

Background: The transcription factor T-bet is pivotal for initiation of Th1-related immunoactivation. Identification of novel genes directly regulated by T-bet is crucial.
Results: Genome-wide analysis and subsequent experiments revealed that T-bet up-regulates IL-36γ/IL-1F9 in myeloid cells.
Conclusion: IL-1-related IL-36γ is a direct T-bet target in myeloid cells.
Significance: Observations suggest that IL-36γ, besides IFNγ, contributes to T-bet functions in immunopathology.
Keywords: Dendritic Cells, Gene Regulation, Innate Immunity, Interleukin, Macrophages, Interleukin-1, Interleukin-36, T-bet
Abstract
By concerted action in dendritic (DC) and T cells, T-box expressed in T cells (T-bet, Tbx21) is pivotal for initiation and perpetuation of Th1 immunity. Identification of novel T-bet-regulated genes is crucial for further understanding the biology of this transcription factor. By combining siRNA technology with genome-wide mRNA expression analysis, we sought to identify new T-bet-regulated genes in predendritic KG1 cells activated by IL-18. One gene robustly dependent on T-bet was IL-36γ, a recently described novel IL-1 family member. Promoter analysis revealed a T-bet binding site that, along with a κB site, enables efficient IL-36γ induction. Using knock-out animals, IL-36γ reliance on T-bet was extended to murine DC. IL-36γ expression by human myeloid cells was confirmed using monocyte-derived DC and M1 macrophages. The latter model was employed to substantiate dependence of IL-36γ on endogenous T-bet in human primary cells. Ectopic expression of T-bet likewise mediated IL-36γ production in HaCaT keratinocytes that otherwise lack this transcription factor. Additional experiments furthermore revealed that mature IL-36γ has the capability to establish an inflammatory gene expression profile in human primary keratinocytes that displays enhanced mRNA levels for TNFα, CCL20, S100A7, inducible NOS, and IL-36γ itself. Data presented herein shed further light on involvement of T-bet in innate immunity and suggest that IL-36γ, besides IFNγ, may contribute to functions of this transcription factor in immunopathology.
Introduction
T-box expressed in T cells (T-bet, Tbx21)2 is regarded as a transcription factor crucial for Th1 lineage commitment and subsequent Th1-like immunoactivation that culminates in ample production of the corresponding signature cytokine IFNγ (1–3). The impressive potency of this transcription factor to determine T cell behavior is highlighted by enforced IFNγ production in committed Th2 cells that underwent ectopic expression of T-bet (3). Increased susceptibility of T-bet knock-out mice for Salmonella (4) or Mycobacterium tuberculosis (5) infections is manifest and in accordance with the pivotal role of Th1-like immunoactivation for anti-bacterial host defense. However, T-bet expression is not confined to T cells but has been detected likewise in additional hematopoietic cell types, among others, IFNγ-activated human monocytes and monocyte-derived dendritic cells (DC) (6). DC derived from T-bet-deficient mice actually display diminished capability to produce IFNγ and to install Th1 differentiation (7, 8). Accordingly, Th1 priming is enhanced in the context of T-bet-overexpressing human DC acting on naïve T cells (9). Altogether, T-bet promotes Th1 immune responses by apparently acting in a coordinated manner on the level of DC and T cells. The regulatory role of T-bet concerning Th17 development has been recently evaluated. Notably, T-bet has the capability to directly restrain Th17-mediated immunopathology (10) likely by decreasing expression of the Th17 signature transcription factor retinoic acid-binding receptor γ (11). Moreover, initiation of T-bet expression in Th17 cells may also favor transdifferentiation into Th1 cells, again strengthening the latter path of T cell polarization under the influence of T-bet (12). Current data furthermore indicate that T-bet expression is up-regulated in human inflammatory/M1 macrophages (13, 14).
On the whole, T-bet knock-out mice display reduced disease severity in models of autoimmunity. Specifically, T-bet deficiency has been associated with amelioration in experimental autoimmune encephalitis (15) and collagen-induced arthritis (CIA) (16). Because T-bet has been associated foremost with IFNγ bioactivity, those observations at first sight do not concur with seemingly protective properties of IFNγ in those aforementioned models of autoimmunity (17, 18) and beyond that with some context-specific anti-inflammatory characteristics of this cytokine (19). Thus, it is tempting to speculate that T-bet-inducible genes apart from IFNγ may play a significant role in the pathophysiology of autoimmune inflammation.
Recently, we reported on robust induction of T-bet mRNA, protein, and biological activity (20) by IL-18 (IL-1F4) (21, 22) in the predendritic acute myelogenous leukemia-derived cell line KG1 (23). Based on this experimental system we set out to identify novel T-bet-inducible genes by combining siRNA technology with genome-wide mRNA expression analysis. We herein introduce the recently described proinflammatory IL-1 cytokine family member IL-36γ (IL-1F9) (24) as a novel T-bet-regulated gene in cells of myeloid origin.
EXPERIMENTAL PROCEDURES
Reagents
Human granulocyte-macrophage colony-stimulating factor, IFNγ, IL-4, and TNFα were purchased from Peprotech Inc. (Frankfurt, Germany). IL-2, IL-12, IL-18, mature IL-36γ (amino acids 18–169), and pro-IL-36γ as well as an anti-IL-4 antibody were from R&D Systems (Wiesbaden, Germany). IL-1β was from Invitrogen. LPS (Serotype R515, TLR grade) and phorbol-12-myristate-13-acetate (PMA) were from Enzo Life Sciences (Lörrach, Germany). SB203580 and IKK-VII were from Calbiochem. Ionomycin was purchased from Sigma, and monensin was obtained from BD Biosciences. Anti-CD3 and anti-CD28 antibodies were from Beckman Coulter (Marseille, France).
Affymetrix Oligonucleotide Microarray Analysis
For affymetrix microarray analysis, KG1 cells were transfected by electroporation (Bio-Rad) (20) either with 2.0 μg of siRNA-Tbet targeting T-bet (5′-GGAAGUUUCAUUUGGGAAA-3′, nt 916–934, NM_013351; Eurofins MWG Operon, Ebersberg, Germany) or with 2.0 μg of siRNAc, a negative control (Silencer® Negative Control siRNA, #4611, Applied Biosystems, Weiterstadt, Germany). After 5 h of rest, transfected cells were stimulated with IL-18 (10 ng/ml) for 12 h, which mediates robust T-bet expression in KG1 cells (20). Total RNA from five independently performed experiments was isolated (TriReagent, Sigma) according to the manufacturer's instructions and pooled in equal shares. Analysis of differential gene expression using the GeneChip® HG-U133A 2.0 Array (Affymetrix, Santa Clara, CA) was performed by the BioChip laboratory of the University Duisburg-Essen (Germany). Mock transfection denotes an electroporation/transfection procedure without siRNA/nucleic acid. The microarray analysis that initiated the studies presented herein has been submitted to the NCBI GEO data repository and obtained the GEO accession number GSE37595.
