

Background: Lipoxygenases (LOXs) generate eicosanoids in inflammation.
Results: Monocyte/macrophage LOXs generate novel phospholipid-esterified eicosanoids containing ketoeicosatetraenoic acid or hydroperoxyeicosatetraenoic acid. They activate peroxisome proliferator-activated receptor-γ transcriptional activity and are found in cystic fibrosis bronchoalveolar fluid.
Significance: LOXs generate esterified eicosanoids in vitro and in vivo.
Conclusion: These new lipids represent new families of bioactive mediators.
Keywords: Eicosanoid, Innate Immunity, Lipoxygenase Pathway, Macrophages, Mass Spectrometry (MS), Monocytes, Phospholipid
Abstract
12/15-Lipoxygenases (LOXs) in monocytes and macrophages generate novel phospholipid-esterified eicosanoids. Here, we report the generation of two additional families of related lipids comprising 15-ketoeicosatetraenoic acid (KETE) attached to four phosphatidylethanolamines (PEs). The lipids are generated basally by 15-LOX in IL-4-stimulated monocytes, are elevated on calcium mobilization, and are detected at increased levels in bronchoalveolar lavage fluid from cystic fibrosis patients (3.6 ng/ml of lavage). Murine peritoneal macrophages generate 12-KETE-PEs, which are absent in 12/15-LOX-deficient mice. Inhibition of 15-prostaglandin dehydrogenase prevents their formation from exogenous 15-hydroxyeicosatetraenoic acid-PE in human monocytes. Both human and murine cells also generated analogous hydroperoxyeicosatetraenoic acid-PEs. The electrophilic reactivity of KETE-PEs is shown by their Michael addition to glutathione and cysteine. Lastly, both 15-hydroxyeicosatetraenoic acid-PE and 15-KETE-PE activated peroxisome proliferator-activated receptor-γ reporter activity in macrophages in a dose-dependent manner. In summary, we demonstrate novel peroxisome proliferator-activated receptor-γ-activating oxidized phospholipids generated enzymatically by LOX and 15-prostaglandin dehydrogenase in primary monocytic cells and in a human Th2-related lung disease. The lipids are a new family of bioactive mediators from the 12/15-LOX pathway that may contribute to its known anti-inflammatory actions in vivo.
Introduction
Lipoxygenases (LOXs)2 are non-heme iron lipid-peroxidizing enzymes that catalyze the oxygenation of polyunsaturated fatty acids to their corresponding hydroperoxy derivatives (1). Although LOXs are best known for generation of free acid eicosanoids, we showed that they also generate four phospholipid-esterified eicosanoids comprising hydroxyeicosatetraenoic acid (HETE) attached to phosphatidylethanolamine (PE) that regulate cytokine generated in monocytes and macrophages (2, 3). These lipids also act as regulators of PE-binding protein 1 to induce mitogen-activated protein kinase activation in epithelial cells and recently were shown to orchestrate clearance of apoptotic cells during inflammation and maintain immunologic tolerance (4, 5). Thus, phospholipid-esterified lipids from this pathway are distinct from free eicosanoids and are significant signaling molecules in their own right.
In terms of free eicosanoids, LOXs generate hydroperoxyeicosatetraenoic acids (HpETEs) as primary products that are reduced by cellular glutathione peroxidase to secondary products, including HETEs. These can be further oxidized to form electrophilic ketoeicosatetraenoic acids (KETEs). This oxidation of the primary alcohol to a carbonyl is catalyzed by 5-hydroxyeicosanoid dehydrogenase, generating 5-KETE in neutrophils and 15-hydroxyprostaglandin dehydrogenase (15-PGDH), which generates 15-KETE in monocytes (6–8) Electrophilic eicosanoids are currently of significant interest because they contain an α,β-unsaturated carbonyl and can readily adduct to proteins via Michael addition, resulting in transcriptional activation, e.g. of PPARγ and Nrf2 (9, 10). Certain non-enzymatically generated oxidized phospholipids, including hexadecyl azelaoyl phosphatidylcholine, also undergo Michael addition (11). Thus, we sought to discover whether LOXs could be a source of enzymatically generated electrophilic phospholipid oxidation products specifically containing KETEs as functional groups.
Herein, a targeted lipidomic strategy was used to identify, characterize, and quantify four KETE-PE lipids generated by human and murine monocytic cells. The lipids were observed in vitro and in vivo in human lung disease, and along with HETE-PEs, we also show that they can activate PPARγ transcriptional activity. Thus, they represent a new class of oxidized phospholipid generated enzymatically by primary cells that could contribute to the immune regulatory actions of mammalian LOXs in health and disease.
EXPERIMENTAL PROCEDURES
Materials
Human recombinant interleukin 4 (IL-4) was from Promega. LymphoprepTM was from Axis-Shield, Oslo, Norway. 1-Stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine was from Avanti Polar Lipids Inc., Alabaster, AL. 15-Hydroxyprostaglandin dehydrogenase inhibitor CAY10397, A23187, Type V soybean lipoxidase, triphenylphosphine, dichloromethane, and Dess-Martin periodinane were from Sigma-Aldrich. HPLC grade hexane, methanol, chloroform, 2-propanol, and water were from Fisher Scientific. HETE- and KETE-PEs were generated as described (12). All other reagents were from Sigma unless otherwise stated.
Isolation and Activation of Murine Peritoneal Macrophages
All animal experiments were performed in accordance with the United Kingdom Home Office Animals (Scientific Procedures) Act, 1986. Wild-type female C57BL/6 mice (8–12 weeks) were from Charles River, UK; kept in constant temperature cages (20–22 °C); and given free access to water and standard chow. Naïve peritoneal macrophages were isolated by lavage into 2 ml of ice-cold PBS. 4 × 106 cells/ml of Krebs buffer (50 mm HEPES, 100 mm NaCl, 5 mm KCl, 1 mm NaH2PO4, 1 mm CaCl2, 2 mm glucose) were stimulated with A23187 (10 μm) at 37 °C for 15–180 min. For reduction reactions, cells were incubated at room temperature with 1 mm SnCl2 for 10 min.
Peritoneal macrophages were also isolated from PPRE-EGFP transgenic mice generated as described previously (13); PPRE-EGFP consists of three tandem consensus peroxisome proliferator-response element sites (AGGACAAAGGTCA) coupled to the gene encoding enhanced green fluorescent protein (EGFP). Mice were injected intraperitoneally with 2 ml of sterile thioglycolate (3%, w/v). Peritoneal exudates were collected after 3 days by lavage as described above.
Isolation and Activation of Human Monocytes
The use of healthy human monocytes was approved by the School of Medicine Research Ethics Committee, Cardiff University. Human monocytes were isolated from buffy coats (Welsh Blood Service) as described previously (2). 4 × 106 cells/ml of Krebs buffer were stimulated with A23187 (10 μm) at 37 °C for 15–180 min. For reduction, cells were incubated at room temperature with 1 mm SnCl2 for 10 min. In some experiments, 15-LOX induction was not required, and monocytes were recovered by scraping following adhesion. For some experiments, 15-HpETE-PE or 15-HETE-PE (1 μm) and 15-PGDH inhibitor CAY10397 (100 μm) were added to cells prior to or following activation of cells. In some experiments, cells were homogenized with a manual glass Teflon homogenizer using 10 strokes.
Analysis of Bronchoalveolar Fluid (BAL) from Patients with Lung Disease
We obtained samples of BAL fluid from patients with cystic fibrosis, bronchiectasis, primary ciliary dyskinesia, cystic fibrosis newborn screen, and persistent bacterial bronchitis who were undergoing a clinically indicated fiberoptic bronchoscopy as described previously (14). The study was approved by the local research ethics committee, and all procedures were performed with informed, parental consent and age-appropriate assent from the child where appropriate. Samples were stored at −80 °C until lipid extraction as detailed below.
