

Background: GIV/Girdin is a GEF for Gαi and a metastasis-related protein, which is required for cancer invasion.
Results: STAT3 directly binds the GIV promoter and triggers GIV transcription. The GEF function of GIV enhances activation of STAT3.
Conclusion: STAT3 up-regulates GIV transcription, and GIV enhances STAT3 activation.
Significance: Insights gained are crucial for devising both therapeutic and prognostic options at the crossroads between STAT3 and GIV.
Keywords: Breast Cancer, Cell Migration, Gene Transcription, Guanine Nucleotide Exchange Factor (GEF), STAT3, Girdin
Abstract
Gα-interacting vesicle-associated protein (GIV) is a guanine nucleotide exchange factor that modulates key signaling pathways during a diverse set of biological processes, e.g. wound healing, macrophage chemotaxis, tumor angiogenesis, vascular repair, and cancer invasion/metastasis. We recently demonstrated that GIV is a metastasis-related protein, which serves both as a therapeutic target and as a biomarker for prognostication in cancer patients. Here we report the discovery that GIV is a direct target of the transcription factor signal transducer and activator of transcription-3 (STAT3), which is commonly known as a central regulator of tumor metastasis. We identified a single STAT3-binding site on the GIV promoter that was necessary and sufficient for transcriptional activation of GIV during wound healing and cancer invasion. Immunohistochemical analysis of breast carcinomas showed significant correlation between STAT3 activation and elevated GIV expression. Furthermore, we provide evidence that GIV positively autoregulates its own transcription by enhancing STAT3 activation via its guanine nucleotide exchange factor activity. Our findings provide mechanistic insights into how STAT3 activation is directly integrated with the receptor tyrosine kinase-GIV-G protein signaling axis. The forward feedback regulation we describe here between GIV and STAT3 may have profound therapeutic implications for cancer and epithelial regeneration/repair and could help invent novel approaches in treating and prognosticating cancer.
Introduction
Gα-interacting vesicle-associated protein (GIV5; also known as Girdin) is a multidomain signal transducer and a novel guanine nucleotide exchange factor (GEF) that triggers cell migration via its ability to scaffold key signaling molecules, i.e. trimeric G proteins, growth factor receptors, PI3K, Akt, and phosphoinositides, with the actin cytoskeleton (1). Mechanistically, GIV influences cell migration by directly interacting with growth factor receptors and modulating some of the major receptor-initiated signaling pathways via its catalytic GEF domain (2). An intact GEF domain triggers cells to preferentially migrate because signals that primarily trigger motility (i.e. motogenic PI3K → Akt, PLCγ1 → inositol 1,4,5-trisphosphate, and diacylglycerol pathways) are enhanced, whereas signals that primarily trigger mitosis (i.e. mitogenic MAPK → extracellular signal-regulated kinase (ERK) and STAT5 pathways) are suppressed. By contrast, the absence of the GEF domain generates an opposite, mirror image signaling profile, which inhibits cell migration and instead triggers cells to divide (2). Consistent with the ability of GIV to modulate several major signaling pathways downstream of both growth factor receptor tyrosine kinases (RTKs) and G protein-coupled receptors (1), we and others have shown that GIV serves as a common modulator of signals during a diverse set of biological processes, e.g. epithelial wound healing, macrophage chemotaxis (3), development (4, 5), neuronal migration (6, 7), vascular repair (8), autophagy (9), tumor angiogenesis (10), tumor cell migration (3, 11), and cancer invasion/metastasis (3, 12, 13).
The first clue that GIV might play a role in cancer invasion came from our finding that GIV-dependent activation of Gαi is essential for Akt enhancement and actin remodeling during tumor cell migration (14). The role of GIV in tumor invasion/metastasis in vivo was further substantiated when its depletion was found to markedly impair metastasis in mouse models (13) and inhibit VEGF-mediated neoangiogenesis (10); the latter is a prerequisite for tumor progression. We demonstrated (2) that expression of GIV mRNA and protein is deregulated in breast and colorectal cancers. In poorly invasive cancer cells and in early staged, preinvasive colorectal carcinomas, GIV is down-regulated by alternative splicing, but in highly invasive cancer cells and late staged invasive carcinomas, GIV is highly expressed at levels ∼20–50-fold above normal. Striking differences in GIV expression have also been reported among primary tumors and cancer cells of other human carcinomas (e.g. breast, colon, lung, skin, pancreas, and uterine cervical) (2, 12, 13). Subsequent work from us and others has demonstrated that GIV behaves as a bona fide metastasis-related protein (12) in that its overexpression predicts invasiveness and prognosticates unfavorable outcome for patients with colorectal cancer (12), breast cancer (15, 16), and glioblastomas (17). Despite the breadth of information available on the biological relevance of GIV in healing wounds, invading carcinomas, and tumor angiogenesis, it remains unexplored which transcription factor regulates GIV expression during any of these processes. Because evidence supporting the role of GIV in cancer metastasis continues to accumulate, the task of identifying the transcription factor that targets GIV becomes both urgent and critical.
Transcription factors are encoded by a large number of oncogenes and tumor suppressor genes (18) that control gene expression patterns and signaling pathways in cancer. It is well accepted that aberrant activation of transcription factors can trigger oncogenesis and cancer progression via a myriad of mechanisms such as changes in gene expression, protein stability, and protein-protein interactions (18). Among the transcription factors that are predicted to bind the stretch of DNA that corresponds to the promoter region of GIV, signal transducer and activator of transcription-3 (STAT3) stands out as the most likely candidate for various reasons. (a) Although GIV behaves as a metastasis-related protein, the STAT3 signaling pathway is referred to as the “central regulator of tumor metastasis” (19). (b) STAT3 regulates a common set of genes involved in wound healing and cancer (20), the two biological processes in which GIV is strongly implicated. (c) A recent study (21) using gene expression profiling in prostate cancer cells showed that STAT3 influences cell migration and proliferation much like GIV (2); i.e. STAT3 inhibits tumor growth (decreases transcription of genes that enhance growth) and favors invasion (increases transcription of genes that are implicated in metastasis). (d) Targeted depletion of STAT3, but not STAT5, in prostate cancer cells specifically reduced the transcription of GIV gene (ccdc88a) in the above study (21). Based on these clues, we hypothesized that STAT3 regulates GIV transcription during epithelial wound healing and tumor invasion/metastasis.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Unless otherwise indicated, all reagents were of analytical grade and obtained from Sigma-Aldrich. Cell culture media and epidermal growth factor (EGF) were purchased from Invitrogen. Custom-designed oligos were obtained from Valuegene (San Diego, CA). Silencer Negative Control scrambled (Scr) siRNA and STAT3 siRNA were purchased from Ambion and Santa Cruz Biotechnology, respectively. Antibodies against GIV that were used in this work include rabbit serum anti-GIV coiled coil immunoglobulin G (for immunoblotting only) (22), affinity-purified GIV coiled coil immunoglobulin G (catalog number ABT80 from EMD Millipore for immunohistochemistry and immunoblotting). Mouse mAbs against STAT3 (Santa Cruz Biotechnology), c-myc (Cell Signaling Technology), anti-histone (histone H3 lysine 9 trimethylation (H3K9me3)) (Millipore), and tubulin (Sigma) were purchased from commercial sources. Rabbit polyclonal antibodies against phospho-Tyr-705 and phospho-Ser-727 STAT3 (Cell Signaling Technology), Gαi3 (Santa Cruz Biotechnology), phospho-Akt-Ser-473 (Cell Signaling Technology), pan-Gβ (Santa Cruz Biotechnology), and phospho-ERK1/2 (Cell Signaling Technology) were obtained commercially. DAPI and anti-mouse Alexa Fluor 594-coupled goat secondary antibody for immunofluorescence were purchased from Invitrogen. Goat anti-rabbit and goat anti-mouse Alexa Fluor 680 or IRDye 800 F(ab′)2 for immunoblotting were from LI-COR Biosciences (Lincoln, NE).
Expression Vectors and Mutagenesis
myc-tagged STAT3 in pcDNA3.1 and luciferase reporter vectors (pGL4 and pTSKL) were generous gifts from Eugene Chin (Brown University) and Christopher K. Glass (University of California, San Diego), respectively. STAT3-C (A662C and A664C), STAT3-Y705F, and STAT3-KR (K685R) constructs were generated by site-directed mutagenesis using a QuikChange kit (Stratagene, San Diego, CA) according to the manufacturer's protocols. His-STAT3 was generated by cloning STAT3 from myc-STAT3 in pcDNA3.1 into pET-28b vector (Novagen). The human GIV promoter containing the 5′ region of the GIV gene (a genomic DNA fragment of ∼1.2-kb size) was cloned into pGL4.10[luc2] vector between KpnI and HindIII. pGL4-GIV(Δ+39/+58) was generated by deleting the predicted STAT3-binding site, which is located between positions +39 and +58. pTSKL-GIV(+39/+58) minimal promoter was cloned by ligating the predicted STAT3-binding region (+39/+58) into a pTSKL plasmid between HindIII and BamHI.
Cell Culture and Transfection
HeLa, DLD-1, and MDA-MB 231 cells were maintained according to ATCC guidelines. HeLa cells were used in the generation of stable cell lines expressing various STAT3 constructs at ∼2-fold above endogenous levels exactly as described previously (2). The 21T breast cell lines (16N, NT, and MT2) were obtained from Arthur B. Pardee (Dana-Farber Cancer Institute and Harvard Medical School, Boston, MA) and maintained as described previously (32, 39). Transfection was performed using Genejuice (Novagen) for DNA plasmids or Oligofectamine (Invitrogen) for siRNA oligos according to the manufacturers' protocols.
Scratch Wound Assays
To monitor gene expression and STAT3 activation in scratch wound assays, HeLa cells were plated at near confluence; transfected with plasmid DNA, siRNA oligos, or both as indicated in various experiments; and grown for an additional 36–48 h until fully confluent. These confluent cultures were subjected to high density scratch wounding using a sterilized, metal, 250-toothed wounding comb exactly as we have done previously (2, 3, 14, 23–25).