Cultivation of KG1 Cells and HaCaT Keratinocytes
The human acute myelogenous leukemia-DC cell line KG1 was obtained from the German Collection of Microorganisms and Cell Cultures (Braunschweig, Germany). Cells were cultivated in RPMI 1640 supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated FCS (Invitrogen). For experiments, cells were seeded at a density of 3 × 106 cells/ml in 6-well polystyrene plates (Greiner, Frickenhausen, Germany) using the aforementioned medium. HaCaT keratinocytes (Prof. Stefan Frank, University Hospital Goethe-University Frankfurt) were maintained in DMEM supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FCS. For experiments, HaCaT keratinocytes were seeded on 6-well polystyrene plates in the aforementioned culture medium. All incubations were performed at 37 °C and 5% CO2.
Ectopic Expression of T-bet and Overexpression of IL-36γ in HaCaT Keratinocytes
Full-length IL-36γ was cloned in pCDNA3-vector (HindIII, EcoRI) and sequenced (Seqlab, Göttingen, Germany). Overexpression of IL-36γ in HaCaT keratinocytes was used herein as a positive control for detection of the cytokine by Western blot analysis. For transfection experiments 2 μg of the CMV6-XL4-Tbet vector (Origene Technologies, Rockville, MD), of control vector (CMV6-XL4), or of pCDNA3-IL-36γ vector were transfected using Nucleofector Technology according to the manufacturer's instructions (Lonza, Cologne, Germany). After a 20-h incubation upon the indicated conditions, total RNA (T-bet-transfection) and cellular lysates (T-bet and IL-36γ transfection) were obtained as previously described (25).
Generation of Human DC, Inflammatory Macrophages, and Th1-primed T Cells
For isolation of peripheral blood mononuclear cells, written informed consent was obtained from healthy donors, and blood was taken. The procedure was approved by the “Ethik Kommission” of the University Hospital Goethe-University Frankfurt (Geschäfts-Nr.: 170/1998). Healthy donors had abstained from taking drugs for 2 weeks before the study. Peripheral blood mononuclear cells were isolated from peripheral blood using Histopaque-1077 (Sigma) according to the manufacturer's instructions. The CD14+ cell population of peripheral blood mononuclear cells was isolated by using CD14 antibody-conjugated magnetic microbeads (Miltenyi, Bergisch-Gladbach, Germany). After isolation, CD14+ cell purity was determined by FACS analysis (FACS Canto, BD Biosciences) using mouse monoclonal anti-human CD14-phycoerythrin (PE) (eBioscience, Frankfurt, Germany). Enrichment of CD14+ cells was >94.5%. Cells were resuspended in RPMI 1640 supplemented with 10% heat-inactivated FCS and seeded at a density of 8 × 105 cells/0.75 ml on 12-well polystyrene plates (Greiner). CD14+ cells were further used to generate monocyte-derived DC or macrophages, respectively. For generation of inflammatory macrophages (26), CD14+ cells were cultured in the presence of granulocyte-macrophage colony-stimulating factor (10 ng/ml)/TNFα (10 ng/ml) for 7 days and LPS (10 μg/ml) during a final maturation period of 3 days (26). Medium was changed on days 3 and 5. For differentiation into DC, CD14+ cells were cultured in the presence of granulocyte-macrophage colony-stimulating factor (50 ng/ml)/IL-4 (20 ng/ml) for 7 days (27). 75% of spent medium was exchanged for fresh medium and fresh cytokines on days 3 and 5. T-bet-siRNA or siRNAc was transfected into macrophages using Lipofectamine 2000 (Invitrogen). For each reaction 20 nm concentrations of indicated siRNA was transfected into cells according to the manufacturer's instructions. The transfection was stopped after 4 h. After 3h of rest, cells were used as unstimulated control or were further stimulated as indicated.
For Th1 differentiation, CD4+ T cells were isolated using the Naïve CD4+ T cell isolation kit II according to the manufacturer's instructions (Miltenyi). To assess successful CD4+ isolation, FACS analysis was performed with the following antibodies: mouse monoclonal anti-human CD4-PE-Cy7 (BD Biosciences) and mouse monoclonal anti-human CD45RA-FITC (Biolegend, Fell, Germany). Enrichment of CD4+ cells was >97.2%. To induce Th1 cell differentiation, cells were resuspended in RPMI 1640 supplemented with 1% human serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 50 μm 2-mercaptoethanol and seeded onto 6-well plates coated with anti-CD3 or anti-CD3/anti-CD28 antibodies (coating with 4 μg/ml of each antibody). Cells were kept in the presence of IL-2 (20 ng/ml), IL-12 (20 ng/ml), and anti-IL-4 (10 ng/ml) for 2 days. After this time period cells were transferred to new culture plates without medium change. After 3 more days, cells were harvested, and T-bet expression was analyzed by Western blot analysis. To confirm successful Th1 differentiation, IFNγ expression was evaluated by intracellular FACS-analysis. To that end, cells were stimulated on day 5 with PMA (50 μg/ml), ionomycin (1 μg/ml), and monensin (2 μm) for 5 h. Thereafter, analysis was performed using the following antibody: mouse monoclonal anti-human IFNγ-FITC (BD Biosciences). Percentage of IFNγ-positive cells after treatment was 37.8 ± 3.9%. Th0 denotes T cells immediately harvested for Western blot analysis after isolation by the Naïve CD4+ T cell isolation kit II.
Isolation of Murine Splenic DC
Isolation of splenocytes from wild-type (C57BL/6J) and T-bet knock-out mice (B6.129S6-Tbx21tm1Glm/J (stock number 004648; The Jackson Laboratory) and further DC isolation was performed according to the protocol from Miltenyi Biotec. For each experiment (n = 3), splenocytes of 2 wild-type and 2 T-bet knock-out mice were pooled for further analysis. 0.5 × 106 DC were seeded on 12-well polystyrene plates in RPMI 1640 culture medium supplemented with 10% FCS. CD11c+ cell isolation was evaluated by FACS analysis (FACS Canto) with the following antibodies: hamster monoclonal anti-mouse CD11c-FITC (Miltenyi) and rat monoclonal anti-mouse MHC-II (I-A/I-E)-PE (eBioscience). Enrichment of CD11c+ cells was 91.7 ± 2.8% and 92.9 ± 1.2% for wild-type and T-bet knock-out mice, respectively.
Preparation of Human Primary Keratinocytes
Human foreskin specimens were obtained from a resident physician (board-certified specialist for urology) located in metropolitan Frankfurt. This procedure was approved by the Ethik Kommission of the University Hospital Goethe-University Frankfurt (Geschäfts-Nr.: 162/12). Foreskin tissue was cut in small pieces and incubated with dispase (PromoCell, Heidelberg, Germany) overnight at 4 °C. Thereafter, the epidermis was separated from the dermis, incubated in Hepes, and trypsinized for 30 min at 37 °C. The reaction was stopped by adding trypsin inhibitor. To prepare a single cell suspension, a nylon cell strainer (70 μm; BD Biosciences) was used. Keratinocytes were resuspended in primary keratinocyte cell culture medium (PromoCell) supplemented with 100 units/ml penicillin, 100 μg/ml streptomycin, and 50 μg/ml gentamycin and seeded at a density of 6 × 105 cells/ml in 6-well polystyrene plates (Greiner). For gene expression studies, cells were stimulated with cytokines at a density of 80–90%.