Lipid Extraction
1,2-Dimyristoyl-PE (10 ng) and 15-HETE-d8 (10 ng) were added to each sample before extraction as an internal standard. Where stated, hydroperoxides were reduced to the corresponding alcohol by adding 1 mm SnCl2 or 1 mm triphenylphosphine for 10 min at room temperature. Lipids were extracted by adding 1 m acetic acid, 2-propanol, hexane (2:20:30, v/v) to the sample at a ratio of 2.5 ml of solvent to 1 ml of sample; vortexing; and then adding 2.5 ml of hexane. Following vortexing and centrifugation (1500 rpm for 5 min), lipids were recovered in the upper hexane layer. The samples were then re-extracted by the addition of an equal volume of hexane followed by further vortexing and centrifugation. The combined hexane layers were then dried under vacuum and analyzed for HpETE-PE, HETE-PE, and KETE-PEs using LC/MS/MS (as described below and in Ref. 12).
Precursor Scanning Tandem Mass Spectrometry
Lipids were separated on a C18 Luna 3-μm, 150 × 2-mm column (Phenomenex) using a gradient of 50–100% B over 10 min followed by 30 min at 100% B (Solvent A, methanol:acetonitrile:water, 1 mm ammonium acetate, 60:20:20; Solvent B, methanol, 1 mm ammonium acetate) with a flow rate of 200 μl/min. Precursor scanning LC/MS/MS in negative mode for precursors of 317.2 was carried out using a 4000 Q-Trap (Applied Biosystems) from 650 to 850 atomic mass units over 6 s with a declustering potential of −130 to −140 V, a linear ion trap fill time of 200 ms, and Q0 trapping.
KETE-, HETE-, and HpETE-PE Quantitation Using LC/MS/MS
Samples were separated as above for precursor scanning but in multiple reaction monitoring mode as described recently (12). Transitions monitored were for parent ions of m/z 736.6, 762.6, 764.6, and 780.6 [M − H]− fragmenting to daughter ions with m/z 317.2 (KETE) or 153.2 (12-KETE); m/z 738.6, 764.6, 766.6, and 782.6 [M − H]− fragmenting to daughter ions with m/z 219.2 (15-HETE) or 179.1 (12-HETE); or m/z 754.6, 780.6, 782.6, and 798.6 [M − H]− fragmenting to daughter ions with m/z 317.2 (HpETE). Standard curves were generated using internal standards (1,2-dimyristoyl-PE) and different synthetic primary standards (12). Products were quantified by LC/MS/MS electrospray ionization on an Applied Biosystems 4000 Q-Trap. Acquisition of product ion spectra was triggered during elution of ions of interest with the instrument operating in ion trap mode.
Free KETE Determination Using LC/MS/MS
Samples were separated on a C18 Spherisorb ODS2 5-μm, 150 × 4.6-mm column (Waters) using a gradient of 50–90% B over 10 min (A, water:acetonitrile:acetic acid, 75:25:0.1; B, methanol:acetonitrile:acetic acid, 60:40:0.1) with a flow rate of 1 ml/min. Products were quantitated by LC/MS/MS electrospray ionization on an Applied Biosystems 4000 Q-Trap using parent-to-daughter transitions of m/z 317.2 (KETE, [M − H]−) to m/z 113.2 (15-KETE) and m/z 327.2 to 226.2 for 15-HETE-d8 with collision energies of −20 to −30 V. Products were identified and quantified by isotopic dilution mass spectrometry using standard curves generated with 15-KETE and 15-HETE-d8 run in parallel under the same conditions.
Reaction of 15-KETE-PE with Glutathione (GSH)
15-KETE-PE (160 μm) was incubated with 62 mm GSH (final concentration; GSH stock made at 250 mm at pH 9 in water) in methanol:dichloromethane:water (60:20:20). Samples were separated on a Gemini 3-μm C18 20 × 2-mm110A mercury column (Phenomenex) using a gradient of 50–100% B over 3 min followed by 1.5 min at 100% B (Solvent A, methanol:acetonitrile:water, 1 mm ammonium acetate, 60:20:20; Solvent B, methanol, 1 mm ammonium acetate) at a flow rate of 750 μl/min. Products were analyzed by LC/MS/MS electrospray ionization on an Applied Biosystems 4000 Q-Trap in negative mode using parent-to-daughter transitions of m/z 780.6 → 283.2 (15-KETE-PE fragmentation of the sn1 fatty acid stearic acid) and an m/z of 543.5 fragmenting to m/z 283.2 (15-KETE-PE-GSH adduct detected as a double charged ion) (declustering potential, 70 V; collision energy, −45 eV).
Transfection and Reporter Gene Assays
HEK293 cells at ∼85% confluence in 24-well plates were transiently co-transfected using Lipofectamine 2000 (Invitrogen) with a plasmid containing the luciferase gene under the control of three tandem PPAR-response elements (3×PPRE TK-luciferase) in pGL3-Basic vector (Promega, Madison, WI) and FLAG-PPARγ1 in pcDNA3.1 vector (Invitrogen) (500 μg and 50 ng of each, respectively). 50 ng of the Renilla luciferase control reporter vector (pRL-TK) was used as an internal control. Twenty-four hours after transfection, cells were serum-deprived (1% FBS) and then treated with various stimuli as indicated for an additional 12 h. The reporter Dual-Luciferase assay kit (Promega) was used to measure the luciferase activity of cells according to the manufacturer's instructions (Victor II, PerkinElmer Life Sciences).
Western Blot
Peritoneal macrophages isolated from PPRE-EGFP mice were treated with 2.5 μm 15-HETE-PE, 15-KETE-PE, SAPE, and their non-esterified derivatives for 12 h. Cell extracts were separated on a 10% SDS-polyacrylamide gel, and proteins were transferred to nitrocellulose membranes. Detection of EGFP was obtained by immunoblotting using an anti-GFP antibody (Sigma). Membranes were probed with anti-GAPDH antibody (Santa Cruz Biotechnology) to ensure equal loading. Images were obtained using the Odyssey Imager (LI-COR Biosciences). For CD36 expression on human monocytes, 4.5 × 106 cells/well were seeded in 24-well plates in culture medium and incubated at 37 °C for 2 h for monocytes to adhere. Non-adherent cells were removed using medium. Cells were stimulated for 6 h with SAPE, 15-HETE-PE, or 15-KETE-PE at 2.5 μm or vehicle control (methanol). Wells were washed two times with PBS, and lysates were generated for each well individually. Protein concentration was determined by nanodrop, and 20 μg was loaded onto a 4–12% polyacrylamide gel. Proteins were transferred to a nitrocellulose membrane; blocked with 5% milk in PBS, 0.05% Tween; and incubated overnight with anti-human CD36 (R&D Systems) or anti-human β-actin. Blots were incubated with the relevant secondary antibody conjugated to HRP and developed to film.
Statistical Analysis
Data were analyzed using Student's t test with p < 0.05 being considered significant (n = 3, mean ± S.E.). Experiments using monocytes and macrophages were repeated at least three independent times on different cell isolates.
RESULTS
Human Monocytes Generate Four 15-KETE-PEs
Lipid extracts from IL-4-treated human monocytes activated for 15 min with calcium ionophore were analyzed using precursor scanning LC/MS/MS for m/z 317.2, the carboxylate anion of KETE [M − H]−. This revealed several major ions, including m/z 780, 764, 762, and 736 (Fig. 1A). These correspond to the predicted KETE adducts of PE analogous to 15-HETE-PEs reported previously in terms of sn1 fatty acid composition, namely 18:0a, 18:0p, 18:1p, and 16:0p/KETE-PE (Scheme 1) where two are acyl-linked and two are plasmalogen-linked at sn1 (2). A second series was also observed (m/z 754, 782, and 798) that corresponds to analogous HpETE-PEs because the hydroperoxide group readily loses water during collision-induced dissociation to yield an ion with m/z 317.2 (not shown). A fourth HpETE-PE is expected at m/z 780 isobaric with 18:0a/KETE-PE. To confirm proposed KETE-PE structures, standards were synthesized (12). Both HPLC retention times and product ion spectra of 18:0a, 18:0p, 18:1p, and 16:0p/15-KETE-PE standards matched those of monocyte-derived products (Fig. 1, B–E). Multiple daughter ions arising from KETE were detected, including 317.2, 219.2, 113.1, and 273.2 (15) (Scheme 1 and Fig. 1). Furthermore, m/z 780.6 shows a daughter ion at 283 (18:0a), whereas sn1 fatty acids are absent in the others consistent with their proposed plasmalogen structures (Scheme 1 and Fig. 1). All spectra show ions generated by neutral loss of the KETE carboxylate anion [M − 318]− or of the KETE ketene [M − 300]−. These are observed as product ions for parents with m/z 780.6 at m/z 762 and m/z 480, respectively, and further confirm the predicted structures (Fig. 1 and Scheme 1).