Lysis and Immunoblotting
Lysates were prepared by resuspending the cells in lysis buffer (20 mm HEPES, pH 7.2, 5 mm magnesium acetate, 125 mm potassium acetate, 0.4% Triton X-100, 1 mm dithiothreitol (DTT) supplemented with phosphatase (Sigma) and protease (Roche Applied Science) inhibitor mixtures), passed through a 28-gauge needle at 4 °C, and cleared 10,000 × g for 10 min before use in subsequent experiments. Equal amounts of total cellular proteins were separated by 10% SDS-PAGE and transferred to PVDF membranes (Millipore, Billerica, MA). Immunoblotting and quantification were carried out as described previously (25) by dual color infrared imaging using an Odyssey imaging system (LI-COR Biosciences). All individual images were processed with ImageJ software (National Institutes of Health) and assembled for presentation with Photoshop and Illustrator software (both Adobe).
RNA Isolation and Quantitative PCR (qPCR)
Total RNA was isolated using an RNeasy kit (Qiagen) according to the manufacturer's protocol. First strand cDNA was synthesized using Superscript II reverse transcriptase (Invitrogen) followed by ribonuclease H treatment (Invitrogen) prior to performing quantitative real time PCR. Reactions omitting reverse transcriptase were performed in each experiment as negative controls. Primer sequences are available upon request. Reactions were then run on a real time PCR system (ABI Prism 7300, Applied Biosystems). Gene expression was detected with SYBR Green (Invitrogen), and relative gene expression was determined by normalizing to GAPDH using the ΔCT method.
Chromatin Immunoprecipitation
These assays were performed essentially as described (26). Briefly, cells were cross-linked by addition of formaldehyde to 1% final concentration in media and incubation at room temperature for 10 min, neutralized with 125 mm glycine, and then subjected to sonication using a Bioruptor (Diagenode) to fragment the chromatin to obtain 200–500-bp fragments. Sonicated chromatin was precleared with a mixture containing 50% protein A/G bead slurry (Pierce), salmon sperm DNA, and BSA. Precleared chromatin was incubated with specific antibodies (myc, H3K9me3, and STAT3) and their respective IgG controls. Protein A/G bead slurry was then added to pull down the antibody-bound chromatin, which was eluted using sodium bicarbonate buffer containing SDS and DTT. Eluted chromatin was decross-linked, and protein was removed by treating with proteinase K. Purified immunoprecipitated chromatin was subjected to quantitative real time PCR amplification using multiple sets of primers. The sequences of these primers are available upon request. Input chromatin was used as a control.
Expression and Purification of Recombinant STAT3
His-tagged recombinant STAT3 protein was expressed and purified from Escherichia coli strain BL21(DE3) (Invitrogen) using HisPurTM Cobalt Resin (Thermo Scientific). Briefly, cultures of transformed bacteria were induced overnight at 20 °C with 1 mm isopropyl β-d-1-thiogalactopyranoside. The bacterium pellet from 500 ml of culture was resuspended in 50 ml of lysis buffer (50 mm NaH2PO4, 300 mm NaCl, 1 mg/ml lysosome, 10 mm imidazole, 2× protease inhibitor mixture (Complete EDTA-free, Roche Diagnostics)). After sonication (6 × 30 s,1 min between cycles), lysates were centrifuged at 12,000 × g at 4 °C for 30 min. Solubilized protein was affinity-purified on HisPur Cobalt Resin. Protein was eluted with elution buffer (50 mm NaH2PO4, 300 mm NaCl, 250 mm imidazole), dialyzed overnight against PBS, and stored at −80 °C.
Electrophoretic Mobility Shift Assay (EMSA)
EMSA was carried out as described previously (27). The following 32P-labeled oligonucleotide pair containing the putative STAT3-binding site at position +33/+62 in the human GIV promoter was used as a probe in EMSA: forward, 5′-TGTGCCAGGGATTCCCAGAGACGCGCTGAA-3′; reverse, 5′-TTCAGCGCGTCTCTGGGAATCCCTGGCACA-3′ (the STAT3-binding sequence is underlined). Recombinant His-STAT3 was incubated with 32P-labeled oligonucleotide probe in 20 μl of buffer containing 10 mm HEPES, pH 7.8, 50 mm KCl, 1 mm EDTA, 5 mm MgCl2, 10% glycerol, 5 mm DTT, 1 mg/ml bovine serum albumin, and 1 mm Na3VO4. After a 15-min incubation at room temperature, the samples were separated on a 6% native polyacrylamide gel. For competition analyses, His-STAT3 was incubated with cold probe (unlabeled oligonucleotide) for 15 min at room temperature before addition of the labeled oligonucleotides.
Luciferase Assay
These assays were performed as described previously (28). Briefly, transfection of various luciferase reporter constructs was carried out using Genejuice according to the manufacturer's protocol, and cytosolic fractions were prepared 48 h after transfection. In siRNA experiments, 300 nm scrambled control or STAT3-targeting siRNA oligonucleotides was added to cells first followed by transfection with the various luciferase reporters after 48 h, and then cells were harvested after another 24 h to carry out luciferase assays. Luciferase activity was analyzed using a microplate luminometer (Turner Biosystem) and normalized to β-galactosidase activity by colorimetric assay at A570 as an internal control for transfection efficiency.
Immunohistochemistry
Forty-five deidentified breast cancer specimens (from the Department of Pathology, China Medical University, China) of known histological type and grade were analyzed by immunohistochemistry using anti-GIV (1:500) and anti-phospho-STAT3 (pSTAT3) (1:300) rabbit polyclonal antibodies. Briefly, formalin-fixed, paraffin-embedded tissue sections of 4-μm thickness were cut and placed on glass slides coated with 3-aminopropyltriethoxysilane followed by deparaffinization and hydration. Heat-induced epitope retrieval was performed using citrate buffer (pH = 6) in a pressure cooker. Tissue sections were incubated with 3% hydrogen peroxidase for 40 min to block endogenous peroxidase activity followed by incubation with primary antibodies overnight in a humidified chamber at 4 °C. Immunostaining was visualized with a labeled streptavidin-biotin using 3,3′-diaminobenzidine as a chromogen and counterstained with hematoxylin. All the samples were first quantitatively analyzed and scored based on two independent criteria. 1) The intensity of staining was scored on a scale of 0–3 where 0 = no staining, 1 = light brown, 2 = brown, and 3 = dark brown. 2) The percentage of cells that stained positive within the tumor was scored on a scale of 0–4 where 0 = 0%, 1 = ≤10%, 2 = 11–50%, 3 = 51–75%, and 4 = >75%. Subsequently, each tumor sample was assigned a final score that is the product of its (intensity of staining) × (percentage of tumor cells that stained positive). Tumors were categorized as negative when their final score was <3 and positive when their final score was ≥3.
Immunofluorescence
Cells were fixed at room temperature with 3% paraformaldehyde for 20–25 min, permeabilized (0.2% Triton X-100) for 45 min, and incubated for 1 h each with primary and then secondary antibodies as described previously (2, 23). Samples were examined with a Zeiss Axiophot microscope (Carl Zeiss Inc.) with a 63× aperture objective (Zeiss Plan Neofluar, 1.30 numerical aperture), and images were collected with an ORCA-ER camera (Hamamatsu) and Volocity software. All individual images were processed with ImageJ software (National Institutes of Health) and assembled for presentation with Photoshop and Illustrator software (both Adobe).
Statistical Analysis
Data are expressed as the mean ± S.D. Unless stated otherwise, statistical significance was determined using Student's t test and was achieved when the p value was ≤0.05. A χ2 test was used to analyze the significance of pSTAT3 or GIV staining and the various histological types of tumors. Fisher's exact test was used to analyze the strength of correlation between pSTAT3 and GIV staining. Statistical significance was assessed with the Wilcoxon signed rank test: *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
RESULTS
GIV Expression Is Up-regulated during Epithelial Wound Healing and Cancer Progression
We and others have demonstrated previously that GIV is required for cell migration during epithelial wound healing and tumor invasion (11, 13). Because wound healing and tumor invasion are dynamic events that require regulation of genes involved in cell proliferation, migration, invasion, stromal regeneration, and neovascularization in a timely fashion (29) and because GIV regulates more than one of these processes (1–3, 10, 11, 13), we asked whether the expression of full-length GIV mRNA and/or protein is altered during the course of epithelial wound healing and cancer progression. GIV mRNA expression was rapidly induced when confluent monolayers of HeLa epithelial cells were subjected to multiple scratch wounds (>10,000 wounds/60 cm2; see “Experimental Procedures”), peaked at 6 h, and decreased at 24 h, the latter coinciding with wound closure (Fig. 1A). GIV protein also increased ∼2.3-fold during the course of 24 h, lagging behind the peak mRNA levels (Fig. 1A). Next we investigated whether a similar induction in GIV expression also occurs during human dermal wound healing by analyzing genome-wide microarray expression data from human skin graft donor sites (30). Skin samples were biopsied immediately before and after harvesting the graft as well as at 3 and 7 days postharvest, representing early and later phases of wound healing. Expression of GIV mRNA peaked at 3 days after grafting by ∼2.25-fold and decreased by 7 days, the latter coinciding with re-epithelialization of the denuded surface (Fig. 1B). Similarly, microarray expression analysis in injured murine skin following a second degree scald burn (31) also showed induction in GIV mRNA after injury (supplemental Fig. S1). These findings demonstrate that GIV expression is up-regulated during epithelial wound healing.
FIGURE 1.