Evaluation of Human IL-36γ mRNA by RNase Protection Assay
Total RNA isolated using Tri-Reagent (Sigma) was used for RNase protection assay, performed as previously described (28). Briefly, IL-36γ DNA probe was cloned into the transcription vector pBluescript II KS (+) (Stratagene, Heidelberg, Germany). After linearization, an antisense transcript was synthesized in vitro with T3 RNA polymerase (Roche Diagnostics) and [α-32P]UTP (800 Ci/mmol; PerkinElmer Life Sciences. RNA samples were hybridized at 42 °C overnight with 100,000 cpm of the labeled antisense transcript. Hybrids were digested with RNase A and T1 (both from Roche Diagnostics) for 1 h at 30 °C. Under these conditions every single mismatch was recognized by the RNases. Protected fragments were separated on 5% polyacrylamide, 8 m urea gels and analyzed using phosphoimaging (Fuji, Straubenhardt, Germany). Individual gene expression of IL-36γ was evaluated on the basis of GAPDH housekeeping gene expression. The cDNAs correspond to nt 112–380 of IL-36γ (NM_019618) and nt 961–1071 of GAPDH (M33197) of the published sequences.
Evaluation of IL-36γ and T-bet mRNA by Standard and IL-36γ, T-bet, TNFα, IL-12p35, IL-23p19, CCL20, iNOS, S100A7, IL-10, and TGF-β1 by Real-time PCR
Total RNA isolated by Tri-Reagent (Sigma) was transcribed using random hexameric primers and Moloney virus reverse transcriptase (Applied Biosystems). The following sequences were performed for standard PCR: 94 °C for 10 min (1 cycle); 94 °C for 30 s, 60 °C (human GAPDH, murine GAPDH, murine T-bet) or 57 °C (human IL-36γ) for 30 s, and 72 °C for 45s; final extension phase at 72 °C for 7 min. Primer sequences and length of resulting amplicons: murine GAPDH, forward 5′-CCTTCATTGACCTCAACTAC-3′; reverse 5′-GGAAGGCCATGCCAGTGAGC-3′, 594 bp, 30 cycles; murine T-bet, forward 5′-AGCATGCCAGGGAACCGC-3′ and reverse 5′-CGGAATCCTTTGGCAAAGGGG-3′, 338 bp, 35 cycles; human GAPDH, forward 5′-ACCACAGTCCATGCCATCAC-3′; reverse 5′-TCCACCACCCTGTTGCTGTA-3′, 452 bp, 25 cycles; human IL-36γ, forward 5′-GTCTATCAATCAATGTGTAAACC-3′ and reverse 5′-ATCTTCTGCTCTTTTAGCTGCA AT-3′, 269 bp, 30 cycles. Identity of amplicons was confirmed by sequencing.
The following predeveloped assay reagents were used for real-time PCR (GAPDH, VIC-dye; all other, 6-carboxyfluorescein, Applied Biosystems): human GAPDH, 4310884E; human IL-36γ, Hs00219742_m1; T-bet, Hs00203436_m1; CCL20, Hs01011368_m1; iNOS, Hs01075529_m1; S100A7, Hs00161488_m1; TNFα, Hs00174128_m1; IL-10, Hs99999035_m1; TGF-β1, Hs99999918_m1; murine GAPDH, 4352339E; murine IL-36γ, Mm00463327_m1. Assay-mix was used from Thermo Scientific (Epsom, Surrey, UK). Real-time PCR was performed on AbiPrism7500 Fast Sequence Detector (Applied Biosystems); one initial step at 95 °C for 5 min was followed by 40 cycles at 95 °C for 2 s and 60 °C for 25s. Detection of the dequenched probe, calculation of threshold cycles (Ct values), and data analysis were performed by the Sequence Detector software. Relative changes in mRNA expression compared with unstimulated control and normalized to GAPDH were quantified by the 2−ddCt method.
Detection of Human IL-36γ and T-bet by Western Blot Analysis
Cell lysates were obtained as previously described (20). For detection of secreted human IL-36γ by Western blot (cultivation of cells without FCS), cell-free supernatants were TCA-precipitated (25). Antibodies were: IL-36γ, goat polyclonal antibody (R&D Systems); T-bet, mouse monoclonal antibody (Santa Cruz Biotechnology); β-actin, mouse monoclonal antibody (Sigma).
Determination of the Human IL-36γ Transcriptional Start Site by RNA Ligase-mediated Rapid Amplification of 5′-cDNA Ends
To identify the IL-36γ transcriptional start site in KG1 cells, Gene Racer- and TOPO TA cloning kits (Invitrogen) were used according to the manufacturer's instructions. For analysis, 12 clones were sequenced that were obtained from three independent rapid amplification of 5′-cDNA ends experiments. All 12 clones gave an identical result. We identified the transcriptional start site at nucleotide 113,452,054 (denoted +1, Fig. 3A) within the sequence given in NC_000002.10 (chromosome 2). All topographic information concerning the human IL-36γ gene locus refers to this transcriptional start site.
FIGURE 3.

T-bet and human IL-36γ promoter activation. A, IL-36γ transcriptional start site is indicated at +1. Cloned promoter fragments and pertinent transcription factor binding sites are depicted (T-box (T) site (−495/−475 nt), κB (N) site (−286/−274 nt)). B, C, E, and F, KG1 cells were transfected with the indicated pGL3-IL-36γ-promoter constructs and Renilla luciferase. Manufacturer's instructions for transfection by DMRIE-C reagent (Invitrogen) recommend PMA (50 μg/ml) as co-stimulus for efficient promoter activation specifically in KG1 cells. Accordingly, PMA was added to plasmid transfection experiments (shown in Fig. 3) including controls. B, C, and F, 2.5 h after transfection cells were kept as control or stimulated with IL-18 (10 ng/ml). After 20 h, luciferase assays were performed. B, means of luciferase activity are shown as -fold induction (compared with control condition transfected with the same plasmid) ± S.D. (n = 5); *, p < 0.05; **, p < 0.01 compared with control; raw data were analyzed by Student's t test. C, cells were transfected with either pGL3-Prom2 with/without siRNA-Tbet or siRNAc or with pGL3-Prom2-TPM (mutated T-box site). Means of luciferase induction by IL-18 compared with control conditions transfected with the same plasmid are shown as % of pGL3-Prom2-transfected cells upon IL-18 stimulation ± S.D. (n = 3); #, p < 0.05 compared with Prom2 upon IL-18 stimulation; $, p < 0.05 compared with Prom2/siRNAc transfection upon IL-18 stimulation. Statistics were performed on -fold induction by Student's t test. D, cells were kept as control or stimulated with IL-18 (10 ng/ml). After 12 h, ChIP was performed. One experiment representative of three independently performed experiments is shown. E, 2.5 h after pGL3-Prom2 transfection, cells were kept as the control or stimulated with IL-18 (10 ng/ml). Where indicated, cells were preincubated (1 h) with IKK-VII (10 μm). After 12 h, luciferase assays were performed. Means of luciferase activity are shown as -fold induction compared with control ± S.D. (n = 4); ***, p < 0.001 compared with control; ###, p < 0.001 compared with IL-18 stimulation; raw data were analyzed by one-way ANOVA with post-hoc Bonferroni correction. F, cells were additionally transfected with pGL3-Prom2-NPM (mutated κB) or pGL3-Prom2-TNPM (double-mutated T-box/κB binding site). Means of luciferase induction (relative to control condition transfected with the same plasmid) are shown as % of pGL3-Prom2-transfected cells upon IL-18 stimulation ± S.D. (n = 3); #, p < 0.05; ##, p < 0.01 compared with pGL3-Prom2 upon IL-18 stimulation; statistics were performed on -fold induction (compared with control condition transfected with the same plasmid) by one-way ANOVA with post-hoc Bonferroni correction.
