fold increase in the expression of SOD-2 (mitochondrial) and a three-fold increase in GPx-1 expression. Thus, culture may be masking treatment effects on gene expression, but ROS accumulation was greater in PHG than control islets even after overnight culture and the presumed increased expression of antioxidant enzymes.
A direct link between decreased islet insulin content and increased islet ROS accumulation is not apparent. No differences in oxidative damage to islet proteins or UCP2 expression (Brand et al. 2010) were detected between control and PHG groups. However, we did find an increase in carbonyl incorporation in PHG skeletal muscle, which indicates higher systemic oxidative stress. Hyperglycemia and oxidative stress have been associated with vascular dysfunction in islets and other cell types (Homo-Delarche et al. 2006; Koukkou et al. 1998; Segar et al. 2009; Teixeira and Andrade 1999), and defects in islet vasculature have been known to decrease insulin secretion (Eberhard et al. 2010; Richards et al. 2010). In the GK/Par spontaneous type 2 diabetes rat model, oxidative stress markers are concentrated in the peri-islet vascular and inflammatory compartments (Lacraz et al. 2009). Defects in islet vasculature might explain the impairment of glucose-stimulated insulin secretion found in the fetus but not in isolated islets, as the isolation procedure would eliminate hemodynamic effects and might also prevent the detection of oxidative damage to peri-islet vasculature.
Although a direct mechanism linking ROS accumulation to impairment of insulin secretion in vivo has not been identified, increased ROS accumulation could represent an islet defect, which, if persistent, will cause oxidative damage and contribute to islet dysfunction in adulthood (Kaneto et al. 1999; Tanaka et al. 1999; Tang et al. 2007). In the GK/Par rat model, islets have greater ROS accumulation prior to the spontaneous onset of diabetes; after diabetes is evident, protective antioxidant enzymes are up regulated and oxidative damage is found in the pancreas (Lacraz et al. 2009). Chronic oxidative stress undermines the signaling potential of ROS and thus blunts insulin secretion function (Pi et al. 2007; Robertson and Harmon 2006). Therefore, we predict that the observed increase in glucose-stimulated ROS accumulation in PHG islets foreshadows overt oxidative stress and the deterioration of islet insulin secretion.
The dextrose infusion rate increased during the two-week PHG treatment in order to maintain chronic hyperglycemia in the pregnant ewe (Figure 2), indicating improved maternal glucose disposal. If the PHG fetuses also have improved insulin sensitivity, they would have greater insulin clearance due to an up regulation of insulin receptors (Flier et al. 1982; Mittelman et al. 2000). Although insulin clearance or insulin sensitivity was not directly measured in the current study, multiple lines of evidence indicate that insulin sensitivity is not a major factor for reduced insulin concentrations in PHG fetuses. No differences were found in insulin to glucose ratios during basal treatment conditions (Figure 3E). Insulin clearance following the arginine bolus was not different between treatments, though it tended (P=0.075) to be lower in PHG (−0.65±0.01 ng/ml) than control (−0.107±0.02 ng/ml) or mPHG (−0.108±0.02) fetuses. Finally, in liver and skeletal muscle, insulin receptor concentrations were not different between control and PHG fetuses (Figure 8). These findings indicate that lower plasma insulin concentrations following the GSIS or GPAIS challenge were due to decreased insulin secretion and not increased clearance, which is also supported by isolated islet experiments (Figure 5).
In the current study, fetal insulin secretion was impaired following a two-week regimen of pulsatile hyperglycemia during late gestation. The PHG treatment was designed to replicate the magnitude and pattern of glycemic control found in pregnant women with diabetes managed under the current clinical guidelines. However, many women do not attain this degree of glycemic control during their pregnancies, and in these cases of fetal exposure to more severe hyperglycemia, the outcomes are likely worse. Islet ROS accumulation appears to occur independently of the impairment in insulin secretion, but if this defect persists, we expect it to cause further damage to islet function. Because of the clinical relevance of the PHG treatment regimen and the resulting diabetic phenotype in fetal offspring, additional work with this model will be useful in investigating the mechanisms for the association of fetal exposure to hyperglycemia and diabetes risk later in life.
ACKNOWLEDGEMENTS
We are grateful to Mandie M. Dunham, Craig S. Weber, and Devora Magier for their technical assistance.