Expression of GIV mRNA and protein is up-regulated during epithelial wound healing and cancer progression. A, confluent HeLa cell monolayers were scratch-wounded and harvested at the indicated time points after wounding, and whole cell lysates were analyzed for full-length GIV mRNA (left) and protein (right) by qPCR and immunoblotting (IB), respectively. Changes in GIV mRNA are expressed as mean ± S.D. (n = 3 separate experiments). B, -Fold changes in GIV mRNA observed immediately after an acute surgical wound of human skin and at 3 and 7 days (d) after wounding were analyzed using the raw microarray data reported in a study by Nuutila et al. (30) investigating the human skin transcriptome during wound healing. All values were normalized to intact normal skin. C, the abundance of GIV mRNA in highly metastatic MDA-MB 231 breast cancer cells (left) and DLD-1 colorectal cancer cells (right) was analyzed by qPCR, normalized to their respective normal controls, and expressed as -fold change in GIV mRNA. D, the abundance of GIV mRNA was analyzed in the isogenic background of the 21T series of breast cancer cells (left) and Ls series of colorectal cancer cells (right) by qPCR, normalized to the least invasive cell line in each series (i.e. 16N in breast and Ls-174T in colon), and expressed as -fold change in GIV mRNA. Error bars represent S.D. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
To determine whether GIV mRNA expression also increases during cancer progression, first we compared the levels of GIV mRNA in highly invasive MDA-MB 231 (breast) and DLD-1 (colorectal) carcinoma cells with their respective normal epithelial control. We demonstrated previously that these highly invasive cell lines express GIV protein, whereas their non-invasive counterparts do not (2). Consistently, GIV mRNA levels in MDA-MB 231 and DLD-1 cells were also elevated by ∼25–35-fold above their respective normal controls (Fig. 1C). To determine whether GIV expression progressively increases with increasing invasiveness of tumor cells during breast and colorectal cancer progression, we took advantage of the isogenic background of the 21T series of human mammary cells (16N, NT, and MT2) (32) and human colon cancer cell lines Ls-174T and Ls-LiM6 (33). 21T cells were derived by successive biopsies from a single breast cancer patient in which 16N was from the normal breast, NT was from the primary tumor (invasive ductal carcinoma), and MT2 was from the metastatic pleural effusions. The human Ls-LiM6 lines were established using a mouse metastasis model by selecting clones with high liver-metastasizing ability from poorly metastatic Ls-174T parent cells and differ only with respect to expression of metastasis-related proteins. We found that the abundance of GIV mRNA progressively increases with increasing invasiveness of 21T breast and Ls colorectal cancer cells (Fig. 1D), indicating that GIV expression increases during breast and colorectal cancer invasion. Furthermore, a survey of Gene Expression Omnibus (GEO) profiles revealed that GIV mRNA is significantly increased during the metastatic progression of a variety of solid tumors, including breast, colon, lung, melanomas, renal, gastric, pancreatic, esophageal, and thyroid (supplemental Fig. S2 and legend). We conclude that GIV expression is up-regulated during two multistep, complex, but related biological processes, wound healing and cancer progression.
STAT3 Is Required for the Induction of GIV Expression during Wound Healing and in Invasive Cancer Cells
Next we asked whether STAT3 affects GIV expression during wound healing and in invasive cancer cells. To this end, we either overexpressed or depleted STAT3 in HeLa cells and analyzed GIV expression by immunoblotting and qPCR. Overexpression of STAT3 in HeLa cells increased GIV mRNA and protein levels compared with vector controls (Fig. 2A), indicating that STAT3 can augment GIV expression. Conversely, siRNA targeted depletion of endogenous STAT3 in HeLa cells reduced GIV mRNA and protein expression compared with scrambled controls (Fig. 2B), indicating that STAT3 is essential for maintaining the steady-state levels of GIV expression in HeLa cells. Furthermore, scratch wound-induced up-regulation of GIV mRNA was seen in HeLa cells stably expressing the constitutively active STAT3-C (34) but not in cells expressing the constitutively inactive, dominant negative STAT3-K685R (35) mutant, indicating that activation of STAT3 is required for up-regulation of GIV during wound healing (Fig. 2C). Exogenous STAT3 could also induce GIV expression in HeLa cells in the absence of phosphorylation at Tyr-705 (supplemental Fig. S3), indicating that tyrosine phosphorylation of STAT3 is not a prerequisite for the induction of GIV. As observed in the case of HeLa cells, depletion of endogenous STAT3 in the invasive cancer cell lines MDA-MB 231 and DLD-1 also resulted in a decrease of GIV mRNA levels (by ∼40%) and a concomitant reduction in the levels of GIV protein (Fig. 2, D and E). The observed decrease in GIV expression in STAT3-depleted cells could not be due to off-target effects of siRNA oligos because exogenous expression of siRNA-resistant STAT3 in cells treated with STAT3 siRNA rescued the levels of GIV mRNA and protein (Fig. 2, F and G). Taken together, these results indicate that STAT3 positively regulates GIV expression during wound healing and in invasive cancer cells.
FIGURE 2.

STAT3 is required for up-regulation of GIV expression during wound healing and in invasive cancer cells. A, overexpression of myc-STAT3 increases both GIV mRNA and protein. Whole cell lysates of HeLa cells transiently overexpressing myc-STAT3 or vector control were analyzed for GIV protein (top) and mRNA (bottom) expression by immunoblotting (IB) and qPCR, respectively. Up-regulation of GIV protein (top) is specific because Gαi3 or tubulin remains unchanged. -Fold changes in GIV mRNA (bottom) are expressed as mean ± S.D. (n = 3). B, depletion of STAT3 reduces both GIV protein and mRNA levels in HeLa cells. HeLa cells were treated with Scr or STAT3 siRNA and analyzed for GIV protein (top) and mRNA (bottom) expression by immunoblotting and qPCR, respectively. Results are expressed as mean ± S.D. (n = 3). C, activation of STAT3 is required for up-regulation of GIV mRNA during wound healing. Whole cell lysates of HeLa cells stably expressing myc-tagged constructs, either the constitutively active STAT3-C mutant, the constitutively inactive, dominant negative STAT3-KR mutant, or vector control, were analyzed for myc-STAT3 and tubulin by immunoblotting (top) and for GIV mRNA by qPCR (bottom). -Fold changes in GIV mRNA are expressed as mean ± S.D. (n = 3). The p value was calculated by comparing GIV mRNA levels in HeLa cells expressing STAT3-C and STAT3-KR at 6 h postwounding. D and E, depletion of STAT3 reduces both GIV protein and mRNA in highly invasive MDA-MB 231 (D) and DLD-1 (E) cells. MDA-MB 231 and DLD-1 cells were treated with Scr or STAT3 siRNA, and whole cell lysates were analyzed for GIV protein (left) and mRNA (right) by immunoblotting and qPCR, respectively. Both GIV protein and mRNA levels were reduced in MDA-MB 231 and DLD-1 cells depleted of STAT3. Results are expressed as mean ± S.D. (n = 3). F and G, expression of siRNA-resistant mouse STAT3 in HeLa cells depleted of endogenous STAT3 increases both GIV mRNA (F) and protein (G) levels. HeLa cells were treated with control (Scr) or anti-human (h) STAT3 siRNA, transfected with vector or myc-STAT3 (mouse) as indicated, and subsequently analyzed for GIV mRNA by qPCR (F) and for GIV, STAT3, and tubulin by immunoblotting. Error bars represent S.D. *, p < 0.05; **, p < 0.01.
STAT3 Binds the GIV Promoter and Activates Its Transcription
To discern whether GIV is a transcriptional target of STAT3, we performed chromatin immunoprecipitation (ChIP) in HeLa cells expressing myc-tagged STAT3 at 0, 6, and 24 h after scratch wounding (Fig. 3, A and B). Immunoprecipitate-bound chromatin was analyzed for STAT3 occupancy by quantitative PCR. Primers for multiple amplicons were designed to cover a 3-kb region within the GIV gene that includes the GIV promoter demarcated with high H3K4me3 and low H3K4me1, a histone signature for the promoter region (Fig. 3A). We found the highest enrichment of STAT3 using primers that amplify from −100 to +250 relative to the transcription start site of GIV (Fig. 3A, primer set 4). Consistent with this, we found a putative STAT3-binding site +39/+58 from the transcription start site using two commonly used prediction software programs, JASPAR and TFBIND. Additionally, STAT3 recruitment to the GIV promoter was triggered by scratch wounding (Fig. 3C): the binding is minimal in cells of an intact monolayer (0 h), peaks at 6 h postwounding, and subsequently decreases at 24 h post-wounding, suggesting that STAT3 binds GIV during the early response after wounding and releases the GIV promoter during the late response. This maximal STAT3 recruitment at 6 h postwounding coincides with the timing for peak expression of GIV mRNA we observed previously (Fig. 1A). An identical pattern was also seen when chromatin immunoprecipitation was carried out in HeLa cells using a monoclonal anti-STAT3 antibody (Santa Cruz Biotechnology) targeting endogenous STAT3 (supplemental Fig. S4), thereby validating our chromatin immunoprecipitation results using anti-myc IgG and overexpression of myc tagged-STAT3. Next we asked whether STAT3 binding to the GIV promoter corresponds to a decrease of transcriptional repressive histone marks, which in turn allows up-regulation of GIV transcription by modulating chromatin. Indeed, when we checked the levels of such a bona fide repressive histone mark, H3K9me3, on the GIV promoter, we found decreased H3K9me3 at 6 h postwounding, coinciding with maximal STAT3 binding. Interestingly, the H3K9me3 level seemed to be restored at 24 h of wounding, coinciding with the release of STAT3 from the GIV promoter. These results strongly indicate that both STAT3 occupancy and a concomitant loss of the H3K9me3 mark correlate directly with GIV expression. A similar regulation of GIV by STAT3 was observed in MDA-MB 231 (Fig. 3D) and DLD-1 (Fig. 3E) cancer cells: STAT3 binds the promoter region of GIV in both cell lines, and this binding coincides with a reduction in the H3K9me3 marks. These findings demonstrate that STAT3 binds to an identical region within the GIV promoter in three different cell lines and can activate transcription via the GIV promoter by modulating the repressive histone profile in that region.
FIGURE 3.

STAT3 binds directly to the GIV promoter. A, top, a schematic representation of STAT3 binding elements on GIV gene is shown. Confluent monolayers of HeLa cells expressing myc-tagged STAT3 were scratch-wounded; harvested at 0, 6, and 24 h postwounding; and analyzed for myc-STAT3 occupancy within a stretch of an ∼3-kb region that includes the GIV promoter and parts of the gene body (indicated by gray shading) by ChIP using anti-myc or control IgGs followed by qPCR using primer pairs that systematically cover the 3-kb region. Shown here are the qPCR results of four such primer pairs, numbered 1–4, demonstrating sites of myc-STAT3 occupancy at different time points during wound healing expressed as -fold enrichment over control IgG. Bottom, a snapshot of the coding region of GIV gene from the UCSC Genome Browser is shown layered below by epigenetic histone marks H3K4me1, which is characteristic of enhancers, and H3K4me3, which is characteristic of promoters. myc-STAT3 binds the GIV gene exclusively at 6 h after wounding and specifically occupies its promoter region (highest enrichment seen with primer pair 4) as determined by its high H3K4me3 and low H3K4me1 associations (demarcated by interrupted gray lines). B, whole cell lysates of HeLa cells used in the above assay were analyzed for expression of myc-STAT3 at 0, 6, and 24 h after scratch wounding by immunoblotting. C–E, increased STAT3 occupancy coincided with a reciprocal loss of the repressive H3K9me3 modification in HeLa (C), MDA-MB 231 (D), and DLD-1 (E) cells. Chromatin immunoprecipitation assays were carried out in HeLa cells (C) after scratch wounding as described earlier and in highly invasive MDA-MB 231 breast cancer (D) and DLD-1 colorectal cancer (E) cell lines overexpressing myc-STAT3 or control vector as indicated using anti-myc (left), anti-H3K9me3 (right), and their respective control IgGs and analyzed for STAT3 and H3K9me3 occupancy by qPCR. F, His-STAT3 binds directly to the GIV promoter as determined by EMSA. Recombinant His-STAT3 formed protein-DNA complexes (shift in lane 2) when incubated with DNA probes harboring the putative STAT3-binding site in the GIV promoter. These complexes are absent in the presence of excess cold probe (lane 3). Error bars represent S.D. *, p < 0.05; **, p < 0.01; ****, p < 0.001.