Cloning of the Human IL-36γ Promoter, Transient Transfection of KG1 Cells, and Luciferase Reporter Assays
Using human genomic DNA (KG1 cells), we amplified the 5′-flanking region of IL-36γ (NM_019618) using Pfu polymerase (Invitrogen) to generate IL-36γ promoter fragments. For that purpose the following primers (excluding an additional flanking BglII cloning/restriction site) were used: Prom1 (2209 bp), forward 5′-AGCATTGACCAGACTGTACTC-3′; Prom2 (1630 bp), forward 5′-TAGGGTGAAAAGTAAGAGAC-3′. The reverse primer for both fragments (excluding an additional flanking HindIII cloning/restriction site) was 5′-AGTGTGGTTGTCTCAGCACCT-3′. Both promoter fragments end 5′ adjacent to the adenine nucleotide of the IL-36γ translational start site. Fragments were cloned into pGL3-Basic (Promega, Mannheim, Germany) and sequenced (Seqlab). Site-directed mutagenesis was performed using the QuikChange site-directed mutagenesis kit (Stratagene, Amsterdam, Netherlands) to generate promoter fragments that show dysfunctional putative T-bet and nuclear factor-κB (NF-κB) binding sites. The following primers were used: pGL3-Prom2-TPM (−501/−464 nt relative to IL-36γ transcriptional start site) forward 5′-CCTGGCTTTCCATTCAGGAAAGGCCTTAGGTGGGGTAG-3′; pGL3-Prom2-NPM (−299/−255 nt) forward 5′-GGAGAGCTGAACTCTGGGAAATTAGCTTAGTTTCCCCTTTATGAG-3′; the identity of the mutants was confirmed by sequencing. Plasmids were transiently transfected into KG1 cells using DMRIE-C reagent (Invitrogen) according to the manufacturer's instructions. Those specifically recommend the addition of PMA (50 μg/ml) as co-stimulus for efficient promoter activation in KG1 cells. Accordingly, PMA was added to all cultures including control cultures at the aforementioned concentration. 0.1 μg of pRL-TK (Promega) coding for Renilla luciferase was cotransfected. For siRNA/Luciferase experiments, KG1 cells were additionally transfected (along with the plasmids) with 100 nm either siRNA-Tbet or siRNAc. After 2.5 h of rest, transfected cells were used as control or were stimulated as described in the figure legends. Thereafter, luciferase activity was determined using the dual reporter gene system (Promega) and an automated chemiluminescence detector (Berthold, Bad Wildbad, Germany).
ChIP
Chromatin immunoprecipitation was performed as described previously (25). For immunoprecipitation, 3 μg of an IgG control or T-bet antibody (4B10, Santa Cruz Biotechnology) were used. To amplify the IL-36γ promoter fragment containing the T-box site under investigation (−495/−475 nt relative to the IL-36γ transcriptional start site), the following primers were used: forward, 5′-GAGGGTACCTGAAACTAGGATTGC-3′; reverse, 5′-GGCTAAAGGCATCCTGTAAGAGC-3′. PCR conditions were: 95 °C for 10 min (1 cycle); 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 45 s for 40 cycles; and a final extension phase at 72 °C for 7min. The identity of amplicon (216 nt) was confirmed by sequencing.
Analysis of Cytokine Release by ELISA
Human IL-8 and IFNγ (BD Biosciences) levels in cell-free cell culture supernatants were determined by ELISA according to the manufacturers' instructions.
Statistical Analysis
Data are shown as the means ± S.E. (DC, macrophages, splenocytes) or means ± standard deviation (S.D.) (KG1 cells, HaCaT keratinocytes) and are presented as -fold induction, % of siRNAc upon IL-18 stimulation, and % of Prom2 upon IL-18 stimulation. Statistical analysis was performed either by unpaired Student's t test (two-tailed) or one-way ANOVA with post-hoc Bonferroni correction (GraphPad 5.0) as indicated in the legends.
RESULTS
Genome-wide mRNA Expression Analysis during T-bet Silencing Identifies IL-36γ as T-bet-regulated Gene in KG1 Cells
Recently, we observed induction of T-bet expression and biological activity in predendritic KG1 cells under the influence of IL-18. In this cellular model, T-bet is activated independently from IFNγ by concerted action of p38 MAPK and IκB kinase (IKK)/NF-κB pathways (20). To classify T-bet expression in activated KG1 cells, protein expression of this transcription factor was compared between IL-18-stimulated KG1 cells and human Th1 cells. We observed in 4 of 5 experiments that Th1 cells expressed higher levels of T-bet protein as compared with activated KG1 cells. Notably, T-bet protein was in all cases detectable by Western blot analysis in IL-18-stimulated KG1 cells and Th1 cells, respectively (Fig. 1A). Because T-bet is capable of mediating gene expression in KG1 cells (20), this experimental system was utilized herein to identify novel T-bet-inducible genes. To that end, KG1 cells were transfected with either control-siRNA (siRNAc) or siRNA targeting T-bet (siRNA-Tbet) in five independently performed experiments. Concurring with previous data (20), silencing of IL-18-induced T-bet was evident on mRNA (Fig. 1B) and protein level. In fact, T-bet protein induction was essentially absent in cells transfected with siRNA-Tbet (Fig. 1C). In further accordance with previous work (20) we observed that T-bet silencing associated with impaired immediate IL-18-induced IFNγ production (Fig. 1D). Pooled total RNA from this same set of experiments was evaluated by genome-wide analysis using the Affymetrix GeneChip® Array System (GSE37595). Among the genes robustly regulated by T-bet was IL-36γ (IL1F9), a previously described novel member of the IL-1 cytokine family (24, 29–31). Because our group is particularly interested in the biology of the IL-1 cytokine family and information on T-bet regarding IL-36γ is lacking, we subsequently focused on detailed analysis of mechanisms directing expression of this cytokine. Fig. 1E displays down-regulation of T-bet and IL-36γ mRNA by siRNA-Tbet in the pooled total mRNA population as detected by gene chip analysis and real-time PCR, respectively. Down-regulation of IL-36γ expression by siRNA-Tbet in unpooled mRNA of this particular set of experiments is shown in Fig. 1F.