FUNDING The project described was supported by Competitive Advantage Award CAA 0230-08 from the Science Foundation of Arizona, Award Number R01 DK084842 (Principle Investigator S.W. Limesand) from the National Institute of Diabetes and Digestive and Kidney Diseases, and an Endocrine Society Bridge Grant (Principle Investigator S.W. Limesand). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Diabetes and Digestive and Kidney Diseases or the National Institutes of Health. A.S. Green was supported by T32 HL7249 and F32 DK088514. A.R. Macko was supported by T32 HL7249.
Footnotes
DECLARATION OF INTEREST All authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported
REFERENCES
- Aerts L, Holemans K, Van Assche FA. Maternal diabetes during pregnancy: consequences for the offspring. Diabetes Metab Rev. 1990;6:147–167. doi: 10.1002/dmr.5610060303. [DOI] [PubMed] [Google Scholar]
- Aerts L, van Assche FA. Rat foetal endocrine pancreas in experimental diabetes. J Endocrinol. 1977;73:339–346. doi: 10.1677/joe.0.0730339. [DOI] [PubMed] [Google Scholar]
- Aerts L, Van Assche FA. Endocrine pancreas in the offspring of rats with experimentally induced diabetes. J Endocrinol. 1981;88:81–88. doi: 10.1677/joe.0.0880081. [DOI] [PubMed] [Google Scholar]
- Aerts L, Vercruysse L, Van Assche FA. The endocrine pancreas in virgin and pregnant offspring of diabetic pregnant rats. Diabetes Res Clin Pract. 1997;38:9–19. doi: 10.1016/s0168-8227(97)00080-6. [DOI] [PubMed] [Google Scholar]
- American Diabetes Association Gestational Diabetes Mellitus. Diabetes Care. 2004;27:S88–90. doi: 10.2337/diacare.27.2007.s88. [DOI] [PubMed] [Google Scholar]
- Araki E, Oyadomari S, Mori M. Impact of endoplasmic reticulum stress pathway on pancreatic beta-cells and diabetes mellitus. Exp Biol Med (Maywood) 2003;228:1213–1217. doi: 10.1177/153537020322801018. [DOI] [PubMed] [Google Scholar]
- Bassett JM, Hanson C. Prevention of hypoinsulinemia modifies catecholamine effects in fetal sheep. Am J Physiol Regul Integr Comp Physiol. 2000;278:R1171–R1181. doi: 10.1152/ajpregu.2000.278.5.R1171. [DOI] [PubMed] [Google Scholar]
- Bihoreau MT, Ktorza A, Kervran A, Picon L. Effect of gestational hyperglycemia on insulin secretion in vivo and in vitro by fetal rat pancreas. Am J Physiol. 1986;251:E86–E91. doi: 10.1152/ajpendo.1986.251.1.E86. [DOI] [PubMed] [Google Scholar]
- Boloker J, Gertz SJ, Simmons RA. Gestational diabetes leads to the development of diabetes in adulthood in the rat. Diabetes. 2002;51:1499–1506. doi: 10.2337/diabetes.51.5.1499. [DOI] [PubMed] [Google Scholar]
- Brand MD, Parker N, Affourtit C, Mookerjee SA, Azzu V. Mitochondrial uncoupling protein 2 in pancreatic beta-cells. Diabetes Obes Metab. 2010;12(Suppl 2):134–140. doi: 10.1111/j.1463-1326.2010.01264.x. [DOI] [PubMed] [Google Scholar]; Carpenter MW, Coustan DR. Criteria for screening tests for gestational diabetes. Am J Obstet Gynecol. 1982;144:768–773. doi: 10.1016/0002-9378(82)90349-0. [DOI] [PubMed] [Google Scholar]
- Carver TD, Anderson SM, Aldoretta PA, Esler AL, Hay WW., Jr Glucose suppression of insulin secretion in chronically hyperglycemic fetal sheep. Pediatr Res. 1995;38:754–762. doi: 10.1203/00006450-199511000-00020. [DOI] [PubMed] [Google Scholar]
- Carver TD, Anderson SM, Aldoretta PW, Hay WW., Jr Effect of low-level basal plus marked “pulsatile” hyperglycemia on insulin secretion in fetal sheep. Am J Physiol. 1996;271:E865–E871. doi: 10.1152/ajpendo.1996.271.5.E865. [DOI] [PubMed] [Google Scholar]