To determine whether STAT3 directly binds to the predicted region of the GIV promoter identified in the ChIP assays, we generated a radiolabeled pair of DNA probes harboring the putative STAT3-binding site on the GIV promoter and used them in an in vitro EMSA with recombinant His-STAT3. We found that STAT3 associates in complex with GIV promoter probes, contributing to a gel shift (Fig. 3F). This complex/shift was lost when STAT3 was preincubated in the presence of excess unlabeled probe. These results demonstrate that STAT3 directly binds to the predicted region on the GIV promoter.
Next we asked whether binding of STAT3 to the promoter region of GIV activates transcription of the GIV gene. To this end, we used luciferase reporter assays driven by the GIV promoter with or without the STAT3-binding site. Four luciferase constructs were generated (Fig. 4A and supplemental Fig. S5A): 1) pGL4-GIV containing ∼1.2 kb of the wild type GIV promoter, 2) pGL4-GIV(Δ+39/+58) carrying ∼1.2 kb of the GIV promoter without the STAT3-binding site (+39 to +58), 3) pGL4-GIV mutant in which the consensus for STAT3-binding site is selectively mutated, and 4) pTSKL-GIV(+39/+58) carrying only the STAT3-binding site with a weak minimal promoter. To examine whether binding of STAT3 to the GIV promoter activates its transcription, we carried out luciferase assays in HeLa, MDA-MB 231, and DLD-1 cells transiently expressing pGL4-GIV promoter with or without depletion of endogenous STAT3. Depletion of endogenous STAT3 resulted in a significant decrease of luciferase activity in all three cell lines (Fig. 4B), indicating that STAT3 is required for activation of GIV transcription. Furthermore, cotransfection of pGL4-GIV with the constitutively active STAT3-C, but not the constitutively inactive STAT3-KR mutant, significantly increased the expression of luciferase via the GIV promoter in all three cell lines (Fig. 4C), indicating that activation of STAT3 is essential for induction of GIV transcription.
FIGURE 4.

Binding of STAT3 to the GIV promoter increases transcription of GIV. A, a schematic diagram of the firefly luciferase reporter constructs used in this work is shown. +39 to +58 denotes the STAT3-binding site independently predicted by two programs. TSS, transcription start site. B, depletion of STAT3 reduced luciferase/GIV promoter activity in HeLa (left), MDA-MB 231 (middle), and DLD-1 (right) cells. All three cell lines were treated with Scr or STAT3 siRNA, subsequently transfected with the GIV promoter-luciferase reporter construct pGL4-GIV, harvested, analyzed for luciferase activation via the GIV promoter with a luminometer, and normalized to β-galactosidase activity by a colorimetric assay as described under “Experimental Procedures.” Results are expressed as mean ± S.D. (n = 3). C, STAT3-C, but not STAT3-KR, enhanced luciferase/GIV promoter activity in HeLa (left), MDA-MB 231 (middle), and DLD-1 (right) cells. All three cell lines were co-transfected with GIV promoter-luciferase reporter construct pGL4-GIV and the active (STAT3-C) or inactive (STAT3-KR) mutants of STAT3 or control vector and analyzed for luciferase activity via the GIV promoter as in B. Results are expressed as mean ± S.D. (n = 3). D, deletion of the STAT3-binding site (+39/+58) from the GIV promoter-luciferase reporter pGL4-GIV reduced luciferase/GIV promoter activity in HeLa (left), MDA-MB 231 (middle), and DLD-1 (right) cells. All three cell lines were transfected with either the GIV promoter-luciferase reporter pGL4-GIV or with the GIV promoter-luciferase reporter pGL4-GIV(Δ+39/+58), in which the STAT3-binding site is deleted, and analyzed for luciferase activity via the GIV promoter as in B. Results are expressed as mean ± S.D. (n = 3). E, depletion of STAT3 reduced luciferase activity in HeLa (left), MDA-MB 231 (middle), and DLD-1 (right) cells expressing pTSKL-GIV(+39/+58), which carries the thymidine kinase minimal promoter and the STAT3-binding site. All three cell lines were treated with Scr or STAT3 siRNA, subsequently transfected with pTSKL-GIV(+39/+58), and analyzed for luciferase activity as in B. Results are expressed as mean ± S.D. (n = 3). F, STAT3-C, but not STAT3-KR, enhanced luciferase activity in HeLa (left), MDA-MB 231 (middle), and DLD-1 (right) cells expressing pTSKL-GIV(+39/+58). All three cell lines were co-transfected with pTSKL-GIV(+39/+58) and the active (STAT3-C) or inactive (STAT3-KR) mutant of STAT3 or control vector and analyzed for luciferase activity as in B. Results are expressed as mean ± S.D. (n = 3). Error bars represent S.D. *, p < 0.05; **, p < 0.01.
To discern whether the STAT3-binding site in the GIV promoter is required for transcriptional activation of GIV, we followed two approaches. First, we compared luciferase activity in HeLa, MDA-MB 231, and DLD-1 cells transiently expressing the pGL4-GIV(Δ+39/+58) construct (which lacks the STAT3-binding site) with those expressing the pGL4-GIV. The lack of the STAT3-binding site reduced luciferase activity in all three cell lines (Fig. 4D) and abolished the ability of STAT3-C to enhance GIV gene expression (supplemental Fig. S5B), indicating that the STAT3-binding site between +39 and +58 of the GIV promoter is required for its activation. As a second approach, when HeLa cells transiently expressing the pGL4-GIV mutant construct (in which the STAT3-binding consensus sequence is selectively mutated; supplemental Fig. S5A) were compared with those expressing the pGL4-GIV, we found that disruption of the STAT3-binding consensus reduced luciferase activity by ∼4-fold (supplemental Fig. S5C), indicating that STAT3 requires the binding consensus sequence to enhance GIV promoter activity. Next we asked whether the STAT3-binding site from the GIV promoter is sufficient for STAT3-mediated transcription of GIV by carrying out luciferase assays in HeLa, MDA-MB 231, and DLD-1 cells transiently expressing the minimal promoter-luciferase construct pTSKL(+39/+58), which contains the STAT3-binding site. Addition of the STAT3-binding site from +39/+58 of the GIV promoter was sufficient to increase luciferase activity driven by a minimal thymidine kinase promoter in all three cell lines (supplemental Fig. S6). As seen in the case of pGL4-GIV, depletion of STAT3 by siRNA resulted in a significant decrease in luciferase activity from pTSKL(+39/+58) in all three cell lines (Fig. 4E). Furthermore, cotransfection of pTSKL(+39/+58) with the constitutively active STAT3-C, but not the constitutively inactive STAT3-KR mutant, significantly increased the expression of luciferase in all three cell lines (Fig. 4F), indicating that the STAT3-binding site is the minimal GIV promoter sequence that directs expression of the luciferase reporter gene. These results demonstrate that binding of STAT3 between +39 and +58 of the GIV promoter is both necessary and sufficient for the activation of GIV transcription.
STAT3 Activation Correlates with GIV Expression in Breast Carcinomas
GIV expression in breast and colorectal tumors and tumor-derived cell lines has recently been shown to positively correlate with increasing tumor aggressiveness (2, 12, 16) and to serve as a poor prognosticator and a predictor of distant metastasis (12, 15). Similarly, separate studies have established that STAT3 is constitutively activated in breast and colorectal tumors and tumor-derived cell lines and that elevated levels of tyrosine 705-phosphorylated STAT3 (Tyr(P)-705 STAT3) in patients with breast and colorectal cancer correlated with poor outcome (36–38). We asked whether increasing GIV expression during breast cancer progression that we previously observed in 21T series tumor cells (12) is also accompanied by enhanced STAT3 activation as determined by tyrosine phosphorylation at Tyr-705. We found that phosphorylated STAT3 progressively increased with increasing GIV protein (Fig. 5A) and mRNA expression (Fig. 1D), which correlates with their previously documented (39) increasing invasiveness and metastatic potential. Next we analyzed the expression of GIV and nuclear translocation of pSTAT3(Tyr-705) proteins in breast tumors in situ by immunohistochemistry. We analyzed a total of 45 samples (15 carcinomas in situ, 15 invasive ductal carcinomas without lymph node metastasis, and 15 invasive ductal carcinomas with lymph node metastasis). The expression of GIV protein as determined by a composite score of the intensity of staining and the percentage of tumor cells that stained positive (see “Experimental Procedures”) increased significantly with cancer progression: 26% in the carcinoma in situ group, 53% in the invasive tumors without lymphatic spread, and 73% in the invasive tumors with lymphatic spread (Fig. 5B and Table 1). Similarly, the abundance of pSTAT3 protein in the nucleus also increased significantly with cancer progression: 33% in the carcinoma in situ group, 60% in the invasive tumors without lymphatic spread, and 86% in the invasive tumors with lymphatic spread (Fig. 5B and Table 2). As expected, pSTAT3, which represents the active pool of STAT3, predominantly showed a nuclear staining pattern. These results indicate that expression of GIV and activation of STAT3 are both increased significantly during breast cancer progression. Furthermore, Fisher's exact test demonstrates a positive association between GIV expression and the abundance of nuclear pSTAT3 (p = 0.04) (Table 3), indicating that GIV is highly expressed often in those breast carcinomas in which STAT3 activation is also enhanced. These results suggest that STAT3 activation may trigger up-regulated expression of GIV during metastatic progression of cancer and are consistent with our prior results that activation of STAT3 is required for transcriptional activation of the GIV gene in invasive cancer cells.
FIGURE 5.