FIGURE 1.

Human monocytes generate four 15-KETE-PEs. Human monocytes were isolated cultured with IL-4 as described under “Experimental Procedures.” Cells were activated in Krebs buffer with 10 μm A23187 for 15 min at 37 °C. Lipids were extracted and analyzed using reverse phase LC/MS/MS as described. A, precursor scan for [M − H]− 317.2 shows four major ions generated by activated human monocytes. Four prominent ions are circled. B–E, representative chromatograms and MS/MS spectra of lipids detected as parent → 317.2 for the four lipids compared with synthetic standards. Standards were made as described (12) and run under conditions identical to those used for human monocyte lipid extracts. MS/MS spectra were acquired in ion trap mode at the apex of elution for each lipid. cps, counts/s.
SCHEME 1.

Structures of 15-KETE-PEs generated by activated human monocytes (A) and 12-KETE-PEs detected in murine peritoneal lavage (B).
15-KETE-PEs were detected in human monocytes without activation of 15-LOX with Ca2+ ionophore causing significant elevations within 15 min and returning to base-line levels by 3 h (Fig. 2, A–D). Non-esterified 15-KETE generation was also observed on ionophore activation within 15 min (Fig. 2E). In contrast to esterified 15-KETE, the free eicosanoid continued to increase in concentration, reaching an average of 2.16 ± 0.12 ng/106 cells after 3 h (Fig. 2E). Notably, at 15 min, total esterified KETE levels (2.2 ng/106 cells) were higher than non-esterified 15-KETE (1.2 ng/106 cells). Thus, esterified but not free KETEs appear to be metabolized by the cells from 15 min onward.
FIGURE 2.

15-KETE-PEs and 15-KETE are generated in response to ionophore by human monocytes. Monocytes were isolated and activated with 10 μm A23187 for 15 min at 37 °C as described under “Experimental Procedures” for varying times before lipid extraction. A–D, generation of 15-HETE-PEs over time. 15-KETE-PEs were monitored as parent → 317.2 and quantified as described under “Experimental Procedures” using LC/MS/MS. E, generation of free 15-KETE over time. 15-KETE was quantified as described in under “Experimental Procedures” using LC/MS/MS at m/z 317.2 → 113.1 (n = 3, mean ± S.E.). Error bars represent S.E.
Murine Peritoneal Macrophages Generate 12-KETE-PEs
Lipid extracts from murine peritoneal lavage were extracted and analyzed using precursor scanning LC/MS/MS as for human monocytes. This revealed four major ions at m/z 780.8, 764.8, 762.8, and 736.8 corresponding to the same m/z generated by human cells (Fig. 3A). As before, ions corresponding to HpETE-PEs were also detected (754, 780, 782, and 798). To confirm the identity of the KETE-PEs, 18:0a/-, 18:0p/-, 18:1p/-, and 16:0p/12-KETE-PEs were generated as described (12). The retention time (Fig. 3, B–E) and MS/MS spectra of the 18:0a/18:0p/18:1p/16:0p/12-KETE-PE standards matched those of murine macrophage-generated lipids (Fig. 3, B–E). In particular, several KETE daughter ions are seen, including 273.2, 219.1, 179.1, and 153.1 (as identified by the Lipid Maps resource). The m/z 179.1 ion was not always apparent as sensitivity was limited by lack of material. The m/z 153.1 ion reflects fragmentation at the C12 ketone, indicating the 12-KETE-PE positional isomer (Fig. 3, B–E). Finally, 12-KETE-PEs were absent from lipid extracts of macrophages from 12/15-LOX−/− mice (not shown).
FIGURE 3.

Murine macrophages generate four 12-KETE-PEs. Murine peritoneal macrophages were isolated from WT mice (8–12 weeks) by lavage with ice-cold PBS. Cells were activated at 37 °C for 15 min with 10 μm A23187, and then lipids were extracted and analyzed using LC/MS/MS as described under “Experimental Procedures.” A, precursor scan for [M − H]− 317.2 shows four major ions generated by murine macrophages. Four prominent ions are circled. B–E, representative chromatograms and MS/MS spectra of lipids detected as parent → 317.2 for the four lipids compared with synthetic standards. Standards were made as described (12) and run under conditions identical to those used for murine macrophage lipid extracts. MS/MS spectra were acquired in ion trap mode at the apex of elution for each lipid. cps, counts/s.
Because purified single isomer 12-KETE-PEs were not available, a quantitative assay could not be established (12). Thus, we used the parent → 317 daughter ion to compare -fold changes on activation by plotting the analyte:internal standard ratio (peak areas). As for 15-KETE-PEs in human monocytes, 12-KETE-PEs were basally present in peritoneal lavage prior to cell activation with levels increasing 2–3-fold on ionophore activation, reaching a maximum at 15–30 min before returning to base line by 3 h (Fig. 4, A–D).
FIGURE 4.

12-KETE-PEs are generated in response to ionophore by murine macrophages. A–D, generation of 12-HETE-PEs over time. Peritoneal macrophages were isolated from WT mice by lavage and pooled. Cells were activated with 10 μm A23187, and at defined time points, lipids were extracted and analyzed using LC/MS/MS. 12-KETE-PEs were monitored as parent → 317.2 as described under “Experimental Procedures” and are expressed as the ratio of integrated area to internal standard (A/IS) (n = 3, mean ± S.E.). Error bars represent S.E. cps, counts/s.
15-PGDH in Human Monocytes Converts 15-HETE-PE to 15-KETE-PE
In monocytes, 15-PGDH catalyzes oxidation of free 15-HETE to 15-KETE (7). To determine whether this enzyme can oxidize intact phospholipids, monocyte lysates were incubated with/without the 15-PGDH inhibitor CAY10397 (100 μm) followed by addition of 18:0a/15-HETE-PE (1 μm). Monocyte lysates were utilized because HETE-PEs are not well incorporated into intact cells during short time frames, and it was important to ensure access to intracellular enzyme (data not shown). After 15 min, lipids were extracted, and conversion of 18:0a/15-HETE-PE to 18:0a/15-KETE-PE was determined (Fig. 5A). Unactivated monocytes generated 251 pg of 15-KETE-PE/2 × 106 cells. 15-KETE-PE levels were significantly enhanced by prior activation with calcium ionophore, reaching 478 pg of KETE-PE/2 × 106 cells. Finally, 15-KETE-PE formation was partially inhibited by CAY10397 (Fig. 5A).
FIGURE 5.

15-KETE-PE can be generated by monocytes via 15-PGDH oxidation of 15-HETE-PE, and 15-KETE-PEs are detected in human disease samples. A, generation of 15-KETE-PE via 15-PGDH. Human monocytes were isolated as described under “Experimental Procedures” and used immediately without induction of 15-LOX. Cells were activated for 15 min at 37 °C with 10 μm A23187 and then homogenized. Samples were incubated with/without 15-PGDH inhibitor CAY10397 (100 μm) for 15 min at 37 °C, then 15-HETE-PE (1 μm) was added, and homogenates were incubated for 15 min (37 °C). Lipids were then extracted and analyzed using LC/MS/MS following multiple reaction monitoring transitions with m/z 317.2 as a daughter ion quantified as described under “Experimental Procedures” (n = 3, mean ± S.E.). B–E, 15-KETE-PEs are detected in BAL fluids from cystic fibrosis patients. Lipids in BAL samples were extracted as described under “Experimental Procedures” and analyzed for 15-KETE-PEs using LC/MS/MS using m/z 317.2 as a daughter ion. Samples are as follows: control (n = 8), CF (n = 17), bronchiectasis (Bronch; n = 5), primary ciliary dyskinesia (PCD; n = 4), CF newborn screen (CF NBS; n = 9), and persistent bacterial bronchitis (PBB; n = 12). The line indicates the mean. *, p < 0.05; **, p < 0.01 based on Student's t test. Error bars represent S.E.