- Cederberg J, Basu S, Eriksson UJ. Increased rate of lipid peroxidation and protein carbonylation in experimental diabetic pregnancy. Diabetologia. 2001;44:766–774. doi: 10.1007/s001250051686. [DOI] [PubMed] [Google Scholar]
- Chen X, Fahy AL, Green AS, Anderson MJ, Rhoads RP, Limesand SW. beta2-Adrenergic receptor desensitization in perirenal adipose tissue in fetuses and lambs with placental insufficiency-induced intrauterine growth restriction. J Physiol. 2010;588:3539–3549. doi: 10.1113/jphysiol.2010.192310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Combs CA, Gunderson E, Kitzmiller JL, Gavin LA, Main EK. Relationship of fetal macrosomia to maternal postprandial glucose control during pregnancy. Diabetes Care. 1992;15:1251–1257. doi: 10.2337/diacare.15.10.1251. [DOI] [PubMed] [Google Scholar]
- Coughlan MT, Vervaart PP, Permezel M, Georgiou HM, Rice GE. Altered placental oxidative stress status in gestational diabetes mellitus. Placenta. 2004;25:78–84. doi: 10.1016/S0143-4004(03)00183-8. [DOI] [PubMed] [Google Scholar]
- Dabelea D, Hanson RL, Lindsay RS, Pettitt DJ, Imperatore G, Gabir MM, Roumain J, Bennett PH, Knowler WC. Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes. 2000;49:2208–2211. doi: 10.2337/diabetes.49.12.2208. [DOI] [PubMed] [Google Scholar]
- de Veciana M, Major CA, Morgan MA, Asrat T, Toohey JS, Lien JM, Evans AT. Postprandial versus Preprandial Blood Glucose Monitoring in Women with Gestational Diabetes Mellitus Requiring Insulin Therapy. N Engl J Med. 1995;333:1237–1241. doi: 10.1056/NEJM199511093331901. [DOI] [PubMed] [Google Scholar]
- Eberhard D, Kragl M, Lammert E. ‘Giving and taking’: endothelial and beta-cells in the islets of Langerhans. Trends Endocrinol Metab. 2010;21:457–463. doi: 10.1016/j.tem.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Eriksson UJ, Borg LA. Diabetes and embryonic malformations. Role of substrate-induced free-oxygen radical production for dysmorphogenesis in cultured rat embryos. Diabetes. 1993;42:411–419. doi: 10.2337/diab.42.3.411. [DOI] [PubMed] [Google Scholar]
- Flier JS, Minaker KL, Landsberg L, Young JB, Pallotta J, Rowe JW. Impaired in vivo insulin clearance in patients with severe target-cell resistance to insulin. Diabetes. 1982;31:132–135. doi: 10.2337/diab.31.2.132. [DOI] [PubMed] [Google Scholar]
- Gauguier D, Bihoreau MT, Picon L, Ktorza A. Insulin secretion in adult rats after intrauterine exposure to mild hyperglycemia during late gestation. Diabetes. 1991;40(Suppl 2):109–114. doi: 10.2337/diab.40.2.s109. [DOI] [PubMed] [Google Scholar]
- Gautier JF, Wilson C, Weyer C, Mott D, Knowler WC, Cavaghan M, Polonsky KS, Bogardus C, Pratley RE. Low acute insulin secretory responses in adult offspring of people with early onset type 2 diabetes. Diabetes. 2001;50:1828–1833. doi: 10.2337/diabetes.50.8.1828. [DOI] [PubMed] [Google Scholar]
- Gillmer MD, Beard RW, Brooke FM, Oakley NW. Carbohydrate metabolism in pregnancy. Part I. Diurnal plasma glucose profile in normal and diabetic women. Br Med J. 1975;3:399–402. doi: 10.1136/bmj.3.5980.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmartin AB, Ural SH, Repke JT. Gestational diabetes mellitus. Rev Obstet Gynecol. 2008;1:129–134. [PMC free article] [PubMed] [Google Scholar]
- Green AS, Macko AR, Rozance PJ, Yates DT, Chen X, Hay WW, Jr., Limesand SW. Characterization of glucose-insulin responsiveness and impact of fetal number and gender on insulin response in the sheep fetus. Am J Physiol Endocrinol Metab. 2011 doi: 10.1152/ajpendo.00572.2010. Epub ahead of print 2011/02/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green AS, Rozance PJ, Limesand SW. Consequences of a compromised intrauterine environment on islet function. J Endocrinol. 2010;205:211–224. doi: 10.1677/JOE-09-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Xu J, Long YS, Epstein PN, Liu YQ. Rat maternal diabetes impairs pancreatic beta-cell function in the offspring. Am J Physiol Endocrinol Metab. 2007;293:E228–236. doi: 10.1152/ajpendo.00479.2006. [DOI] [PubMed] [Google Scholar]