Expression of GIV protein increases during metastatic progression of breast carcinomas. A, whole cell lysates of 21T series of human mammary cells (16N, NT, and MT2) were analyzed for GIV, phospho-Tyr-705 STAT3 (p STAT3), total STAT3 (t STAT3), Gαi3, and tubulin by immunoblotting (IB). B, the abundance of GIV (top) and pSTAT3 (bottom) was analyzed in equal numbers of human breast carcinomas representing three stages during metastatic progression, carcinoma in situ (left), invasive ductal carcinoma without lymph node metastasis (middle), and invasive ductal carcinoma with lymph node metastasis (right). Shown here are representative images of three tumors from separate patients. GIV shows nuclear and cytosolic staining, whereas pSTAT3 is predominantly nuclear. Although carcinomas in situ stained mostly negative for both GIV and pSTAT3, invasive ductal carcinomas frequently stained positive for both (see Tables 1–3). Both the intensity of staining and the percentage of tumor cells that stained positive for GIV and pSTAT3 increased with tumor progression. Insets display a region in the field of the tumor enlarged by digital magnification. Original magnifications, 20×.
TABLE 1.
GIV expression as determined by cytosolic and nuclear staining (GIV-positive) of tumor epithelium correlates with tumor aggressiveness
Tumors were scored as either negative or positive for GIV staining as described under “Experimental Procedures.” The p value was determined by χ2 test. LN, lymph node; pos, positive; neg, negative.
| Histologic type | n | GIV-pos | GIV-neg | Percent GIV-pos | χ2 value | p value |
|---|---|---|---|---|---|---|
| Carcinoma in situ | 15 | 4 | 11 | 26.67 | 6.581 | 0.037 |
| Invasive ductal carcinoma without LN metastasis | 15 | 7 | 8 | 53.33 | ||
| Invasive ductal carcinoma with LN metastasis | 15 | 11 | 4 | 73.33 |
TABLE 2.
STAT3 activity as determined by the presence of nuclear pSTAT3 (pSTAT3-positive) in tumor epithelium correlates with tumor aggressiveness
Tumors were scored as either negative or positive for pSTAT3(Tyr-705) staining as described under “Experimental Procedures.” The p value was determined by χ2 test. LN, lymph node; pos, positive; neg, negative.
| Histologic type | n | pSTAT3-pos | pSTAT3-neg | Percent pSTAT3-pos | χ2 value | p value |
|---|---|---|---|---|---|---|
| Carcinoma in situ | 15 | 5 | 10 | 33.33 | 8.889 | 0.028 |
| Invasive ductal carcinoma without LN metastasis | 15 | 9 | 6 | 60 | ||
| Invasive ductal carcinoma with LN metastasis | 15 | 13 | 2 | 86.67 |
TABLE 3.
STAT3 activity and GIV expression in breast carcinomas correlate significantly
The contingency analysis (Fisher's exact test) comparing GIV expression and STAT3 activation (as determined by the presence of nuclear pSTAT3(Tyr-705)) in 45 breast carcinomas representing various clinical stages of the disease is shown. LN, lymph node; pos, positive; neg, negative.
| n | GIV-pos | GIV-neg | r value | p value | |
|---|---|---|---|---|---|
| pSTAT3-neg | 18 | 8 | 10 | .03 | 0.0446 |
| pSTAT3-pos | 27 | 20 | 7 |
GIV Enhances STAT3 Activation via Its GEF Motif
We previously reported (2) that the C terminus of GIV directly binds and modulates EGF receptor signaling at the plasma membrane and demonstrated that the presence or absence of the C terminus of GIV, more specifically an intact GEF motif within the C terminus, is a key determinant of the downstream signaling programs and tumor cell phenotype. When the GEF motif of GIV is intact, GIV couples RTKs to Gαi, these G proteins are activated via the GEF function of GIV in the vicinity of ligand-activated RTKs, and the motogenic PI3K-Akt and PLCγ1 signaling pathways are preferentially enhanced. When the GEF motif of GIV is disrupted by a specific mutagenesis, RTKs are no longer coupled to activation of Gαi, and mitogenic MAPK and STAT5 signaling pathways are preferentially propagated. Of note, in the absence of the entire GIV molecule (i.e. when depleted by siRNA), RTK signaling is inhibited across all downstream pathways examined (2). Given the specificity of the GEF motif of GIV as a key determinant of Gαi activity and RTK signaling programs in cells, we next asked whether and how the presence or absence of an intact GIV GEF motif alters STAT3 signaling. To this end, we used a previously characterized set of HeLa cell lines (2) stably expressing siRNA-resistant GIV constructs, either GIV-WT (HeLa-GIV-WT) or the GEF-deficient F1685A mutant (HeLa-GIV-FA); depleted them of endogenous GIV by siRNA; and analyzed their ability to enhance STAT3 phosphorylation in response to EGF stimulation or scratch wounding. Consistent with our previous report (2), HeLa-GIV-WT, but not HeLa-GIV-FA, enhanced Akt phosphorylation, whereas ERK activation was sustained longer (30 min) in HeLa-GIV-FA as compared with HeLa-GIV-WT cells. In the case of STAT3, its phosphorylation at both Tyr-705 and Ser-727 peaked at 5 min and was enhanced ∼3-fold more in HeLa-GIV-WT cells compared with a blunt response in HeLa-GIV-FA cells (Fig. 6, A and B). HeLa-GIV-WT, but not HeLa-GIV-FA, cells also enhanced the phosphorylation of STAT3 at Tyr-705 and Ser-727 and thereby its activity after scratch wounding (Fig. 6C). Furthermore, nuclear translocation of STAT3, which occurs after its activation (35), was seen exclusively in HeLa-GIV-WT, but not in HeLa-GIV-FA, cells (Fig. 6D and supplemental Fig. S7). Collectively these results indicate that the GEF motif of GIV is required for enhancement of STAT3 activity.
FIGURE 6.

GIV enhances STAT3 activation via its GEF motif. A–C, HeLa-GIV-WT, but not HeLa-GIV-FA, cells enhance STAT3 activation in response to EGF and scratch wounding. A, serum-starved HeLa WT and FA cell lines were stimulated with 50 nm EGF for the indicated time periods, and whole cell lysates were analyzed for total (t) and phospho (p)-Tyr-705 and -Ser-727 STAT3, Akt, ERK1/2, and tubulin by immunoblotting (IB). B, kinetics of EGF-initiated STAT3 signaling in HeLa WT and FA cell lines in A was determined by phospho-Tyr-705 STAT3:total STAT3 ratios at each time point after EGF stimulation and expressed as absolute values. The p value was calculated by comparing STAT3 activity in HeLa-GIV-WT and HeLa-GIV-FA cells at 6 h postwounding. C, monolayers of HeLa-GIV-WT and HeLa-GIV-FA cells were scratch-wounded and subsequently harvested at 6 h. Whole cell lysates were prepared and analyzed for Tyr(P)-705 and Ser(P)-727 STAT3 and tubulin by immunoblotting. D, upon activation, STAT3 localizes to the nucleus in HeLa-GIV-WT, but not in HeLa-GIV-FA, cells after ligand stimulation. HeLa cell lines were stimulated with 50 nm EGF for 5 min and costained for STAT3 (red) and the nucleus (DAPI; blue). Bar, 10 μm. E and F, HeLa-GIV-WT, but not HeLa-GIV-FA cells up-regulate GIV mRNA (E) and protein (F) levels after scratch wounding. Confluent monolayers of HeLa WT and FA cell lines were scratch-wounded, harvested at the indicated time points, and analyzed for GIV mRNA and protein by qPCR and immunoblotting, respectively. The p value in E was calculated by comparing GIV mRNA in HeLa-GIV-WT and HeLa-GIV-FA cells at 6 h postwounding. G–I, the GEF function of GIV is required for induction of STAT3 target genes Bcl-2, MMP2, and MMP9 after scratch wounding. Monolayers of HeLa-GIV-WT and HeLa-GIV-FA cells were scratch-wounded and subsequently harvested at 6 h. mRNA levels of Bcl-2 (G), MMP2 (H), and MMP9 (I) were analyzed by qPCR. Error bars represent S.D. *, p < 0.05; **, p < 0.01.
Because the GEF motif of GIV enhances STAT3 activation and GIV is a direct transcriptional target of STAT3, we asked whether GIV can regulate its own expression via its GEF motif by modulating STAT3 activity. We found that scratch wound-induced up-regulation of GIV mRNA (Fig. 6E) and protein (Fig. 6F; by ∼2.2-fold) was abolished in HeLa-GIV-FA cells as compared with HeLa-GIV-WT cells and vector-transfected control cells, indicating that the GEF motif enhances the induction of GIV expression during wound healing. We hypothesized that, besides GIV, expression of other STAT3 target genes that are known to regulate tumor invasion/metastasis may also be affected in HeLa cell lines. That is indeed the case because we found that the levels of expression of Bcl-2, MMP-2, and MMP-9, three metastasis-related genes (40–42), were higher in HeLa-GIV-WT compared with HeLa-GIV-FA cells after scratch wounding (Fig. 6, G–I). Based on these findings, we propose a working model wherein GIV may autoregulate its own expression and the expression of a variety of other metastasis-related genes targeted by STAT3 via modulation of STAT3 activation by its GEF motif (Fig. 7, A and B).
FIGURE 7.
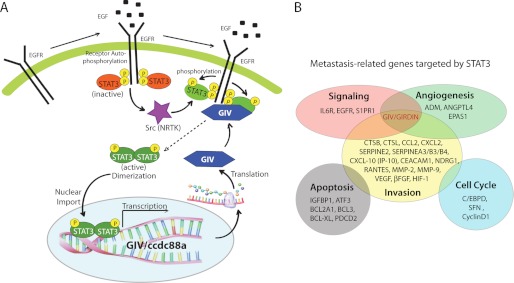
Implications of the forward feedback mutual regulation between GIV and STAT3. A, proposed model for the interplay between GIV and STAT3 during wound healing and cancer invasion. Upon ligand stimulation of EGF receptor (EGFR) and other growth factor receptors, inactive STAT3 (red oval) is recruited to the cytoplasmic tail of the activated receptor and subsequently activated (green oval) by phosphorylation at Tyr-705 by receptor and non-receptor tyrosine kinases (purple star). STAT3 dimerizes, enters the nucleus, and activates transcription of GIV gene (ccdc88a). GIV (blue hexagon) enhances receptor-initiated PI3K signals and triggers cytoskeletal rearrangement during wound healing and cancer invasion. GIV also directly binds to the receptor tail (2) and enhances STAT3 activation via its GEF motif, thereby creating a positive/forward feedback loop (interrupted arrow). B, GIV is a multifunctional, direct transcriptional target of STAT3. A schematic representation of various direct transcriptional targets of STAT3 is shown in which each target gene is grouped according to its gene ontology classification. GIV, which is a cytoskeleton-associated protein that has intrinsic enzyme activity (i.e. GEF), has to date been implicated in signal enhancement downstream of multiple RTKs and G protein-coupled receptors (1), angiogenesis (10), and cell invasion (13). Because GIV enhances STAT3 activation, it could regulate the expression of one or more of the genes shown here that are direct targets of STAT3.