15-KETE-PEs Are Detected in BAL Fluid from Cystic Fibrosis Patients
BAL fluid was obtained from patients with a range of lung diseases, including cystic fibrosis, bronchiectasis, primary ciliary dyskinesia, cystic fibrosis (newborn screen), and persistent bacterial bronchitis (Fig. 5, B–E). Little or no KETE-PE was detected in BAL from healthy volunteers; however, samples from cystic fibrosis patients exhibited significantly elevated levels of all four 15-KETE-PEs. Levels in BAL from patients with bronchiectasis were similar to healthy volunteers, whereas individual BAL samples from other conditions revealed only modest and inconsistent increases in 15-KETE-PEs (Fig. 5, B–E).
Reaction of 15-KETE-PE with GSH
15-KETE possesses an α,β-unsaturated carbonyl, a functional group that can support Michael addition with nucleophiles such as protein amines and thiol functional groups (16, 17). Thus, the electrophilic reactivity of 15-KETE-PE was characterized by following the reaction with glutathione. The formation of thiol adducts with 15-KETE-PE were detected as double charged species with m/z of 543.5 [M − 2H]2− with the parent phospholipid disappearing on GSH reaction (Fig. 6A). Adducts also formed under more physiologically relevant conditions of pH 7.4 with 6 mm GSH; however, this occurred more slowly and was incomplete at up to 90 min (not shown). Although 15-KETE-PE has two electrophilic centers at C11 and C13, double GSH adducts were not detected, indicating a more favorable addition at the γ carbon or steric hindrance of addition at the γ carbon after addition at the ϵ carbon (not shown). Fig. 6B shows the proposed structure of the GSH-15-KETE-PE adduct. MS/MS spectra reveal fragments characteristic of 15-KETE-PE, including 283.2, 317.2, and 113.2, and a number of ions known to arise from GSH, including 306.0, 128.0, 143.0, 160.0, 167.0, 177.0, 179.0, 210.1, and 254.0 (Fig. 6C).
FIGURE 6.

15-KETE-PE forms a covalent adduct with glutathione in vitro. A, chromatograms showing formation of 15-KETE-PE-GSH with loss of 15-KETE-PE over time. 15-KETE-PE (160 μm) was incubated at 22 °C in methanol:dichloromethane:GSH (GSH in water at pH 9) (60:20:20) with a final GSH concentration of 62 mm as described under “Experimental Procedures.” Sample was injected under flow onto a mercury LC column and monitored in negative mode using m/z 283.2 (18:0) as a daughter ion for both species. The disappearance of the parent phospholipid was determined by monitoring m/z 780.6 → 283.2. The GSH-15-KETE-PE adduct (molecular weight, 1089) was detected as a doubly charged ion with the multiple reaction monitoring transition 543.5 → 283.2. B, proposed structure of the 15-KETE-PE-GSH adduct with fragmentation pattern. C, MS/MS spectrum of 15-KETE-PE-GSH adduct. An MS/MS spectrum was obtained at the apex of the peak in the negative mode for 15-KETE-PE-GSH in ion trap mode. cps, counts/s.
15-HETE-PE and 15-KETE-PEs Activate PPARγ in Macrophages and Induce CD36 in Human Monocytes
In macrophages, activation of 12/15-lipoxygenases generates PPARγ ligands (18). Thus, it is of interest to study whether esterified eicosanoids such as 15-KETE-PE and 15-HETE-PE similarly elicit PPARγ transcriptional activity. PPRE-reporter assays in HEK293 cells co-transfected with PPARγ showed a dose-dependent activation of PPARγ transcriptional activity in response to 15-KETE-PE or 15-HETE-PE (Fig. 7A). Equivalent concentrations of 18:0a/20:4-PE were ineffective, indicating that activation of PPARγ requires oxidation (Fig. 7A). PPARγ activation was alternatively monitored in thioglycollate-elicited peritoneal macrophages obtained from PPRE-EGFP transgenic reporter mice (Fig. 7B). Western blot analysis indicated that esterified 15-HETE and 15-KETE (2.5 μm) but not equimolar concentrations of 18:0a/20:4-PE increased GFP intensity. The thiazolidinedione rosiglitazone served as a bona fide positive control for maximal PPARγ activation (Fig. 7, C and D). In human monocytes, Western blot analysis indicated that 2.5 μm 15-HETE- or 15-KETE-PE for 6 h elevated the expression of the PPARγ-inducible protein CD36 (Fig. 7, E and F).
FIGURE 7.

Macrophage PPARγ-stimulating activity of 15-HETE-PE and 15-KETE-PE. A, HEK293 cells were transfected with 1 μg of 3×PPRE-luciferase (Luc), 50 ng of pRL-TK, and 50 ng of pcDNA-FLAG-PPARγ using Lipofectamine 2000 for 24 h. Cells were serum-deprived (1% FBS) and treated with the lipids for 12 h. Rosiglitazone was used as positive control, and SAPE was used as an unoxidized lipid control (n = 4, mean ± S.E.). n = 3–4; *, p < 0.05; **, p < 0.01 versus SAPE based on Student's t test B, thioglycollate-elicited peritoneal macrophages were cultured from PPRE-EGFP reporter transgenic mice and treated with a 1 μm concentration of each lipid for 12 h. The panels show representative fluorescent images. The bar chart shows the average pixel intensity of 10 different cells in each photo calculated using ImageJ. C, EGFP expression in peritoneal macrophages from PPRE-EGFP transgenic mice was detected by Western blot. Protein was extracted after 12-h treatment with a 2.5 μm concentration of the lipids using an anti-GFP antibody. The same membrane was analyzed with anti-GAPDH antibody to confirm equal loading. The white line shows where other bands were removed for clarity, but the rosiglitazone (Rosi) lane was from the same gel analyzed at the same time. D, GFP lanes were analyzed for pixel intensity using ImageJ and then normalized to GAPDH lanes. E, CD36 expression in primary human monocytes was detected using Western blot after 6-h incubation with a 2.5 μm concentration of the lipids. Lysates were separately analyzed for actin to confirm protein loading. F, CD36 lanes were analyzed for area of bands using ImageJ and then normalized to actin. Error bars represent S.E.
HpETE-PE Is a Precursor for HETE-PE Generation by Monocytes and Macrophages
Our previous studies showed that human monocytes and murine macrophages generate 15-HETE- and 12-HETE-PE lipids, respectively (2, 3). However, in those studies, lipid extracts had been reduced using SnCl2 so that the total flux of LOX-dependent esterified H(p)ETE-PE was determined. To determine HpETE-PE versus HETE-PE levels, temporal generation of HETE-PE by both human and murine monocytic cells was compared with and without chemical reduction. Thus, HpETE-PE levels would be indicated by the difference between levels before and after SnCl2 treatment. Reduction caused a 2- or 4–5-fold increase in HETE-PE for human monocytes or murine macrophages, respectively (Fig. 8, A–H). Use of the hydroperoxide-specific triphenylphosphine as the reductant in place of SnCl2 yielded similar results (Fig. 9A).
FIGURE 8.

Chemical reduction of cell extracts significantly increases detection of 15- or 12-HETE-PE in human monocytes and murine macrophages. A–D, time course of 15-HETE-PE generation by activated human IL-4-treated monocytes with/without SnCl2 reduction of lipid extracts. Human monocytes were isolated, cultured, and then activated at 37 °C with 10 μm A23187, and at defined time points (15–180 min), some samples were reduced by incubation with 1 mm SnCL2 for 10 min at 22 °C followed by lipid extraction (n = 3, mean ± S.E.). E–H, time course of 12-HETE-PE generation by activated murine peritoneal macrophages with/without SnCl2 reduction of lipid extracts. Murine peritoneal macrophages were isolated from WT mice (8–12 weeks) by lavage with ice-cold PBS and pooled. Cells were activated at 37 °C with 10 μm A23187, and at defined time points (15–180 min), some samples were reduced by incubation with 1 mm SnCl2 for 10 min at 22 °C followed by lipid extraction (n = 3, mean ± S.E.). Error bars represent S.E.