- Holemans K, Aerts L, Van Assche FA. Evidence for an insulin resistance in the adult offspring of pregnant streptozotocin-diabetic rats. Diabetologia. 1991;34:81–85. doi: 10.1007/BF00500377. [DOI] [PubMed] [Google Scholar]
- Homo-Delarche F, Calderari S, Irminger JC, Gangnerau MN, Coulaud J, Rickenbach K, Dolz M, Halban P, Portha B, Serradas P. Islet inflammation and fibrosis in a spontaneous model of type 2 diabetes, the GK rat. Diabetes. 2006;55:1625–1633. doi: 10.2337/db05-1526. [DOI] [PubMed] [Google Scholar]
- Isles TE, Dickson M, Farquhar JW. Glucose tolerance and plasma insulin in newborn infants of normal and diabetic mothers. Pediatr Res. 1968;2:198–208. doi: 10.1203/00006450-196805000-00007. [DOI] [PubMed] [Google Scholar]
- Jackson BT, Piasecki GJ, Cohn HE, Cohen WR. Control of fetal insulin secretion. Am J Physiol Regul Integr Comp Physiol. 2000;279:R2179–R2188. doi: 10.1152/ajpregu.2000.279.6.R2179. [DOI] [PubMed] [Google Scholar]
- Jovanovic-Peterson L, Peterson CM, Reed GF, Metzger BE, Mills JL, Knopp RH, Aarons JH. Maternal postprandial glucose levels and infant birth weight: the Diabetes in Early Pregnancy Study. The National Institute of Child Health and Human Development--Diabetes in Early Pregnancy Study. Am J Obstet Gynecol. 1991;164:103–111. doi: 10.1016/0002-9378(91)90637-7. [DOI] [PubMed] [Google Scholar]
- Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, Hanafusa T, Matsuzawa Y, Yamasaki Y, Hori M. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes. 1999;48:2398–2406. doi: 10.2337/diabetes.48.12.2398. [DOI] [PubMed] [Google Scholar]
- Kaneto H, Kawamori D, Matsuoka TA, Kajimoto Y, Yamasaki Y. Oxidative stress and pancreatic beta-cell dysfunction. Am J Ther. 2005;12:529–533. doi: 10.1097/01.mjt.0000178773.31525.c2. [DOI] [PubMed] [Google Scholar]
- Kaneto H, Xu G, Song KH, Suzuma K, Bonner-Weir S, Sharma A, Weir GC. Activation of the hexosamine pathway leads to deterioration of pancreatic beta-cell function through the induction of oxidative stress. J Biol Chem. 2001;276:31099–31104. doi: 10.1074/jbc.M104115200. [DOI] [PubMed] [Google Scholar]
- Karlsson K, Kjellmer I. The outcome of diabetic pregnancies in relation to the mother’s blood sugar level. Am J Obstet Gynecol. 1972;112:213–220. doi: 10.1016/0002-9378(72)90118-4. [DOI] [PubMed] [Google Scholar]
- Kervran A, Guillaume M, Jost A. The endocrine pancreas of the fetus from diabetic pregnant rat. Diabetologia. 1978;15:387–393. doi: 10.1007/BF01219648. [DOI] [PubMed] [Google Scholar]
- Kinalski M, Sledziewski A, Telejko B, Zarzycki W, Kinalska I. Antioxidant therapy and streptozotocin-induced diabetes in pregnant rats. Acta Diabetol. 1999;36:113–117. doi: 10.1007/s005920050153. [DOI] [PubMed] [Google Scholar]
- King KC, Oliven A, Kalhan SC. Functional enteroinsular axis in full-term newborn infants. Pediatr Res. 1989;25:490–495. doi: 10.1203/00006450-198905000-00013. [DOI] [PubMed] [Google Scholar]
- Koukkou E, Ghosh P, Lowy C, Poston L. Offspring of normal and diabetic rats fed saturated fat in pregnancy demonstrate vascular dysfunction. Circulation. 1998;98:2899–2904. doi: 10.1161/01.cir.98.25.2899. [DOI] [PubMed] [Google Scholar]