DISCUSSION
Here we demonstrate that transcription of GIV is up-regulated during epithelial wound healing and metastatic progression of cancer, and the key finding in this work is the identification of STAT3 as one of the major and specific transcription factors that up-regulates GIV expression during both processes. Using three different models of epithelial wounding, scratch wounding on a monolayer of HeLa epithelial cells, surgical skin graft wounding in human, and burn injury in murine skin, we showed here that GIV mRNA and protein expression is up-regulated during wound healing. We also demonstrated a similar increase in GIV expression during cancer progression using two different approaches in two different types of cancers, isolated tumor cell lines (21T series of breast cancer, MDA-MB 231 breast cancer, and DLD-1 colorectal cancer cells) and breast tumors in situ. By analysis of microarray databases, we extended our initial observation of GIV induction in breast (current work) and colorectal cancer (2, 12) progression to other solid tumor types. We conclude that GIV is up-regulated during two biological processes, epithelial wound healing and cancer progression.
We also demonstrate that GIV expression is up-regulated due to increased transcription mediated directly by STAT3. We demonstrate that STAT3 is recruited to an identical region of the GIV promoter in three different cell lines, HeLa epithelial cells responding to scratch wounds and the breast and colorectal cancer cells MDA-MB 231 and DLD-1, respectively. Using all three cell lines, we showed that levels of STAT3 had a direct bearing on the levels of GIV mRNA and protein; i.e. overexpression of STAT3 increased GIV expression, whereas depletion of STAT3 decreased GIV expression. We also demonstrated that activation of STAT3 is essential for up-regulation of GIV expression upon scratch wounding and in invasive cancer cells. That a mutant STAT3 deficient in phosphorylation at Tyr-705 also enhanced GIV expression is consistent with others' findings that STAT3 can activate gene transcription independently of phosphorylation at Tyr-705 (43, 44). We conclude that STAT3 up-regulates GIV expression in epithelial cells responding to wounding as well as in highly invasive breast and colorectal cancer cells.
Considerable evidence has independently implicated STAT3 (20) and GIV (1) as major players during wound healing and cancer invasion/metastasis, two processes with common pathophysiology (29). Although STAT3 exerts its effects via its wide range of transcriptional targets (see Fig. 7B legend), the multidomain signal transducer GIV affects an overlapping set of cellular processes by modulating signals triggered by multiple members of two diverse classes of receptors, G protein-coupled and growth factor receptors (1). Thus, by demonstrating here that STAT3 directly regulates GIV transcription, we have discovered another novel molecular link between healing wounds and invading cancers.
The clinical significance of our discovery is 2-fold both in the realm of diagnostics and therapeutics. The diagnostic significance stems from the fact that both STAT3 (36–38) and GIV (2, 12, 15, 16) play significant roles in metastatic progression of breast and colorectal cancers and have been shown to serve as valuable independent prognosticators of survival among cancer patients. It is possible that a diagnostic platform that combines both may perform even better. Because STAT3 can be activated by cytokines released during inflammation and injury (45), tumors that are strongly positive for active STAT3 and GIV could represent those that are driven by inflammation and bear the worst prognosis (46). The therapeutic significance lies in the fact that STAT3 has long been recognized as a powerful therapeutic target in multiple cancers (18, 47–50), and small molecule inhibitors of STAT3 have already been tested in several clinical trials (51). Our discovery that GIV is a direct transcriptional target of STAT3 implies that STAT3 inhibitors at least in part work via repression of GIV expression and suppression of GIV-dependent signaling.
It is well accepted that the classic mechanism by which STAT3 regulates gene transcription is by direct interaction with the promoter region of target genes (52). Consistently, we found that STAT3 regulates GIV transcription by directly binding a single site on the GIV promoter in three different cell types, HeLa, MDA-MB 231, and DLD-1. Sequence comparisons indicate that this binding site for STAT3 on the GIV promoter is non-canonical: it deviates from the consensus, but key resemblances to the core of the consensus are preserved, which allowed it to be picked up by prediction programs. This is not unexpected because STAT3 is commonly found to activate the transcription of target genes via such non-canonical binding sites on the promoters of other genes, e.g. CCAAT/enhancer-binding protein δ (53) and proopiomelanocortin (54), where inducible STAT3 binding has been detected in vivo by chromatin immunoprecipitation assays. We also demonstrate that STAT3 binding is both necessary and sufficient for transcription activity via the GIV promoter in luciferase assays: targeted deletion or mutagenesis of the binding site reduced transcription activity, and selective expression of the binding site enhanced transcription activity. Validation of the exact binding site for STAT3 on the GIV promoter creates an opportunity for targeted therapy using custom-designed triplex-forming oligos as tools to selectively interfere with the binding of a given transcription factor to one target gene of interest (55). Such a selective approach will allow STAT3 to regulate other target genes while annihilating its ability to up-regulate GIV. Thus, our discovery not only represents a major advance in our understanding of how GIV, a metastasis-related protein, is regulated but also offers a powerful therapeutic target for down-regulating GIV-dependent prometastatic signaling during cancer progression. Of note, depletion of STAT3 did not completely abolish GIV transcription, suggesting that STAT3 may work cooperatively with other transcription factors and cofactor complex to effectively and efficiently fine tune GIV expression in response to a variety of external stimuli. Such cooperation has been well documented in the case of several genes that STAT3 co-regulates with other transcription factors either via binding at distinct sites (56, 57) or on overlapping sites (54). Because GIV is expressed ubiquitously and is implicated in multiple biological processes, i.e. autophagy, development, neuronal migration, vascular repair, etc. (1), it is possible that transcription factors other than STAT3 may regulate GIV expression in different cell types under different circumstances.
We also demonstrate that GIV enhances STAT3 activation via its C-terminal GEF motif. In cells expressing GIV-WT, growth factor signals were modulated via an intact GEF motif, and a persistent high level of STAT3 activation was induced, which is in sharp contrast to the transient and blunted nature of STAT3 activation induced in those cells expressing GIV-FA in which the critical GEF motif is disabled. The effect of the GEF motif of GIV on the STAT3 pathway is in striking contrast to the effect it exerts on another member of the STAT family, STAT5b; the latter was found to be suppressed in the presence of an intact GEF motif and enhanced in its absence (2). This differential STAT signaling we observed in GIV-WT and GIV-FA cells is in keeping with their respective cellular phenotypes (2, 21) and the contrasting transcriptional profiles of STAT3 versus STAT5 (58): cells that express GIV-WT or exhibit enhanced STAT3 signaling migrate/invade rapidly but proliferate slowly, whereas those that express GIV-FA or exhibit enhanced STAT5 signaling proliferate rapidly but migrate/invade slowly. We conclude that GIV, more specifically the presence or absence of the GEF motif of GIV, has a profound effect on the STAT signaling pathway, and it is possible that the phenomenon of migration-proliferation dichotomy we observed previously in GIV-WT and GIV-FA cells (2) is in part contributed by their preferential activation of STAT3 and STAT5, respectively.
The implications of STAT3 activation by the C-terminal GEF motif of GIV are far reaching. First, we demonstrate that activation of STAT3 by GIV in turn affects the expression of GIV, which we show here is a direct transcriptional target of STAT3. We conclude that GIV autoregulates its expression by enhancing STAT3 activity via its GEF motif. Second, we demonstrate that STAT3 activation and GIV expression show statistically significant correlation at every stage of breast cancer progression. In non-invasive tumors during early growth in which the C terminus of GIV is alternatively spliced such that it lacks the GEF motif (2), the levels of STAT3 activation are low. By contrast, in highly invasive tumors later during metastatic progression in which full-length GIV with an intact GEF motif is overexpressed, the levels of STAT3 activation are high. We conclude that specificity and intensity of the GIV-dependent STAT3 axis of signaling, which has diverse biological consequences, are in part determined by the abundance of full-length GIV molecules with intact GEF motifs in the C terminus (1). Third, the ability of GIV to enhance STAT3 activation, which in turn enhances GIV transcription, sets up a platform for mutual regulation: by virtue of its ability to enhance the activation of STAT3, GIV, which exists in varying abundance and as multiple isoforms (some with and some without the C-terminal GEF motif), can affect which cells activate STAT3 and when they activate it. Similarly, by virtue of its ability to directly up-regulate the expression of full-length GIV, STAT3, whose activation is modulated by a variety of cytokines and growth factors, can affect which cells express GIV and when to up-regulate it. Fourth, although expressed ubiquitously at low levels (22), we show here that the abundance of full-length GIV increases under circumstances such as scratch wound injury and during cancer progression. It is likely that increasing copies of the GEF motif of GIV under those circumstances proportionately enhance STAT3 activation, which leads to a further increase in GIV transcription and further persistent activation of STAT3 and so on and so forth within a positive feedback loop. We also demonstrated that another consequence of such a positive feedback loop is the ability of GIV to affect the expression of key STAT3 target genes. GIV may similarly affect other direct and indirect transcriptional targets of STAT3 (see Fig. 7B and legend) and thereby cast a broader influence on signaling networks that drive metastasis via regulation of STAT3, a central regulator of tumor metastasis (19). Of note, some of these transcriptional targets of STAT3, e.g. RTKs (EGF receptor), G protein-coupled receptors (sphingosine 1-phosphate receptor-1), and growth factors (VEGF), are proteins that could further enhance STAT3 signaling via the receptor-GIV-G protein axis. It is noteworthy that the feedback loop between GIV and STAT3 that we report here is not unusual and is showcased by multiple proteins that are direct or indirect targets of STAT3, e.g. sphingosine 1-phosphate receptor-1 (59), p53 (60), β-catenin (61), c-Met/hepatocyte growth factor (62), and Rho kinase ROCK (63).