FIGURE 9.
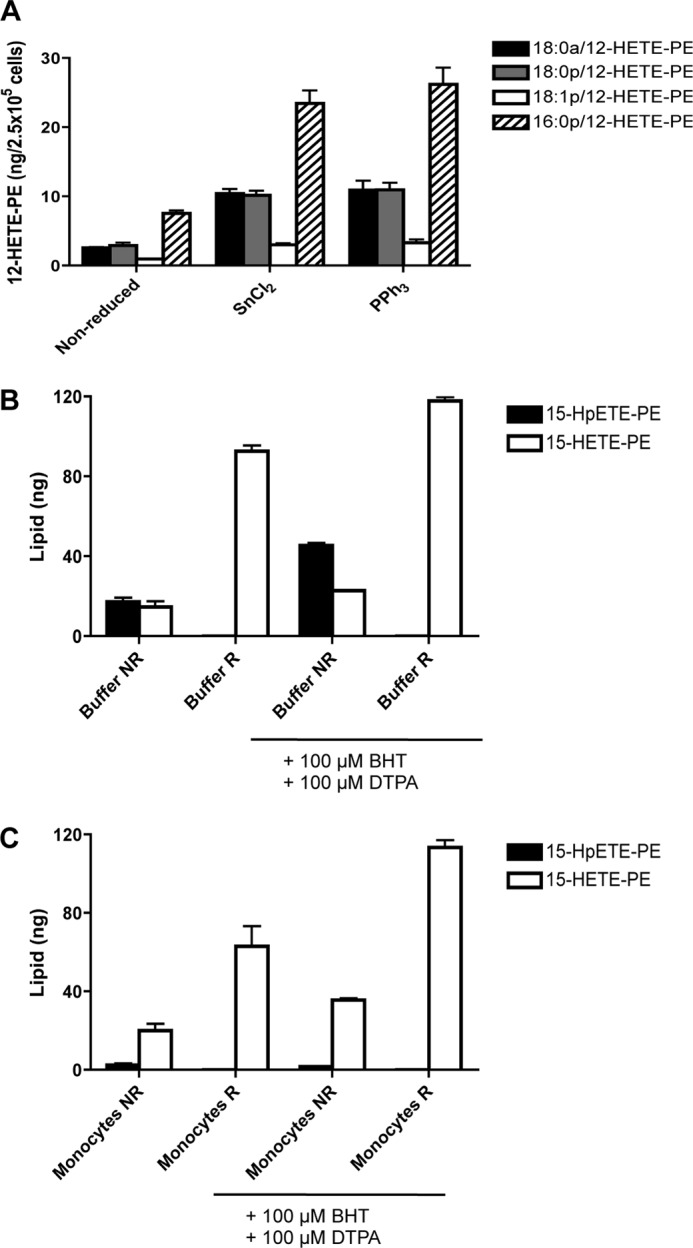
Identification of HpETE-PE as the SnCl2 reducible lipid; however, its decomposition is accelerated during lipid extraction from cells. A, confirmation of 15-HpETE-PE generation by murine macrophages. Total peritoneal cells were recovered by lavage from WT mice and activated for 15 min with 10 μm A23187 at 37 °C. Lipids were reduced with either 1 mm SnCl2 or 1 mg triphenylphosphine (PPh3) for 10 min at 22 °C and then extracted. 12-HETE-PEs were monitored as described under “Experimental Procedures.” B and C, acceleration of HpETE-PE decomposition by cell extracts and protection using antioxidants. Human monocytes were isolated from buffy coats by adhesion. 360 ng of 18:0a/15-HpETE-PE was added to either Krebs buffer (B) or Krebs + cells (4 × 106 cells/ml) (C) in the presence or absence of 100 μm butylated hydroxytoluene (BHT) and 100 μm diethylenetriaminepentaacetic acid (DTPA). This was followed either by immediate organic extraction without reduction (NR) or reduction with 1 mm SnCl2 (R) for 10 min at 22 °C followed by organic extraction. Multiple reaction monitoring transitions 798.6 → 317.2 (15-HpETE-PE) and 782.6 → 219.1 (15-HETE-PE) were monitored, and the lipid recovered was quantified as described under “Experimental Procedures.” Error bars represent S.E.
Next, HpETE-PE was directly measured. First, 18:0a/15-HpETE-PE was synthesized and found to be detected to a high degree of sensitivity using m/z 798.6 → 317.2 by LC/MS/MS (not shown). However, using this assay, very low and variable levels of HpETE-PEs were detected in monocytes or macrophages that were considerably less than expected based on calculations from data in Fig. 8 (not shown). To probe the reasons for this discrepancy, 18:0a/15-HpETE-PE (360 ng) was added either to Krebs buffer or monocytes (not IL-4-treated), and then lipids were extracted and analyzed using LC/MS/MS. When added to Krebs buffer, only ∼20 or 40 ng was recovered in the absence or presence of chelators/antioxidants (diethylenetriaminepentaacetic acid and butylated hydroxytoluene), respectively (Fig. 9B). Furthermore, if added to cells during extraction, virtually all the HpETE-PE decomposed (Fig. 9C). Reduction using SnCl2 prior to extraction increased the levels of HETE-PE detected, although these (around 90 or 120 ng without or with diethylenetriaminepentaacetic acid/butylated hydroxytoluene, respectively) were still only around 50% of added hydroperoxide. Unreduced samples contained small amounts of HETE-PE (Fig. 9C, left bar). While stored in methanolic solution, our standard shows a total absence of HETE-PE (when scanning in Q1 for m/z 782.6); thus, decomposition (including its conversion to the small amount of HETE-PE noted herein) starts as soon as the lipid is added to aqueous solvent (in the 2–5 min before reduction is initiated) and is further accelerated during extraction, particularly in the presence of cellular material. These losses are unlikely to be due to low extraction efficiency because HETE- and KETE-PEs are extracted using this protocol with 94 and 78% efficiencies, respectively (12). Instead, we suggest that HpETE-PE is unstable in aqueous medium and that its decay is further accelerated by cell lysates. Thus, its direct measurement will significantly underestimate the flux of HpETE-PE generation in immune cells. In conclusion, we suggest that the SnCl2-reducible pool generated on activation, which accounts for 50% of the total HETE-PE detected, is instead HpETE-PE that does not survive solvent extraction.
DISCUSSION
Herein, generation of four new oxidized phospholipids that contain a KETE functional group attached to PE is shown (Scheme 1 and Figs. 2 and 4) (19). Along with HETE-PEs, they were found to activate PPARγ transcriptional activity in intact cells. KETE-PEs are generated by human IL-4-treated monocytes and murine peritoneal macrophages and were detected in bronchoalveolar lavage from cystic fibrosis (CF) patients (Fig. 5, B–E). Acute pulmonary inflammation in CF is associated with neutrophil influx and proinflammatory cytokine generation with bone marrow-derived and alveolar macrophages and eosinophils exacerbating this inflammatory response (20, 21). Both these cell types express 15-LOX and thus are potential sources of 15-KETE-PEs in vivo (22). 15-LOX is also induced by the Th2 cytokines IL-4 and IL-13, which are also elevated in CF airways (23, 24). Recent studies indicate that activation of PPARγ-dependent gene expression in CF mice ameliorates disease severity in mice, suggesting that electrophilic 15-LOX products might potentially act to dampen down inflammation in this disorder and that these or homologous molecules may be of therapeutic benefit in treating acute and chronic airway inflammation (25). In our studies, esterified HETEs are almost exclusively found in PE with little or none contained in phosphatidylcholine (2). This is likely due to the enzymatic nature of their generation, which appears selective for one class of phospholipid.