- Lacraz G, Figeac F, Movassat J, Kassis N, Coulaud J, Galinier A, Leloup C, Bailbe D, Homo-Delarche F, Portha B. Diabetic beta-cells can achieve self-protection against oxidative stress through an adaptive up-regulation of their antioxidant defenses. PLoS One. 2009;4:e6500. doi: 10.1371/journal.pone.0006500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappas M, Permezel M, Rice GE. Release of proinflammatory cytokines and 8-isoprostane from placenta, adipose tissue, and skeletal muscle from normal pregnant women and women with gestational diabetes mellitus. J Clin Endocrinol Metab. 2004;89:5627–5633. doi: 10.1210/jc.2003-032097. [DOI] [PubMed] [Google Scholar]
- Lenzen S, Drinkgern J, Tiedge M. Low antioxidant enzyme gene expression in pancreatic islets compared with various other mouse tissues. Free Radic Biol Med. 1996;20:463–466. doi: 10.1016/0891-5849(96)02051-5. [DOI] [PubMed] [Google Scholar]
- Leos RA, Anderson MJ, Chen X, Pugmire J, Anderson KA, Limesand SW. Chronic exposure to elevated norepinephrine suppresses insulin secretion in fetal sheep with placental insufficiency and intrauterine growth restriction. Am J Physiol Endocrinol Metab. 2010;298:E770–778. doi: 10.1152/ajpendo.00494.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limesand SW, Hay WW., Jr Adaptation of ovine fetal pancreatic insulin secretion to chronic hypoglycaemia and euglycaemic correction. J Physiol. 2003;547(1):95–105. doi: 10.1113/jphysiol.2002.026831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limesand SW, Rozance PJ, Smith D, Hay WW., Jr Increased insulin sensitivity and maintenance of glucose utilization rates in fetal sheep with placental insufficiency and intrauterine growth restriction. Am J Physiol Endocrinol Metab. 2007;293:E1716–1725. doi: 10.1152/ajpendo.00459.2007. [DOI] [PubMed] [Google Scholar]
- Limesand SW, Rozance PJ, Zerbe GO, Hutton JC, Hay WW., Jr Attenuated insulin release and storage in fetal sheep pancreatic islets with intrauterine growth restriction. Endocrinology. 2006;147:1488–1497. doi: 10.1210/en.2005-0900. [DOI] [PubMed] [Google Scholar]
- Maechler P, Jornot L, Wollheim CB. Hydrogen peroxide alters mitochondrial activation and insulin secretion in pancreatic beta cells. J Biol Chem. 1999;274:27905–27913. doi: 10.1074/jbc.274.39.27905. [DOI] [PubMed] [Google Scholar]
- Metzger BE, Lowe LP, Dyer AR, Trimble ER, Chaovarindr U, Coustan DR, Hadden DR, McCance DR, Hod M, McIntyre HD, et al. Hyperglycemia and adverse pregnancy outcomes. N Engl J Med. 2008;358:1991–2002. doi: 10.1056/NEJMoa0707943. [DOI] [PubMed] [Google Scholar]
- Mittelman SD, Van Citters GW, Kim SP, Davis DA, Dea MK, Hamilton-Wessler M, Bergman RN. Longitudinal compensation for fat-induced insulin resistance includes reduced insulin clearance and enhanced beta-cell response. Diabetes. 2000;49:2116–2125. doi: 10.2337/diabetes.49.12.2116. [DOI] [PubMed] [Google Scholar]
- Most O, Langer O. GDM women in good glycemic control: which meal-related measure enhances fetal well-being? J Perinat Med. 2007;35:481–485. doi: 10.1515/JPM.2007.130. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Inoguchi T, Sonta T, Maeda Y, Sasaki S, Sawada F, Tsubouchi H, Sonoda N, Kobayashi K, Sumimoto H, et al. Increased expression of NAD(P)H oxidase in islets of animal models of Type 2 diabetes and its improvement by an AT1 receptor antagonist. Biochem Biophys Res Commun. 2005;332:927–933. doi: 10.1016/j.bbrc.2005.05.065. [DOI] [PubMed] [Google Scholar]
- National Research Council . Nutrient Requirements of Small Ruminants: Sheep, Goats, Cervids, and New World Camelids. National Academy Press; Washington, D.C.: 2007. [Google Scholar]