Clues as to how the GEF motif of GIV may affect STAT3 activation are provided by our previous work in which we explored the mechanisms by which the GEF motif of GIV modulates growth factor signaling via activation of Gαi (2). Based on these, we propose (see Fig. 7A and legend) that the recruitment of a Gαi-GIV complex to RTKs enhances tyrosine phosphorylation and subsequent activation of STAT3 by regulating signaling complexes assembled at the cytoplasmic tail of ligand-activated receptors at the plasma membrane. Because a small pool of GIV is also seen inside the nucleus (3), it is possible that GIV could interact with STAT3 inside the nucleus. Whether such STAT3-GIV interaction occurs and whether such interaction directly enhances STAT3 dimerization and/or DNA binding and transcriptional activity of STAT3 remain to be tested. Regardless, our study has identified a positive feedback mechanism that facilitates persistent STAT3 activation frequently observed in healing wounds and invading cancers (64, 65) at least in the setting where GIV-dependent growth factor signaling, which is crucial for cancer progression as well as during wound healing, is the driving force.
In conclusion, our discovery that STAT3 up-regulates GIV transcription and that GIV in turn enhances STAT3 activation via its GEF function provides evidence for and mechanistic insights into how STAT3 activation is directly integrated with the RTK-GIV-G protein signaling axis. The forward feedback regulation we describe here between GIV and STAT3 may have profound biological and therapeutic implications for cancer and epithelial regeneration/repair.
Acknowledgments
We thank M. G. Farquhar (University of California, San Diego) and Mikel Garcia-Marcos (Boston University) for scientific advice and thoughtful comments during the preparation of this manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants 5 T32 GM007198 and T32 GM008666 (to M. L.). This work was also supported by the Burroughs Wellcome Fund, the Doris Duke Charitable Foundation, and American Cancer Society Grant ACS-IRG 70-002 (to P. G.).

This article contains supplemental Figs. S1–S7.
- GIV
- Gα-interacting vesicle-associated protein
- GEF
- guanine nucleotide exchange factor
- RTK
- receptor tyrosine kinase
- MMP
- matrix metalloproteinase
- Bcl-2
- B-cell lymphoma 2
- Scr
- scrambled
- qPCR
- quantitative PCR
- pSTAT3
- phospho-STAT3
- H3K
- histone H3 lysine
- me3
- trimethylation
- me1
- monomethylation
- KR
- K685R
- FA
- F1685A.
REFERENCES
- 1. Ghosh P., Garcia-Marcos M., Farquhar M. G. (2011) GIV/Girdin is a rheostat that fine-tunes growth factor signals during tumor progression. Cell Adh. Migr. 5, 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ghosh P., Beas A. O., Bornheimer S. J., Garcia-Marcos M., Forry E. P., Johannson C., Ear J., Jung B. H., Cabrera B., Carethers J. M., Farquhar M. G. (2010) A Gαi-GIV molecular complex binds epidermal growth factor receptor and determines whether cells migrate or proliferate. Mol. Biol. Cell 21, 2338–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ghosh P., Garcia-Marcos M., Bornheimer S. J., Farquhar M. G. (2008) Activation of Gαi3 triggers cell migration via regulation of GIV. J. Cell Biol. 182, 381–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamaguchi M., Suyari O., Nagai R., Takahashi M. (2010) dGirdin a new player of Akt /PKB signaling in Drosophila melanogaster. Front. Biosci. 15, 1164–1171 [DOI] [PubMed] [Google Scholar]
- 5. Puseenam A., Yoshioka Y., Nagai R., Hashimoto R., Suyari O., Itoh M., Enomoto A., Takahashi M., Yamaguchi M. (2009) A novel Drosophila Girdin-like protein is involved in Akt pathway control of cell size. Exp. Cell Res. 315, 3370–3380 [DOI] [PubMed] [Google Scholar]
- 6. Bradshaw N. J., Porteous D. J. (2012) DISC1-binding proteins in neural development, signalling and schizophrenia. Neuropharmacology 62, 1230–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Enomoto A., Asai N., Namba T., Wang Y., Kato T., Tanaka M., Tatsumi H., Taya S., Tsuboi D., Kuroda K., Kaneko N., Sawamoto K., Miyamoto R., Jijiwa M., Murakumo Y., Sokabe M., Seki T., Kaibuchi K., Takahashi M. (2009) Roles of disrupted-in-schizophrenia 1-interacting protein girdin in postnatal development of the dentate gyrus. Neuron 63, 774–787 [DOI] [PubMed] [Google Scholar]
- 8. Miyake H., Maeda K., Asai N., Shibata R., Ichimiya H., Isotani-Sakakibara M., Yamamura Y., Kato K., Enomoto A., Takahashi M., Murohara T. (2011) The actin-binding protein Girdin and its Akt-mediated phosphorylation regulate neointima formation after vascular injury. Circ. Res. 108, 1170–1179 [DOI] [PubMed] [Google Scholar]
- 9. Garcia-Marcos M., Ear J., Farquhar M. G., Ghosh P. (2011) A GDI (AGS3) and a GEF (GIV) regulate autophagy by balancing G protein activity and growth factor signals. Mol. Biol. Cell 22, 673–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kitamura T., Asai N., Enomoto A., Maeda K., Kato T., Ishida M., Jiang P., Watanabe T., Usukura J., Kondo T., Costantini F., Murohara T., Takahashi M. (2008) Regulation of VEGF-mediated angiogenesis by the Akt/PKB substrate Girdin. Nat. Cell Biol. 10, 329–337 [DOI] [PubMed] [Google Scholar]
- 11. Enomoto A., Murakami H., Asai N., Morone N., Watanabe T., Kawai K., Murakumo Y., Usukura J., Kaibuchi K., Takahashi M. (2005) Akt/PKB regulates actin organization and cell motility via Girdin/APE. Dev. Cell 9, 389–402 [DOI] [PubMed] [Google Scholar]
- 12. Garcia-Marcos M., Jung B. H., Ear J., Cabrera B., Carethers J. M., Ghosh P. (2011) Expression of GIV/Girdin, a metastasis-related protein, predicts patient survival in colon cancer. FASEB J. 25, 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jiang P., Enomoto A., Jijiwa M., Kato T., Hasegawa T., Ishida M., Sato T., Asai N., Murakumo Y., Takahashi M. (2008) An actin-binding protein Girdin regulates the motility of breast cancer cells. Cancer Res. 68, 1310–1318 [DOI] [PubMed] [Google Scholar]
- 14. Garcia-Marcos M., Ghosh P., Farquhar M. G. (2009) GIV is a nonreceptor GEF for Gαi with a unique motif that regulates Akt signaling. Proc. Natl. Acad. Sci. U.S.A. 106, 3178–3183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liu C., Zhang Y., Xu H., Zhang R., Li H., Lu P., Jin F. (2012) Girdin protein: a new potential distant metastasis predictor of breast cancer. Med. Oncol. 29, 1554–1560 [DOI] [PubMed] [Google Scholar]
- 16. Ling Y., Jiang P., Cui S. P., Ren Y. L., Zhu S. N., Yang J. P., Du J., Zhang Y., Liu J. Y., Zhang B. (2011) Clinical implications for girdin protein expression in breast cancer. Cancer Invest. 29, 405–410 [DOI] [PubMed] [Google Scholar]
- 17. Natsume A., Kato T., Kinjo S., Enomoto A., Toda H., Shimato S., Ohka F., Motomura K., Kondo Y., Miyata T., Takahashi M., Wakabayashi T. (2012) Girdin maintains the stemness of glioblastoma stem cells. Oncogene 31, 2715–2724 [DOI] [PubMed] [Google Scholar]
- 18. Darnell J. E. (2005) Validating Stat3 in cancer therapy. Nat. Med. 11, 595–596 [DOI] [PubMed] [Google Scholar]
- 19. Devarajan E., Huang S. (2009) STAT3 as a central regulator of tumor metastases. Curr. Mol. Med. 9, 626–633 [DOI] [PubMed] [Google Scholar]
- 20. Dauer D. J., Ferraro B., Song L., Yu B., Mora L., Buettner R., Enkemann S., Jove R., Haura E. B. (2005) Stat3 regulates genes common to both wound healing and cancer. Oncogene 24, 3397–3408 [DOI] [PubMed] [Google Scholar]
- 21. Gu L., Dagvadorj A., Lutz J., Leiby B., Bonuccelli G., Lisanti M. P., Addya S., Fortina P., Dasgupta A., Hyslop T., Bubendorf L., Nevalainen M. T. (2010) Transcription factor Stat3 stimulates metastatic behavior of human prostate cancer cells in vivo, whereas Stat5b has a preferential role in the promotion of prostate cancer cell viability and tumor growth. The Am. J. Pathol. 176, 1959–1972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Le-Niculescu H., Niesman I., Fischer T., DeVries L., Farquhar M. G. (2005) Identification and characterization of GIV, a novel Gαi/s-interacting protein found on COPI, endoplasmic reticulum-Golgi transport vesicles. J. Biol. Chem. 280, 22012–22020 [DOI] [PubMed] [Google Scholar]
- 23. Lin C., Ear J., Pavlova Y., Mittal Y., Kufareva I., Ghassemian M., Abagyan R., Garcia-Marcos M., Ghosh P. (2011) Tyrosine phosphorylation of the Gα-interacting protein GIV promotes activation of phosphoinositide 3-kinase during cell migration. Sci. Signal. 4, ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Garcia-Marcos M., Kietrsunthorn P. S., Pavlova Y., Adia M. A., Ghosh P., Farquhar M. G. (2012) Functional characterization of the guanine nucleotide exchange factor (GEF) motif of GIV protein reveals a threshold effect in signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 1961–1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mittal Y., Pavlova Y., Garcia-Marcos M., Ghosh P. (2011) Src homology domain 2-containing protein-tyrosine phosphatase-1 (SHP-1) binds and dephosphorylates Gα-interacting, vesicle-associated protein (GIV)/Girdin and attenuates the GIV-phosphatidylinositol 3-kinase (PI3K)-Akt signaling pathway. J. Biol. Chem. 286, 32404–32415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kumar A., Choi K. H., Renthal W., Tsankova N. M., Theobald D. E., Truong H. T., Russo S. J., Laplant Q., Sasaki T. S., Whistler K. N., Neve R. L., Self D. W., Nestler E. J. (2005) Chromatin remodeling is a key mechanism underlying cocaine-induced plasticity in striatum. Neuron 48, 303–314 [DOI] [PubMed] [Google Scholar]