FIGURE 1.

Identification of IL-36γ as T-bet-inducible gene in KG1 cells. A, CD4+ T cells were isolated from peripheral blood and differentiated as described under “Experimental Procedures.” KG1 cells were kept as unstimulated control or stimulated with IL-18 (10 ng/ml) for 12 h. Thereafter, T-bet protein was determined by Western blot analysis (30 μg of total protein/lane). One experimental representative of five independently performed is shown. B, D, and F, KG1 cells were transfected with siRNA directed against T-bet (siRNA-Tbet) or control-siRNA (siRNAc). Thereafter, cells were stimulated with IL-18 (10 ng/ml). After 12 h, total RNA and culture supernatants were obtained. Five independent experiments were performed and evaluated. B, T-bet mRNA expression in unpooled RNA preparations (n = 5) was determined by real-time PCR analysis in the context of siRNA-Tbet or siRNAc transfection. T-bet mRNA was normalized to that of GAPDH and is shown as % of IL-18 stimulation upon siRNAc transfection ± S.D. C, KG1 cells were mock-transfected or transfected with either siRNA-Tbet or siRNAc. After 12 h, T-bet protein was determined in unstimulated or IL-18 stimulated cells by Western blot analysis. After stripping, blots were stained for β-actin. One representative of 5 independently performed experiments is shown. D, IFNγ content was determined by ELISA in supernatants derived from experiments used in B. Data (n = 5) are shown as % of IL-18 stimulation upon siRNAc transfection ± S.D. E, RNA populations (n = 5) used in B were equally pooled and utilized for microarray analysis as outlined under “Experimental Procedures.” Affymetrix Gene Chip results (gray bars) of T-bet and IL-36γ mRNA expression are shown. In addition, Affymetrix Gene Chip results were verified by real-time PCR analysis (black bars) of those same pooled RNA fractions. mRNA expression of T-bet and IL-36γ (normalized to that of GAPDH) is shown as (% of IL-18 stimulation upon siRNAc transfection). F, IL-36γ mRNA in unpooled RNA preparations (used in B; n = 5) was determined by real-time PCR analysis in the context of siRNA-Tbet or siRNAc transfection. T-bet mRNA was normalized to that of GAPDH and is shown as % of IL-18 stimulation upon siRNAc transfection ± S.D. B, D, and F, *, p < 0.05; ***, p < 0.001 compared with IL-18 stimulation upon siRNAc transfection; raw data were analyzed by Student's t test.
Next, regulation of IL-36γ in KG1 cells was investigated on mRNA and protein levels in additional independently performed experiments. First, induction of IL-36γ mRNA in IL-18-stimulated KG1 cells was confirmed using a standard biochemical methodology different from real-time PCR and not depending on additional amplification steps, namely RNase protection assay. Using this technique we confirm IL-36γ mRNA expression by KG1 cells under the influence of IL-18 (Fig. 2A). Further experiments revealed that IL-18, but not IFNγ, mediates IL-36γ mRNA expression in KG1 cells as detected by real-time (Fig. 2B) and standard PCR (Fig. 2B, inset), respectively. mRNA induction by IL-18 translated into IL-36γ protein, which was detectable by Western blot analysis in cell lysates (Fig. 2C, upper panel) and culture supernatants (Fig. 2C, lower panel), respectively. To validate KG1 cell-associated IL-36γ immunoreactivity detected by Western blot analysis, recombinant pro-IL-36γ and lysates from IL-1β/TNFα-stimulated HaCaT keratinocytes as well as those from HaCaT keratinocytes transfected with the pCDNA3-IL-36γ expression plasmid were run on the same gel. Keratinocytes activated by IL-1β/TNFα are supposed to express IL-36γ (31). In fact, immunoreactivity induced by IL-18 in KG1 cells and detected by the polyclonal anti-IL-36γ antibody (R&D Systems) in use co-migrated with immunoreactivity associated with IL-1β/TNFα stimulation or pCDNA3-IL-36γ transfection of HaCaT keratinocytes and, most importantly, with that of recombinant pro-IL-36γ (at ∼18.7 kDa, R&D Systems) (Fig. 2D). Induction of IL-36γ was not detected in KG1 cells exposed to IFNγ (Fig. 2B), an observation concurring with insufficient up-regulation of T-bet by IFNγ in this cell type (Fig. 2E).
FIGURE 2.

IL-18 up-regulates IL-36γ mRNA and protein in KG1 cells; contribution of p38 MAPK and NF-κB. A, KG1 cells were either kept as unstimulated control (C) or stimulated with IL-18 (10 ng/ml). After 12 h, IL-36γ mRNA was analyzed by RNase protection assay (10 μg total RNA/condition). Three independently performed experiments are shown. tR and FP denote tRNA and free probe, respectively. B, KG1 cells were either kept as unstimulated control or stimulated with IL-18 (10 ng/ml) or IFNγ (20 ng/ml) for 12 h. IL-36γ mRNA was assessed by real-time PCR. Target mRNA was normalized to that of GAPDH and is shown as -fold induction compared with unstimulated control ± S.D. (n = 6 for control and IL-18, n = 4 for IFNγ); *, p < 0.05 compared with unstimulated control; raw data were analyzed by one-way ANOVA with post hoc Bonferroni correction. B, inset, additionally, data from one representative experiment was analyzed by standard PCR. 36, IL-36; G, GAPDH. C, KG1 cells were either kept as unstimulated control or stimulated as indicated with IL-18 (10 ng/ml), IFNγ (20 ng/ml), or with the combination TNFα (50 ng/ml)/IL-1β (20 ng/ml) for 20 h (upper panel) or 16 h (lower panel). Upper panel, cellular IL-36γ content was evaluated by Western blot analysis. One experiment representative of three independently performed experiments is shown. Lower panel, IL-36γ in cell lysates or TCA-precipitated supernatants (medium without FCS) of matching samples were evaluated by Western blot analysis. One experiment representative of three independently performed experiments is shown. D, KG1 cells were kept as the unstimulated control or were stimulated with IL-18 (10 ng/ml). HaCaT keratinocytes were either kept as the unstimulated control or were stimulated with IL-1β (20 ng/ml)/TNFα (50 ng/ml) or were transfected with pCDNA3-IL-36γ expression vector as outlined under “Experimental Procedures.” After 12 h (KG1) or 20 h (HaCaT keratinocytes) cells were harvested, and lysates were analyzed for cellular IL-36γ protein by Western blot analysis. Recombinant pro-IL-36γ served as a further positive control. Total protein applied per lane: lysate of cytokine-stimulated KG1/HaCaT cells, 30 μg; pCDNA3-IL-36γ transfection of HaCaT keratinocytes, 0.3 μg; recombinant pro-IL-36γ, 100 pg. E, KG1 cells were either kept as unstimulated control or stimulated with IL-18 (10 ng/ml) or IFNγ (20 ng/ml) for 12 h. T-bet mRNA was assessed by real-time PCR. Target mRNA was normalized to that of GAPDH and is shown as -fold induction compared with unstimulated control ± S.D. (n = 4); ***, p < 0.001 compared with unstimulated control; ###, p < 0.001; raw data were analyzed by one-way ANOVA with post hoc Bonferroni correction. F, cells were either kept as unstimulated control or stimulated with IL-18 (10 ng/ml). Where indicated, cells were preincubated with SB203580 (10 μm) or IKK-VII (10 μm) for 1 h before IL-18 addition. All cultures were adjusted to a final concentration of 0.1% DMSO (vehicle for SB203580, IKK-VII). After 12 h, IL-36γ was assessed by real-time PCR analysis. IL-36γ mRNA was normalized to that of GAPDH and is shown as -fold induction compared with unstimulated control ± S.D. (n = 5); **, p < 0.01 compared with unstimulated control; #, p < 0.05; ##, p < 0.01 compared with stimulation with IL-18 alone; raw data were analyzed by one-way ANOVA with post hoc Bonferroni correction. F, inset, cellular IL-36γ protein (of cultures treated as described in F) was determined by Western blot analysis. One experiment representative of three independently performed experiments is shown. SB, SB203580.