Our studies show that esterified eicosanoids are functionally distinct from their free acid isomers; thus, characterization of esterified KETEs as distinct to their free acid analogs is novel in terms of understanding the immunomodulatory roles of LOXs. Chemically, KETEs are very different from HETEs with the potential to adduct to amino acid residues through Michael addition. In the case of an esterified KETE, this could generate a lipid capable of membrane anchoring. Thus, the identification and characterization of esterified KETEs as a family of lipid signaling mediators in their own right is relevant for macrophage biology and inflammation.
The requirement for 15- and 12/15-LOX in KETE-PE generation is indicated by their regiospecificity and their absence in peritoneal lavage from mice deficient in 12/15-LOX (Figs. 1 and 3 and data not shown). The discovery of this new class of lipids adds to the growing list of esterified eicosanoids generated by LOXs that includes 5-, 12-, and 15-HETE-PEs and -phosphatidylcholines in neutrophils, platelets/murine macrophages, and monocytes, respectively, and 14-hydroxydocosahexanoic acid-PE from thrombin-stimulated human platelets (2, 3, 26–28). These lipid families display diverse bioactivities, including enhancing coagulation, suppressing Toll-like receptor signaling, dampening neutrophil responses, and as shown herein for 15-KETE and 15-HETE-PE activation of PPARγ (Fig. 7) (3, 26, 28).
KETE-, HETE-, and HpETE-PEs generated by 12/15-LOXs are basally detected in monocytes and macrophages but are elevated by ionophore (Figs. 2, 4, and 8) (2, 3). In contrast, esterified eicosanoids are not detected in neutrophils and platelets prior to agonist activation (26, 28). Several lines of evidence suggest that 12/15-LOXs are involved in homeostatic processes, including wound healing and inflammation resolution (29–32). Our observations of basal continuous enzyme turnover in resting cells along with PPARγ stimulation activity are consistent with this.
Previously, we showed that HETE-PEs form via direct oxidation of PE (3). To determine whether generation of KETE-PEs was also via direct oxidation or required arachidonate release and re-esterification of newly formed KETE, we examined its generation in the presence of H218O, looking for analogs with +2 atomic mass units. However, these are isobaric and were found to co-elute with 15-[16O]HETE-PE of which a small amount also forms on macrophage activation (data not shown). However, we note that macrophage activation in H218O did not result in formation of less 12-[16O]KETE-PE. Thus, it is very likely that the KETE-PEs form through direct oxidation of PE without requirement for phospholipase A2 hydrolysis. Analogous KETE-PEs were not detected in human platelets or neutrophils, indicating that these cells do not further oxidize esterified 12- or 5-HETE-PE (26, 28) (data not shown). This infers that the neutrophil 5-PGDH capable of oxidizing 5-HETE to 5-KETE may not be active in utilizing complex substrates such as phospholipid adducts (6, 9). Herein, we showed that inhibition of 15-PGDH partially blocks generation of 15-KETE-PE from exogenous 15-HETE-PE. This suggests that KETE-PEs may form through oxidation of preformed HETE-PE by the enzyme rather than esterification of newly generated KETE (Fig. 5A). However, because inhibition was only partial, we cannot rule out that at least some is generated via homolytic cleavage of the peroxide group via LOX-catalyzed hydroperoxidase activity. We also note that conversion of HETE-PE to KETE-PE is stimulated by ionophore activation of cells before lysis. The mechanism is unknown but could include calcium-dependent post-translational modification of PGDH.
The precursor for KETE-PE generation, HETE-PE, is generated through glutathione peroxidase-dependent reduction of the primary LOX product, HpETE-PE. We previously found that neutrophils and platelets efficiently convert the majority of HpETE-PEs to the more stable HETE-PEs (26, 28). In contrast, in monocytes/macrophages, up to 50% was not endogenously reduced, suggesting slower rates of glutathione peroxidase activity in these cells (Fig. 8). The difficulty in analyzing HpETE-PEs due to their instability during extraction is a major problem when trying to accurately estimate the levels generated (Fig. 9). In monocytes, HETE-PEs form by oxidation of intact phospholipid, whereas in platelets and neutrophils, generation involves rapid esterification of a newly synthesized free HETE into the phospholipid pool (26, 28). We also note that the fate of esterified 12/15- or 15-LOX products in monocytic cells is different from that formed by 12- or 5-LOX in platelets or neutrophils. Specifically, 12- or 5-HETE-PEs are relatively long lived species with little metabolism occurring up to 3 h following generation (26, 28). In activated monocytes/macrophages, HETE-, HpETE-, and KETE-PEs reach a maximal concentration for 30 min with levels returning close to base line by 3 h (Figs. 2, 4, and 8). Potential metabolic events accounting for this short half-life that were discounted included (i) conversion of oxidized PE to phosphatidylcholine or phosphatidylserine via headgroup interconversion, (ii) chain elongation of the sn2 acyl group by addition of C2H4 groups (33), and (iii) hydrolysis of esterified eicosanoid derivatives by endogenous phospholipases (not shown). Levels of free 15-KETEs, which were acutely generated by human monocytes within 15 min, continued to increase over 3 h of activation (Fig. 2). This indicates distinct metabolic pathways for free versus esterified KETEs in monocytes.
Recent studies have indicated an important role for electrophilic free acid lipids in mediating anti-inflammatory signaling through the formation of Michael adducts with proteins via histidine or cysteine. Activation of transcription factors, including PPARγ and Nrf2, by lipids such as 15-deoxyprostaglandin J2, nitrolipids, and carbonyl-containing short and long chain species has been extensively characterized and is proposed to be an important part of the cell response to oxidative stress (11, 34–37). KETE-PEs contain an α,β-unsaturated ketone in the sn2 fatty acid. Thus, a functionally significant reaction that KETE-PE species can undergo is Michael addition with cellular nucleophiles such as the thiol of GSH (Fig. 6) or protein cysteine and histidine residues. Although 15-KETE-PE readily adducted to GSH in vitro, we were unable to increase the rate of reaction at physiological pH using glutathione S-transferase (Fig. 6 and data not shown). Furthermore, in ex vivo test systems, it did not activate Nrf2 or PPARγ through direct ligand binding (data not shown). This suggests that the electrophilic centers may be inaccessible to cytosolic thiols or that phospholipid esterification sterically limits interaction of the electrophilic fatty acid with the multiple reactive thiols of Keap1 and the ligand binding domain Cys-285 of PPARγ. This does not exclude potential electrophilic reactions of KETE-PEs with additional protein targets, notably those of integral membrane or membrane-associated proteins.
Both 15-HETE-PE and 15-KETE-PE dose-dependently activated PPARγ when added to intact cells; thus, they may act indirectly either through phospholipase A2 hydrolysis to release free PPARγ ligands or through upstream modulation of PPARγ expression. Previous reports indicated that oxidized free acid lipids generated by LOXs can activate PPARγ (18). The current thinking is that a number of lipids can act as low affinity physiological activators and that PPARγ steady state activity is a function of the total concentration of all ligands present at any one time in the cell. PPARγ can be activated by many fatty acids, eicosanoids, and other oxidized fatty acids with the more potent lipid stimuli appearing to act via electrophilic addition to the protein (e.g. nitrolipids and 15-deoxyprostaglandin J2) and synthetic agonists (e.g. rosiglitazone) giving the highest activation (38, 39). In the case of LOX, several known activators could be present in monocytes, including free HETEs, hydroxyoctadecadienoic acids, and both HETE- and KETE-PEs as shown here. We note that the negative control (SAPE) did not activate PPARγ; thus, this was specific for the oxidized analogs (Fig. 7A). Monocytes generate ∼2.2 ng of KETE-PEs/106 cells. As average monocyte volumes are 250 μm3 (3, 40) and all the endogenously generated lipids remain cell-associated, we calculate that this equates to a cellular concentration of up to 11 mm, which is clearly considerably higher than that added to the cells. Several oxidized phospholipids, including 15-HETE-PE, can attenuate inflammatory signaling through inhibition of TLR4, a known inhibitor of PPARγ expression and activity (3, 41, 42). The mechanism involves binding to accessory proteins such as CD14 and LPS-binding protein. In this way, dampening down TLR4 may result in elevated PPARγ signaling as noted herein (Fig. 7).