- 27. Park O. K., Schaefer L. K., Wang W., Schaefer T. S. (2000) Dimer stability as a determinant of differential DNA binding activity of Stat3 isoforms. J. Biol. Chem. 275, 32244–32249 [DOI] [PubMed] [Google Scholar]
- 28. Niu G., Wright K. L., Huang M., Song L., Haura E., Turkson J., Zhang S., Wang T., Sinibaldi D., Coppola D., Heller R., Ellis L. M., Karras J., Bromberg J., Pardoll D., Jove R., Yu H. (2002) Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21, 2000–2008 [DOI] [PubMed] [Google Scholar]
- 29. Dvorak H. F. (1986) Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. New Engl. J. Med. 315, 1650–1659 [DOI] [PubMed] [Google Scholar]
- 30. Nuutila K., Siltanen A., Peura M., Bizik J., Kaartinen I., Kuokkanen H., Nieminen T., Harjula A., Aarnio P., Vuola J., Kankuri E. (2012) Human skin transcriptome during superficial cutaneous wound healing. Wound Repair Regen., doi: 10.1111/j.1524-475X.2012.00831.x [DOI] [PubMed] [Google Scholar]
- 31. Feezor R. J., Paddock H. N., Baker H. V., Varela J. C., Barreda J., Moldawer L. L., Schultz G. S., Mozingo D. W. (2004) Temporal patterns of gene expression in murine cutaneous burn wound healing. Physiol. Genomics 16, 341–348 [DOI] [PubMed] [Google Scholar]
- 32. Band V., Zajchowski D., Swisshelm K., Trask D., Kulesa V., Cohen C., Connolly J., Sager R. (1990) Tumor progression in four mammary epithelial cell lines derived from the same patient. Cancer Res. 50, 7351–7357 [PubMed] [Google Scholar]
- 33. Bresalier R. S., Hujanen E. S., Raper S. E., Roll F. J., Itzkowitz S. H., Martin G. R., Kim Y. S. (1987) An animal model for colon cancer metastasis: establishment and characterization of murine cell lines with enhanced liver-metastasizing ability. Cancer Res. 47, 1398–1406 [PubMed] [Google Scholar]
- 34. Liddle F. J., Alvarez J. V., Poli V., Frank D. A. (2006) Tyrosine phosphorylation is required for functional activation of disulfide-containing constitutively active STAT mutants. Biochemistry 45, 5599–5605 [DOI] [PubMed] [Google Scholar]
- 35. Yuan Z. L., Guan Y. J., Chatterjee D., Chin Y. E. (2005) Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 307, 269–273 [DOI] [PubMed] [Google Scholar]
- 36. Diaz N., Minton S., Cox C., Bowman T., Gritsko T., Garcia R., Eweis I., Wloch M., Livingston S., Seijo E., Cantor A., Lee J. H., Beam C. A., Sullivan D., Jove R., Muro-Cacho C. A. (2006) Activation of stat3 in primary tumors from high-risk breast cancer patients is associated with elevated levels of activated SRC and survivin expression. Clin. Cancer Res. 12, 20–28 [DOI] [PubMed] [Google Scholar]
- 37. Morikawa T., Baba Y., Yamauchi M., Kuchiba A., Nosho K., Shima K., Tanaka N., Huttenhower C., Frank D. A., Fuchs C. S., Ogino S. (2011) STAT3 expression, molecular features, inflammation patterns, and prognosis in a database of 724 colorectal cancers. Clin. Cancer Res. 17, 1452–1462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kusaba T., Nakayama T., Yamazumi K., Yakata Y., Yoshizaki A., Inoue K., Nagayasu T., Sekine I. (2006) Activation of STAT3 is a marker of poor prognosis in human colorectal cancer. Oncol. Rep. 15, 1445–1451 [PubMed] [Google Scholar]
- 39. Qiao M., Iglehart J. D., Pardee A. B. (2007) Metastatic potential of 21T human breast cancer cells depends on Akt/protein kinase B activation. Cancer Res. 67, 5293–5299 [DOI] [PubMed] [Google Scholar]
- 40. Ma X. T., Wang S., Ye Y. J., Du R. Y., Cui Z. R. (2003) Relationship of Stat3 and its target gene products with malignancy in human colorectal carcinoma. Ai Zheng 22, 1135–1139 [PubMed] [Google Scholar]
- 41. Xie T. X., Wei D., Liu M., Gao A. C., Ali-Osman F., Sawaya R., Huang S. (2004) Stat3 activation regulates the expression of matrix metalloproteinase-2 and tumor invasion and metastasis. Oncogene 23, 3550–3560 [DOI] [PubMed] [Google Scholar]
- 42. Senft C., Priester M., Polacin M., Schröder K., Seifert V., Kögel D., Weissenberger J. (2011) Inhibition of the JAK-2/STAT3 signaling pathway impedes the migratory and invasive potential of human glioblastoma cells. J. Neurooncol. 101, 393–403 [DOI] [PubMed] [Google Scholar]
- 43. Yang J., Chatterjee-Kishore M., Staugaitis S. M., Nguyen H., Schlessinger K., Levy D. E., Stark G. R. (2005) Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 65, 939–947 [PubMed] [Google Scholar]
- 44. Yang J., Stark G. R. (2008) Roles of unphosphorylated STATs in signaling. Cell Res. 18, 443–451 [DOI] [PubMed] [Google Scholar]
- 45. Grivennikov S. I., Karin M. (2010) Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer. Cytokine Growth Factor Rev. 21, 11–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kuraishy A., Karin M., Grivennikov S. I. (2011) Tumor promotion via injury- and death-induced inflammation. Immunity 35, 467–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin L., Deangelis S., Foust E., Fuchs J., Li C., Li P. K., Schwartz E. B., Lesinski G. B., Benson D., Lü J., Hoyt D., Lin J. (2010) A novel small molecule inhibits STAT3 phosphorylation and DNA binding activity and exhibits potent growth suppressive activity in human cancer cells. Mol. Cancer 9, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Turkson J., Jove R. (2000) STAT proteins: novel molecular targets for cancer drug discovery. Oncogene 19, 6613–6626 [DOI] [PubMed] [Google Scholar]
- 49. Masciocchi D., Gelain A., Villa S., Meneghetti F., Barlocco D. (2011) Signal transducer and activator of transcription 3 (STAT3): a promising target for anticancer therapy. Future Med. Chem. 3, 567–597 [DOI] [PubMed] [Google Scholar]
- 50. Mankan A. K., Greten F. R. (2011) Inhibiting signal transducer and activator of transcription 3: rationality and rationale design of inhibitors. Expert Opin. Investig. Drugs 20, 1263–1275 [DOI] [PubMed] [Google Scholar]
- 51. Page B. D., Ball D. P., Gunning P. T. (2011) Signal transducer and activator of transcription 3 inhibitors: a patent review. Expert Opin. Ther. Pat. 21, 65–83 [DOI] [PubMed] [Google Scholar]
- 52. Darnell J. E., Jr. (1997) STATs and gene regulation. Science 277, 1630–1635 [DOI] [PubMed] [Google Scholar]
- 53. Cantwell C. A., Sterneck E., Johnson P. F. (1998) Interleukin-6-specific activation of the C/EBPδ gene in hepatocytes is mediated by Stat3 and Sp1. Mol. Cell. Biol. 18, 2108–2117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bousquet C., Zatelli M. C., Melmed S. (2000) Direct regulation of pituitary proopiomelanocortin by STAT3 provides a novel mechanism for immuno-neuroendocrine interfacing. J. Clin. Investig. 106, 1417–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Noonberg S. B., Scott G. K., Hunt C. A., Hogan M. E., Benz C. C. (1994) Inhibition of transcription factor binding to the HER2 promoter by triplex-forming oligodeoxyribonucleotides. Gene 149, 123–126 [DOI] [PubMed] [Google Scholar]
- 56. Kwon H., Thierry-Mieg D., Thierry-Mieg J., Kim H. P., Oh J., Tunyaplin C., Carotta S., Donovan C. E., Goldman M. L., Tailor P., Ozato K., Levy D. E., Nutt S. L., Calame K., Leonard W. J. (2009) Analysis of interleukin-21-induced Prdm1 gene regulation reveals functional cooperation of STAT3 and IRF4 transcription factors. Immunity 31, 941–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schiavone D., Dewilde S., Vallania F., Turkson J., Di Cunto F., Poli V. (2009) The RhoU/Wrch1 Rho GTPase gene is a common transcriptional target of both the gp130/STAT3 and Wnt-1 pathways. Biochem. J. 421, 283–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Walker S. R., Nelson E. A., Zou L., Chaudhury M., Signoretti S., Richardson A., Frank D. A. (2009) Reciprocal effects of STAT5 and STAT3 in breast cancer. Mol. Cancer Res. 7, 966–976 [DOI] [PubMed] [Google Scholar]
- 59. Lee H., Deng J., Kujawski M., Yang C., Liu Y., Herrmann A., Kortylewski M., Horne D., Somlo G., Forman S., Jove R., Yu H. (2010) STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 16, 1421–1428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Niu G., Wright K. L., Ma Y., Wright G. M., Huang M., Irby R., Briggs J., Karras J., Cress W. D., Pardoll D., Jove R., Chen J., Yu H. (2005) Role of Stat3 in regulating p53 expression and function. Mol. Cell. Biol. 25, 7432–7440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Armanious H., Gelebart P., Mackey J., Ma Y., Lai R. (2010) STAT3 upregulates the protein expression and transcriptional activity of β-catenin in breast cancer. Int. J. Clin. Exp. Pathol. 3, 654–664 [PMC free article] [PubMed] [Google Scholar]
- 62. Syed Z. A., Yin W., Hughes K., Gill J. N., Shi R., Clifford J. L. (2011) HGF/c-met/Stat3 signaling during skin tumor cell invasion: indications for a positive feedback loop. BMC Cancer 11, 180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sanz-Moreno V., Gaggioli C., Yeo M., Albrengues J., Wallberg F., Viros A., Hooper S., Mitter R., Féral C. C., Cook M., Larkin J., Marais R., Meneguzzi G., Sahai E., Marshall C. J. (2011) ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 20, 229–245 [DOI] [PubMed] [Google Scholar]
- 64. Sano S., Chan K. S., DiGiovanni J. (2008) Impact of Stat3 activation upon skin biology: a dichotomy of its role between homeostasis and diseases. J. Dermatol. Sci. 50, 1–14 [DOI] [PubMed] [Google Scholar]
- 65. Bromberg J. (2002) Stat proteins and oncogenesis. J. Clin. Investig. 109, 1139–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]