- Noda M, Yamashita S, Takahashi N, Eto K, Shen LM, Izumi K, Daniel S, Tsubamoto Y, Nemoto T, Iino M, et al. Switch to anaerobic glucose metabolism with NADH accumulation in the beta-cell model of mitochondrial diabetes. Characteristics of betaHC9 cells deficient in mitochondrial DNA transcription. J Biol Chem. 2002;277:41817–41826. doi: 10.1074/jbc.M207690200. [DOI] [PubMed] [Google Scholar]
- Obenshain SS, Adam PA, King KC, Teramo K, Raivio KO, Raiha N, Schwartz R. Human fetal insulin response to sustained maternal hyperglycemia. N Engl J Med. 1970;283:566–570. doi: 10.1056/NEJM197009102831104. [DOI] [PubMed] [Google Scholar]
- Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death.Differ. 2004;11:381–389. doi: 10.1038/sj.cdd.4401373. [DOI] [PubMed] [Google Scholar]
- Parretti E, Mecacci F, Papini M, Cioni R, Carignani L, Mignosa M, La Torre P, Mello G. Third-trimester maternal glucose levels from diurnal profiles in nondiabetic pregnancies: correlation with sonographic parameters of fetal growth. Diabetes Care. 2001;24:1319–1323. doi: 10.2337/diacare.24.8.1319. [DOI] [PubMed] [Google Scholar]
- Pettitt DJ, Bennet PH, Knowler WC, Baird R, Aleck KA. Gestational diabetes mellitus and impaired glucose tolerance during pregnancy: long term effects on obesity and glucose tolerance in the offspring. Diabetes. 1985;34:119–122. doi: 10.2337/diab.34.2.s119. [DOI] [PubMed] [Google Scholar]
- Pi J, Bai Y, Zhang Q, Wong V, Floering LM, Daniel K, Reece JM, Deeney JT, Andersen ME, Corkey BE, et al. Reactive oxygen species as a signal in glucose-stimulated insulin secretion. Diabetes. 2007;56:1783–1791. doi: 10.2337/db06-1601. [DOI] [PubMed] [Google Scholar]
- Pildes RS, Hart RJ, Warrner R, Cornblath M. Plasma insulin response during oral glucose tolerance tests in newborns of normal and gestational diabetic mothers. Pediatrics. 1969;44:76–83. [PubMed] [Google Scholar]
- Plagemann A, Harder T, Kohlhoff R, Rohde W, Dorner G. Glucose tolerance and insulin secretion in children of mothers with pregestational IDDM or gestational diabetes. Diabetologia. 1997;40:1094–1100. doi: 10.1007/s001250050792. [DOI] [PubMed] [Google Scholar]
- Pribylova J, Kozlova J. Glucose and galactose infusions in newborns of diabetic and healthy mothers. Biol Neonate. 1979;36:193–197. doi: 10.1159/000241227. [DOI] [PubMed] [Google Scholar]
- Richards OC, Raines SM, Attie AD. The role of blood vessels, endothelial cells, and vascular pericytes in insulin secretion and peripheral insulin action. Endocr Rev. 2010;31:343–363. doi: 10.1210/er.2009-0035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson RP. Estimation of beta-cell mass by metabolic tests: necessary, but how sufficient? Diabetes. 2007;56:2420–2424. doi: 10.2337/db07-0742. [DOI] [PubMed] [Google Scholar]
- Robertson RP, Harmon JS. Diabetes, glucose toxicity, and oxidative stress: A case of double jeopardy for the pancreatic islet beta cell. Free Radic Biol Med. 2006;41:177–184. doi: 10.1016/j.freeradbiomed.2005.04.030. [DOI] [PubMed] [Google Scholar]
- Rozance PJ, Limesand SW, Hay WW., Jr Decreased nutrient-stimulated insulin secretion in chronically hypoglycemic late-gestation fetal sheep is due to an intrinsic islet defect. Am J Physiol Endocrinol Metab. 2006;291:E404–411. doi: 10.1152/ajpendo.00643.2005. [DOI] [PubMed] [Google Scholar]