Because p38 MAPK and IKK/NF-κB activation are mandatory for induction of T-bet in KG1 cells (20), blockage of both pathways was investigated. In fact, SB203580, inhibiting p38 MAPK, and IKKVII, inhibiting IKK/NF-κB, suppressed IL-36γ induction under the influence of IL-18. This modulatory action observed on the level of IL-36γ mRNA (Fig. 2F) was mirrored by cellular IL-36γ protein levels (Fig. 2F, inset).
Promoter Studies Reveal a Crucial Function of Specific T-box and κB Sites for Induction of Human IL-36γ
To further delineate molecular mechanisms driving IL-36γ in KG1 cells, a human IL-36γ promoter fragment was cloned that spans from −1576 to +633 nt (immediately adjacent to the translational start site at +634 nt) relative to the transcriptional start site (at +1). Sequence analysis revealed the presence of a non-canonical TATA box, recently entitled GATA box at position −29/−24 nt (5′-GATAAA-3′) in this fragment. Based on this primary fragment (Prom1), a deletion mutant (Prom2) was generated (Fig. 3A), and fragments were analyzed in luciferase reporter assays performed in the context of IL-18-activated KG1 cells. Robust IL-36γ promoter activation was achieved by IL-18 with no significant differences between Prom1 and Prom2 (Fig. 3B). Accordingly, all further experiments were performed using Prom2. Sequence analysis of Prom2 by MatInspector (Genomatix 7.4.4., Munich, Germany) brought to light a potential T-box (T) site (−495/−475 nt) with a monomeric binding mode. This element (Fig. 3A) displayed significant similarity to monomeric binding mode T-box sites of transcription factors related to T-bet, among others Brachyury and Tbx2 (32–35). To further substantiate a role for T-bet in IL-36γ promoter activation, two strategies were pursued. First, luciferase reporter assays were performed upon silencing of T-bet by siRNA. Second, the aforementioned T-box site (−495/−475 nt) was mutated (Prom2-TPM) as outlined under “Experimental Procedures.” Both strategies in fact resulted in significant reduction of IL-36γ promoter activity detected in IL-18-stimulated cells by 55.4% (siRNA-Tbet) and 56.9% (Prom2-TPM) (Fig. 3C). Likewise, enhanced binding of T-bet to this specific promoter site (−495/−475 nt) was proven by ChIP analysis (Fig. 3D). Thus, data presented herein demonstrate that activation of T-bet significantly contributes to IL-36γ induction as detected in IL-18-activated KG1 cells.
Because inhibition of IKK impaired IL-36γ mRNA and protein expression (Fig. 2F), promoter activation was investigated in the presence of IKK-VII. As expected, this inhibitor likewise suppressed IL-36γ promoter activation (Fig. 3E). Sequence analysis (MatInspector) of IL-36γ Prom2 actually revealed a κB site (−286/−274 nt) that was further examined by mutational analysis. Deactivation of this specific κB binding site by mutation (Prom2-NPM) resulted in a significant 79.6% reduction of IL-36γ promoter activity detected in IL-18-stimulated cells. Notably, a Prom2 construct holding a κB (−286/−274 nt)/T-box (−495/−475 nt) double-mutation (Prom2-TNPM) displayed nearly complete loss of inducibility by IL-18 (Fig. 3F). Altogether, data indicate that those two specific κB and T-box sites identified in the present study are crucial for efficient induction of human IL-36γ gene expression.
Analysis of IL-36γ Expression as Detected in Splenic DC Obtained from Wild-type or T-bet-deficient Mice
To further the assumption of T-bet being crucial for IL-36γ regulation in myeloid cells, experiments were performed using splenic DC obtained from wild-type or T-bet knock-out mice, respectively. Cells were stimulated with the Toll-like receptor (TLR)-4 ligand LPS, a prototypic activator of myeloid cells including murine splenic DC (36). Experiments revealed that purified splenic DC activated by LPS displayed a significant 73.1% reduction of IL-36γ mRNA expression upon lack of T-bet (Fig. 4A). Activation of splenic DC by LPS mediated robust T-bet induction in wild-type cells but, as expected, not in cells obtained from T-bet knock-out mice (Fig. 4B).
FIGURE 4.
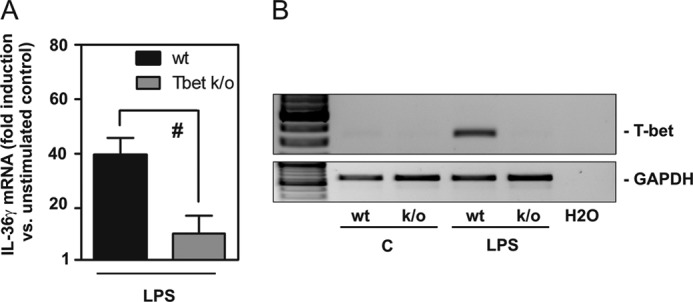
Impaired IL-36γ production in splenic DC obtained from T-bet knock-out mice. A and B, splenic DC isolated from wild-type or T-bet knock-out mice were kept as unstimulated control or stimulated with LPS (1 μg/ml). A, after 14 h IL-36γ mRNA expression was assessed by real-time PCR. mRNA was normalized to that of GAPDH and is shown as -fold induction compared with unstimulated control (C, of the respective genotype) ± S.E. (n = 3); p < 0.05 compared with LPS-stimulated wild-type cells; statistics were performed on -fold induction by Student's t test. B, T-bet mRNA content was evaluated in these same experiments by standard PCR analysis. One representative of the three independently performed experiments is shown.