In summary, this study identifies the cell and organ generation of novel families of esterified eicosanoids acutely generated by 12/15-LOXs. The study highlights a new class of electrophilic α,β-unsaturated ketone-containing phospholipids generated by immune cells that are expected to mediate adaptive anti-inflammatory signaling actions, including the activation of PPARγ-dependent gene expression.
This work was supported, in whole or in part, by National Institutes of Health Grants HL068878 and HL89544 (to Y. E. C. and L. V.); R01-HL058115, R01-HL64937, P30-DK072506, and P01-HL103455 (to B. A.F.); and R01-AT006822-01 (to F. J. S.). This work was also supported by the Wellcome Trust (to V. B. O., C. M. L, N. P., Y. G. D., and V. H.), a European Union Marie Curie fellowship (to C. P. T.), American Heart Association Grant 10SDG4150085 (to L. V.), the American Diabetes Association (to F. J. S.), the Academy of Finland and the Sigrid Juselius Foundation (to A. L. L. and E. K.), and American Diabetes Association Junior Faculty Award 7-08-JF 52 (to F. J. S.). B.A.F. acknowledges financial interest in Complexa, Inc. and Nitromega, Inc.
- LOX
- lipoxygenase
- KETE
- ketoeicosatetraenoic acid
- PE
- phosphatidylethanolamine
- HETE
- hydroxyeicosatetraenoic acid
- HpETE
- hydroperoxyeicosatetraenoic acid
- PPAR
- peroxisome proliferator-activated receptor
- PGDH
- hydroxyprostaglandin dehydrogenase
- PPRE
- peroxisome proliferator-response element
- EGFP
- enhanced green fluorescent protein
- BAL
- bronchoalveolar fluid
- GSH
- glutathione
- TK
- thymidine kinase
- SAPE
- 1-stearoyl-2-arachidonoyl-sn-glycero-3-phosphoethanolamine
- CF
- cystic fibrosis.
REFERENCES
- 1. Kuhn H., Thiele B. J. (1999) The diversity of the lipoxygenase family. Many sequence data but little information on biological significance. FEBS Lett. 449, 7–11 [DOI] [PubMed] [Google Scholar]
- 2. Maskrey B. H., Bermúdez-Fajardo A., Morgan A. H., Stewart-Jones E., Dioszeghy V., Taylor G. W., Baker P. R., Coles B., Coffey M. J., Kühn H., O'Donnell V. B. (2007) Activated platelets and monocytes generate four hydroxyphosphatidylethanolamines via lipoxygenase. J. Biol. Chem. 282, 20151–20163 [DOI] [PubMed] [Google Scholar]
- 3. Morgan A. H., Dioszeghy V., Maskrey B. H., Thomas C. P., Clark S. R., Mathie S. A., Lloyd C. M., Kühn H., Topley N., Coles B. C., Taylor P. R., Jones S. A., O'Donnell V. B. (2009) Phosphatidylethanolamine-esterified eicosanoids in the mouse: tissue localization and inflammation-dependent formation in Th-2 disease. J. Biol. Chem. 284, 21185–21191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Uderhardt S., Herrmann M., Oskolkova O. V., Aschermann S., Bicker W., Ipseiz N., Sarter K., Frey B., Rothe T., Voll R., Nimmerjahn F., Bochkov V. N., Schett G., Krönke G. (2012) 12/15-Lipoxygenase orchestrates the clearance of apoptotic cells and maintains immunologic tolerance. Immunity 36, 834–846 [DOI] [PubMed] [Google Scholar]
- 5. Zhao J., O'Donnell V. B., Balzar S., St Croix C. M., Trudeau J. B., Wenzel S. E. (2011) 15-Lipoxygenase 1 interacts with phosphatidylethanolamine-binding protein to regulate MAPK signaling in human airway epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 108, 14246–14251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Powell W. S., Gravelle F., Gravel S. (1992) Metabolism of 5(S)-hydroxy-6,8,11,14-eicosatetraenoic acid and other 5(S)-hydroxyeicosanoids by a specific dehydrogenase in human polymorphonuclear leukocytes. J. Biol. Chem. 267, 19233–19241 [PubMed] [Google Scholar]
- 7. Wei C., Zhu P., Shah S. J., Blair I. A. (2009) 15-Oxo-eicosatetraenoic acid, a metabolite of macrophage 15-hydroxyprostaglandin dehydrogenase that inhibits endothelial cell proliferation. Mol. Pharmacol. 76, 516–525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lee S. H., Rangiah K., Williams M. V., Wehr A. Y., DuBois R. N., Blair I. A. (2007) Cyclooxygenase-2-mediated metabolism of arachidonic acid to 15-oxo-eicosatetraenoic acid by rat intestinal epithelial cells. Chem. Res. Toxicol. 20, 1665–1675 [DOI] [PubMed] [Google Scholar]
- 9. O'Flaherty J. T., Rogers L. C., Paumi C. M., Hantgan R. R., Thomas L. R., Clay C. E., High K., Chen Y. Q., Willingham M. C., Smitherman P. K., Kute T. E., Rao A., Cramer S. D., Morrow C. S. (2005) 5-Oxo-ete analogs and the proliferation of cancer cells. Biochim. Biophys. Acta 1736, 228–236 [DOI] [PubMed] [Google Scholar]
- 10. Groeger A. L., Cipollina C., Cole M. P., Woodcock S. R., Bonacci G., Rudolph T. K., Rudolph V., Freeman B. A., Schopfer F. J. (2010) Cyclooxygenase-2 generates anti-inflammatory mediators from ω-3 fatty acids. Nat. Chem. Biol. 6, 433–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Davies S. S., Pontsler A. V., Marathe G. K., Harrison K. A., Murphy R. C., Hinshaw J. C., Prestwich G. D., Hilaire A. S., Prescott S. M., Zimmerman G. A., McIntyre T. M. (2001) Oxidized alkyl phospholipids are specific, high affinity peroxisome proliferator-activated receptor γ ligands and agonists. J. Biol. Chem. 276, 16015–16023 [DOI] [PubMed] [Google Scholar]
- 12. Morgan A. H., Hammond V. J., Morgan L., Thomas C. P., Tallman K. A., Garcia-Diaz Y. R., McGuigan C., Serpi M., Porter N. A., Murphy R. C., O'Donnell V. B. (2010) Quantitative assays for esterified oxylipins generated by immune cells. Nat. Protoc. 5, 1919–1931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang J., Villacorta L., Chang L., Fan Z., Hamblin M., Zhu T., Chen C. S., Cole M. P., Schopfer F. J., Deng C. X., Garcia-Barrio M. T., Feng Y. H., Freeman B. A., Chen Y. E. (2010) Nitro-oleic acid inhibits angiotensin II-induced hypertension. Circ. Res. 107, 540–548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tan H. L., Regamey N., Brown S., Bush A., Lloyd C. M., Davies J. C. (2011) The Th17 pathway in cystic fibrosis lung disease. Am. J. Respir. Crit. Care Med. 184, 252–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gulliksson M., Brunnström A., Johannesson M., Backman L., Nilsson G., Harvima I., Dahlén B., Kumlin M., Claesson H. E. (2007) Expression of 15-lipoxygenase type-1 in human mast cells. Biochim. Biophys. Acta 1771, 1156–1165 [DOI] [PubMed] [Google Scholar]
- 16. Murphy R. C., Zarini S. (2002) Glutathione adducts of oxyeicosanoids. Prostaglandins Other Lipid Mediat. 68–69, 471–482 [DOI] [PubMed] [Google Scholar]
- 17. Bowers R. C., Hevko J., Henson P. M., Murphy R. C. (2000) A novel glutathione containing eicosanoid (FOG7) chemotactic for human granulocytes. J. Biol. Chem. 275, 29931–29934 [DOI] [PubMed] [Google Scholar]
- 18. Huang J. T., Welch J. S., Ricote M., Binder C. J., Willson T. M., Kelly C., Witztum J. L., Funk C. D., Conrad D., Glass C. K. (1999) Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature 400, 378–382 [DOI] [PubMed] [Google Scholar]