- Sakai K, Matsumoto K, Nishikawa T, Suefuji M, Nakamaru K, Hirashima Y, Kawashima J, Shirotani T, Ichinose K, Brownlee M, et al. Mitochondrial reactive oxygen species reduce insulin secretion by pancreatic beta-cells. Biochem Biophys Res Commun. 2003;300:216–222. doi: 10.1016/s0006-291x(02)02832-2. [DOI] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- Seaquist ER, Robertson RP. Effects of hemipancreatectomy on pancreatic alpha and beta cell function in healthy human donors. J Clin Invest. 1992;89:1761–1766. doi: 10.1172/JCI115779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segar EM, Norris AW, Yao JR, Hu S, Koppenhafer SL, Roghair RD, Segar JL, Scholz TD. Programming of growth, insulin resistance and vascular dysfunction in offspring of late gestation diabetic rats. Clin Sci (Lond) 2009;117:129–138. doi: 10.1042/CS20080550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund T, Rad NT, Ritterath C, Siebert G, Henrich W, Buhling KJ. Longitudinal changes in the continuous glucose profile measured by the CGMS in healthy pregnant women and determination of cut-off values. Eur J Obstet Gynecol Reprod Biol. 2008;139:46–52. doi: 10.1016/j.ejogrb.2007.12.006. [DOI] [PubMed] [Google Scholar]
- Silverman BL, Metzger BE, Cho NH, Loeb CA. Impaired glucose tolerance in adolescent offspring of diabetic mothers. Relationship to fetal hyperinsulinism. Diabetes Care. 1995;18:611–617. doi: 10.2337/diacare.18.5.611. [DOI] [PubMed] [Google Scholar]
- Sobngwi E, Boudou P, Mauvais-Jarvis F, Leblanc H, Velho G, Vexiau P, Porcher R, Hadjadj S, Pratley R, Tataranni PA, et al. Effect of a diabetic environment in utero on predisposition to type 2 diabetes. Lancet. 2003;361:1861–1865. doi: 10.1016/S0140-6736(03)13505-2. [DOI] [PubMed] [Google Scholar]
- Suhonen L, Hiilesmaa V, Teramo K. Glycaemic control during early pregnancy and fetal malformations in women with type I diabetes mellitus. Diabetologia. 2000;43:79–82. doi: 10.1007/s001250050010. [DOI] [PubMed] [Google Scholar]
- Takahashi H, Tran PO, LeRoy E, Harmon JS, Tanaka Y, Robertson RP. D-Glyceraldehyde causes production of intracellular peroxide in pancreatic islets, oxidative stress, and defective beta cell function via non-mitochondrial pathways. J Biol Chem. 2004;279:37316–37323. doi: 10.1074/jbc.M403070200. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U.S.A. 1999;96:10857–10862. doi: 10.1073/pnas.96.19.10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Tran PO, Harmon J, Robertson RP. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci U S A. 2002;99:12363–12368. doi: 10.1073/pnas.192445199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C, Han P, Oprescu AI, Lee SC, Gyulkhandanyan AV, Chan GN, Wheeler MB, Giacca A. Evidence for a role of superoxide generation in glucose-induced beta-cell dysfunction in vivo. Diabetes. 2007;56:2722–2731. doi: 10.2337/db07-0279. [DOI] [PubMed] [Google Scholar]
- Teixeira AS, Andrade SP. Glucose-induced inhibition of angiogenesis in the rat sponge granuloma is prevented by aminoguanidine. Life Sci. 1999;64:655–662. doi: 10.1016/s0024-3205(98)00607-9. [DOI] [PubMed] [Google Scholar]
- Tiedge M, Lortz S, Drinkgern J, Lenzen S. Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes. 1997;46:1733–1742. doi: 10.2337/diab.46.11.1733. [DOI] [PubMed] [Google Scholar]
- Tsubouchi H, Inoguchi T, Inuo M, Kakimoto M, Sonta T, Sonoda N, Sasaki S, Kobayashi K, Sumimoto H, Nawata H. Sulfonylurea as well as elevated glucose levels stimulate reactive oxygen species production in the pancreatic beta-cell line, MIN6-a role of NAD(P)H oxidase in beta-cells. Biochem Biophys Res Commun. 2005;326:60–65. doi: 10.1016/j.bbrc.2004.10.201. [DOI] [PubMed] [Google Scholar]
- Assche FA, Holemans K, Aerts L. Long-term consequences for offspring of diabetes during pregnancy. Br Med Bull. 2001;60:173–182. doi: 10.1093/bmb/60.1.173. [DOI] [PubMed] [Google Scholar]