T-bet Mediates Expression of IL-36γ in Human Inflammatory M1 Macrophages
T-bet expression has been detected on mRNA or mRNA/protein level in activated murine DC (7), human monocytes or monocyte-derived DC (6), and in human predendritic KG1 cells (20) as well as human inflammatory or M1 macrophages (13, 14) but not in murine macrophages (7). To extend these current observations to human primary cells, experiments were performed using monocyte-derived DC and activated primary macrophages. Fig. 5A demonstrates IL-36γ mRNA induction in both human cell types as detected by RNase protection assay. We furthermore confirm by real-time PCR previous observations (6) on robust T-bet mRNA induction in monocyte-derived DC under the influence of IFNγ, which was used as a single stimulus or in combination with IL-1β plus TNFα. Up-regulation of T-bet mRNA in fact associated with enhanced expression of IL-36γ mRNA in these same experiments (Fig. 5B). However, we were unable to reliably detect T-bet protein in human monocyte-derived DC. Therefore, we focused on inflammatory/activated human macrophages (26) in subsequent experiments. Exposure of these macrophages to IFNγ alone or together with IL-1β/TNFα mediated strong gene induction of TNFα, IL-12-p35, and IL-23-p19 (Table 1) echoing an inflammatory M1 macrophage activation status (37). Under those conditions, T-bet expression was likewise induced (Fig. 5C). Thus, we confirm previous observations (14) indicating that M1 macrophages display increased T-bet mRNA. Notably, robust up-regulation of T-bet protein became likewise apparent under those conditions (Fig. 5D). T-bet protein was accompanied by significant induction of IL-36γ mRNA under the influence of IFNγ as a single stimulus or in combination with IL-1β/TNFα (Fig. 5E). siRNA technology was used to verify the function of endogenous T-bet for IL-36γ regulation in M1 macrophages. Macrophages of four healthy donors were either mock-treated or transfected with siRNAc or siRNA-Tbet, respectively. Thereafter, cellular T-bet and IL-36γ expression was verified in the context of exposure to IL-1β/TNFα/IFNγ. Analysis revealed that silencing of T-bet as shown by mRNA expression (Fig. 5F, left panel) was in all experiments performed associated with suppression of IL-36γ mRNA induction (Fig. 5F, right panel).
FIGURE 5.

Expression of IL-36γ in human monocyte-derived DC and inflammatory M1 macrophages. A–F, monocytes were isolated from human peripheral blood mononuclear cells and further differentiated to DC (A and B) or inflammatory M1 macrophages (A and C–F) as described under “Experimental Procedures.” A, DC and macrophages were either kept as unstimulated control (C, indicated by −) or stimulated with IFNγ (50 ng/ml)/TNFα (50 ng/ml)/IL-1β (20 ng/ml) (indicated by +). Unstimulated and IL-18 (10 ng/ml) stimulated KG1 cells were used as the positive control for IL-36γ mRNA induction as detected by RNase protection assay (8 or 5 μg of total RNA/condition for analysis of DC/macrophages or KG1 cells, respectively). tR and FP denote tRNA and free probe, respectively. B, DC were either kept as unstimulated control or stimulated with IFNγ (50 ng/ml) alone or in combination with TNFα (50 ng/ml) and IL-1β (20 ng/ml). After 14 h, T-bet and IL-36γ mRNA was assessed by real-time PCR analysis. Target mRNA was normalized to that of GAPDH and is shown as -fold induction compared with unstimulated control ± S.E. (n = 7); *, p < 0.05; ***, p < 0.001 compared with unstimulated control; raw data were analyzed by one-way ANOVA with post-hoc Bonferroni correction. C–F, macrophages were either kept as control or stimulated with IFNγ (50 ng/ml) alone or in combination with TNFα (50 ng/ml) and IL-1β (20 ng/ml) for 14 h. C and E, T-bet (C) and IL-36γ (E) mRNA was assessed by real-time PCR. Target mRNA was normalized to that of GAPDH and is shown as -fold induction compared with control ± S.E. (n = 3–5); *, p < 0.05; **, p < 0.01, ***, p < 0.001 compared with control; raw data were analyzed by Student's t test. D, T-bet protein content of cellular lysates was evaluated by Western blot analysis. One experiment representative of three independently performed experiments is shown. F, macrophages were mock-transfected or either transfected with siRNA-Tbet or siRNAc as described under “Experimental Procedures.” Thereafter, cells were further used as control or were stimulated with IFNγ (50 ng/ml), TNFα (50 ng/ml), and IL-1β (20 ng/ml) for another 14 h. T-bet (left panel) and IL-36γ (right panel) mRNA was assessed by real-time PCR analysis. Target mRNA was normalized to that of GAPDH and is shown as -fold induction compared with mock-transfected control (cultivation without IFNγ/TNFα/IL-1β). Four different donors were investigated, and individual results are shown. The different colors refer to individual donors.
TABLE 1.
mRNA induction of signature cytokine genes by human inflammatory macrophages under the influence of IFNγ (50 ng/ml) with/without IL-1β (20 ng/ml)/TNFα (50 ng/ml)
After a 14-h stimulation period, cytokine mRNA, analyzed by real time PCR, was normalized to that of GAPDH and is shown as -fold induction compared to control. Data are expressed as the means ±S.E. Raw data were analyzed by one-way ANOVA with post hoc Bonferroni correction.
| Stimulus | mRNA |
||
|---|---|---|---|
| TNFα | IL-12-p35 | IL-23-p19 | |
| IFNγ | 41.5 ± 13.4a (n = 5) | 46.8 ± 3.4a (n = 4) | 33.5 ± 2.4a (n = 3) |
| IL-1β/TNFα/IFNγ | 44.4 ± 21.4b (n = 4) | 46.4 ± 13.1a (n = 4) | 11.0 ± 2.7a (n = 3) |
a p < 0.05, compared to control.
b p < 0.01 compared to control.
Ectopic Expression of T-bet Enforces IL-36γ Production in HaCaT Keratinocytes
To further broaden the connection between T-bet and IL-36γ, T-bet was transiently expressed in HaCaT keratinocytes. Notably, keratinocytes usually do not display production of T-bet but are capable of producing IL-36γ in response to proinflammatory cytokines (29, 31, 38–40). Compared with transfection with control plasmid (CMV6-XL4), ectopic expression of T-bet achieved by transfection of CMV6-XL4-Tbet enhanced IL-36γ mRNA (Fig. 6A) and protein (Fig. 6B) accumulation, which became apparent in otherwise unstimulated keratinocytes or those under the influence of IL-1β/TNFα. As anticipated, T-bet expression in HaCaT keratinocytes was only observed upon transfection by CMV6-XL4-Tbet (Fig. 6B) but remained undetectable even in cells activated by IFNγ (data not shown). Neither did enforced T-bet initiate IFNγ production by HaCaT keratinocytes nor was constitutive secretion of IL-8 enhanced by transfection with CMV6-XL4-Tbet in this same set of experiments (data not shown). This observation exposes discrete specificity of misplaced T-bet action in HaCaT keratinocytes. Altogether, those data further substantiate the potential of T-bet to mediate IL-36γ production and beyond that indicate the notable capability of this transcription factor to unfold this activity in a rather broad or even misplaced cellular context.
FIGURE 6.