- 19. Dioszeghy V., Rosas M., Maskrey B. H., Colmont C., Topley N., Chaitidis P., Kühn H., Jones S. A., Taylor P. R., O'Donnell V. B. (2008) 12/15-Lipoxygenase regulates the inflammatory response to bacterial products in vivo. J. Immunol. 181, 6514–6524 [DOI] [PubMed] [Google Scholar]
- 20. Bruscia E. M., Zhang P. X., Ferreira E., Caputo C., Emerson J. W., Tuck D., Krause D. S., Egan M. E. (2009) Macrophages directly contribute to the exaggerated inflammatory response in cystic fibrosis transmembrane conductance regulator−/− mice. Am. J. Respir. Cell Mol. Biol. 40, 295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu H., Lazarus S. C., Caughey G. H., Fahy J. V. (1999) Neutrophil elastase and elastase-rich cystic fibrosis sputum degranulate human eosinophils in vitro. Am. J. Physiol. Lung Cell. Mol. Physiol. 276, L28–L34 [DOI] [PubMed] [Google Scholar]
- 22. Levy B. D., Romano M., Chapman H. A., Reilly J. J., Drazen J., Serhan C. N. (1993) Human alveolar macrophages have 15-lipoxygenase and generate 15(S)-hydroxy-5,8,11-cis-13-trans-eicosatetraenoic acid and lipoxins. J. Clin. Investig. 92, 1572–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mantovani A., Sozzani S., Locati M., Allavena P., Sica A. (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 [DOI] [PubMed] [Google Scholar]
- 24. Hartl D., Griese M., Kappler M., Zissel G., Reinhardt D., Rebhan C., Schendel D. J., Krauss-Etschmann S. (2006) Pulmonary TH2 response in Pseudomonas aeruginosa-infected patients with cystic fibrosis. J. Allergy Clin. Immunol. 117, 204–211 [DOI] [PubMed] [Google Scholar]
- 25. Harmon G. S., Dumlao D. S., Ng D. T., Barrett K. E., Dennis E. A., Dong H., Glass C. K. (2010) Pharmacological correction of a defect in PPAR-γ signaling ameliorates disease severity in CFTR-deficient mice. Nat. Med. 16, 313–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Clark S. R., Guy C. J., Scurr M. J., Taylor P. R., Kift-Morgan A. P., Hammond V. J., Thomas C. P., Coles B., Roberts G. W., Eberl M., Jones S. A., Topley N., Kotecha S., O'Donnell V. B. (2011) Esterified eicosanoids are acutely generated by 5-lipoxygenase in primary human neutrophils and in human and murine infection. Blood 117, 2033–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Morgan L. T., Thomas C. P., Kühn H., O'Donnell V. B. (2010) Thrombin-activated human platelets acutely generate oxidized docosahexaenoic-acid-containing phospholipids via 12-lipoxygenase. Biochem. J. 431, 141–148 [DOI] [PubMed] [Google Scholar]
- 28. Thomas C. P., Morgan L. T., Maskrey B. H., Murphy R. C., Kühn H., Hazen S. L., Goodall A. H., Hamali H. A., Collins P. W., O'Donnell V. B. (2010) Phospholipid-esterified eicosanoids are generated in agonist-activated human platelets and enhance tissue factor-dependent thrombin generation. J. Biol. Chem. 285, 6891–6903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gronert K., Maheshwari N., Khan N., Hassan I. R., Dunn M., Laniado Schwartzman M. (2005) A role for the mouse 12/15-lipoxygenase pathway in promoting epithelial wound healing and host defense. J. Biol. Chem. 280, 15267–15278 [DOI] [PubMed] [Google Scholar]
- 30. Kenchegowda S., Bazan N. G., Bazan H. E. (2011) EGF stimulates lipoxin A4 synthesis and modulates repair in corneal epithelial cells through ERK and p38 activation. Invest. Ophthalmol. Vis. Sci. 52, 2240–2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gronert K. (2005) Lipoxins in the eye and their role in wound healing. Prostaglandins Leukot. Essent. Fatty Acids 73, 221–229 [DOI] [PubMed] [Google Scholar]
- 32. Bazan H. E. (2005) Cellular and molecular events in corneal wound healing: significance of lipid signalling. Exp. Eye Res. 80, 453–463 [DOI] [PubMed] [Google Scholar]
- 33. Harkewicz R., Fahy E., Andreyev A., Dennis E. A. (2007) Arachidonate-derived dihomoprostaglandin production observed in endotoxin-stimulated macrophage-like cells. J. Biol. Chem. 282, 2899–2910 [DOI] [PubMed] [Google Scholar]
- 34. Ide T., Egan K., Bell-Parikh L. C., FitzGerald G. A. (2003) Activation of nuclear receptors by prostaglandins. Thromb. Res. 110, 311–315 [DOI] [PubMed] [Google Scholar]
- 35. Kim E. J., Park K. S., Chung S. Y., Sheen Y. Y., Moon D. C., Song Y. S., Kim K. S., Song S., Yun Y. P., Lee M. K., Oh K. W., Yoon D. Y., Hong J. T. (2003) Peroxisome proliferator-activated receptor-γ activator 15-deoxy-Δ12,14-prostaglandin J2 inhibits neuroblastoma cell growth through induction of apoptosis: association with extracellular signal-regulated kinase signal pathway. J. Pharmacol. Exp. Ther. 307, 505–517 [DOI] [PubMed] [Google Scholar]
- 36. Hong J. T., Lee M. K., Park K. S., Jung K. M., Lee R. D., Jung H. K., Park K. L., Yang K. J., Chung Y. S. (2002) Inhibitory effect of peroxisome proliferator-activated receptor γ agonist on ochratoxin A-induced cytotoxicity and activation of transcription factors in cultured rat embryonic midbrain cells. J. Toxicol. Environ. Health A. 65, 407–418 [DOI] [PubMed] [Google Scholar]
- 37. Baker P. R., Lin Y., Schopfer F. J., Woodcock S. R., Groeger A. L., Batthyany C., Sweeney S., Long M. H., Iles K. E., Baker L. M., Branchaud B. P., Chen Y. E., Freeman B. A. (2005) Fatty acid transduction of nitric oxide signaling: multiple nitrated unsaturated fatty acid derivatives exist in human blood and urine and serve as endogenous peroxisome proliferator-activated receptor ligands. J. Biol. Chem. 280, 42464–42475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nagy L., Szanto A., Szatmari I., Széles L. (2012) Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol. Rev. 92, 739–789 [DOI] [PubMed] [Google Scholar]
- 39. Tontonoz P., Spiegelman B. M. (2008) Fat and beyond: the diverse biology of PPARγ. Annu. Rev. Biochem. 77, 289–312 [DOI] [PubMed] [Google Scholar]
- 40. Akiyama Y., Miller P. J., Thurman G. B., Neubauer R. H., Oliver C., Favilla T., Beman J. A., Oldham R. K., Stevenson H. C. (1983) Characterization of a human blood monocyte subset with low peroxidase activity. J. Clin. Investig. 72, 1093–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Necela B. M., Su W., Thompson E. A. (2008) Toll-like receptor 4 mediates cross-talk between peroxisome proliferator-activated receptor γ and nuclear factor-κB in macrophages. Immunology 125, 344–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oskolkova O. V., Afonyushkin T., Preinerstorfer B., Bicker W., von Schlieffen E., Hainzl E., Demyanets S., Schabbauer G., Lindner W., Tselepis A. D., Wojta J., Binder B. R., Bochkov V. N. (2010) Oxidized phospholipids are more potent antagonists of lipopolysaccharide than inducers of inflammation. J. Immunol. 185, 7706–7712 [DOI] [PubMed] [Google Scholar]