Ectopic expression of T-bet in HaCaT keratinocytes induces IL-36γ expression. A and B, HaCaT keratinocytes were transiently transfected with CMV6-XL4-Tbet or CMV6-XL4 control vector as described under “Experimental Procedures.” After transfection procedure and 2 h of rest, cells were further used as unstimulated control or where stimulated with IL-1β (20 ng/ml) and TNFα (50 ng/ml) for 20 h. A, IL-36γ mRNA expression was assessed by real-time PCR. mRNA was normalized to that of GAPDH and is shown as -fold induction compared with CMV6-XL4 transfected unstimulated control ± S.D. (n = 3); ***, p < 0.001 compared with CMV6-XL4 transfected unstimulated control; ###, p < 0.001 compared with CMV6-XL4-transfected IL-1β/TNFα-stimulated cells or CMV6-XL4-Tbet-transfected unstimulated cells; raw data were analyzed by one-way ANOVA with post-hoc Bonferroni correction. Values were log-transformed to reduce skew. B, cellular content of T-bet, β-actin, and IL-36γ in these same experiments was assessed by Western blot analysis. One experiment representative of three independently performed experiments is shown.
Mature IL-36γ Induces an Inflammatory Gene Expression Profile in Human Primary Keratinocytes
As keratinocytes activated by inflammatory cytokines display IL-36γ expression (29, 31, 38–40) and DC/macrophages are supposed to act proximal in the inflammatory cascade, we set out to investigate IL-36γ expression in IL-36γ-stimulated human primary keratinocytes. In fact, mature IL-36γ, but not pro-IL-36γ, stimulated its own expression (Fig. 7A) and moreover that of the prototypic proinflammatory parameters TNFα (Fig. 7B), CCL20 (Fig. 7C), S100A7/psoriasin (Fig. 7D), and iNOS (Fig. 7E). In contrast, anti-inflammatory IL-10 and TGF-β1 were not up-regulated under the influence of mature IL-36γ (data not shown).
FIGURE 7.
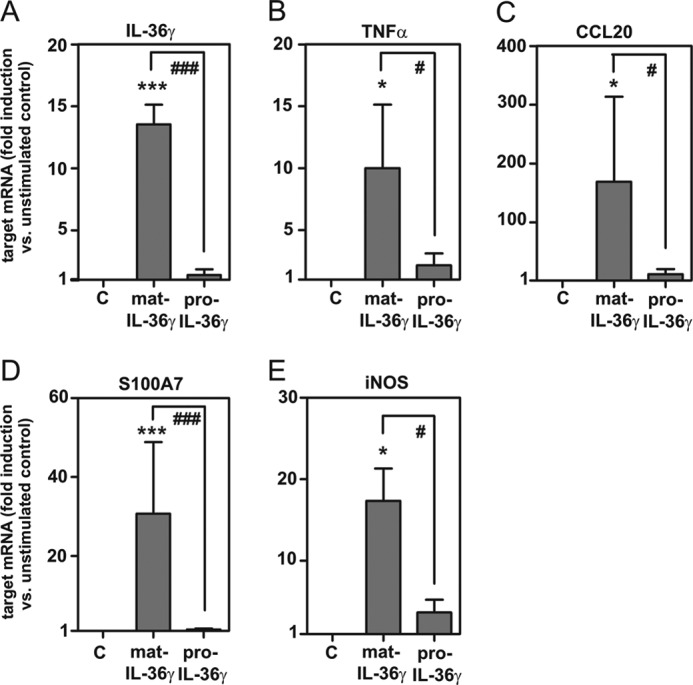
IL-36γ mediates expression of inflammatory genes, including IL-36γ itself, in human primary keratinocytes. Human primary keratinocytes were isolated as described under “Experimental Procedures” and either kept as the unstimulated control (C) or stimulated with mature IL-36γ or pro-IL-36γ (both at 100 ng/ml). After 14 h, IL-36γ mRNA (A, n = 3), TNFα mRNA (B, n = 4), CCL20 (C, n = 4), S100A7 (D, n = 4), and iNOS mRNA (E, n = 4) was assessed by real-time PCR analysis. Target mRNA was normalized to that of GAPDH and is shown as -fold induction compared with unstimulated control ± S.E.; *, p < 0.05; ***, p < 0.001 compared with unstimulated control; #, p < 0.05; ###, p < 0.001; raw data were analyzed by one-way ANOVA with post-hoc Bonferroni correction.
DISCUSSION
Identification of novel genes susceptible to direct activation by T-bet is pivotal for further understanding pathophysiological functions of this crucial transcription factor. Previous studies following this route focused on lymphocytes, particularly T cells, and brought to light novel T-bet-inducible genes that in part visibly connected to Th1 functions. Exemplarily, CXCR3, CCL4, and NKG7 shall be quoted herein (41–43). However, expression of T-bet in activated DC (6, 7) and human monocytes/macrophages (13, 14) likewise suggests a vital role for this transcription factor in myeloid cells and beyond that in innate immunity.
Herein, IL-36γ, a recently described member of the IL-1 cytokine family (24, 30), is introduced as a novel direct target of T-bet action in myeloid cells. By using human predendritic acute myelogenous leukemia-derived KG1 cells, we present for the first time analysis of molecular mechanisms driving IL-36γ gene activation. We demonstrate direct binding of T-bet to a specific T-box site at the IL-36γ promoter that coupled to promoter activation and expression of the cytokine. Moreover, a κB binding site is identified that allowed for T-bet independent IL-36γ induction but likewise cooperated with the T-box site for most efficient cytokine expression. Observations were supported by using primary human DC and inflammatory M1 macrophages as well as splenic DC obtained from wild-type or T-bet knock-out mice. Notably, ectopic expression of T-bet was sufficient to drive IL-36γ in HaCaT keratinocytes as a single stimulus, although low-level basal NF-κB activity operating in this cell line (44) possibly synergized with T-bet for cytokine induction.
IL-36γ displays decisive characteristics of an IL-1 cytokine family member, including a lack of a signal peptide and proteolytic processing as a prerequisite for efficient bioactivity. Notably, the IL-36γ promoter contains at position −29/−24 nt a non-canonical TATA box, recently coined GATA box (5′-GATAAA-3′). Because this element can mediate TATA box-like regulatory activity (45, 46), IL-36 γ in this regard can be grouped together with IL-1β/IL-1F2 that actually displays a canonical TATA box (47). A functional TATA box apparently determines the quantity of IL-1β expression (48). Moreover, IL-36γ is part of the IL1 cluster at the human chromosome 2 (24, 29–31, 49–52). Binding of IL-36γ to heterodimeric IL-36R (IL-1 receptor like 2/IL-1 receptor accessory protein), the common receptor for IL-36α, IL-36β, and IL-36γ, mediates activation of NF-κB, MAPKs, and the IL-8 promoter (29, 50). Altogether this concurs with a proinflammatory cytokine function. Biological activity of IL-36R ligands is controlled by action of IL-36 receptor antagonist (IL-36Ra) and IL-38 (52–54). Studies on IL-36γ production hitherto focused largely on keratinocytes and the skin compartment (29, 31, 39, 40, 52, 53, 55). In fact, IL-36R ligands, including IL-36γ, emerge as crucial component of innate immune responses at host/environment interfaces. Inflammatory stimuli such as IL-1, TNFα, and IL-17 as well as the TLR5 ligand flagellin increase IL-36γ gene expression in primary human keratinocytes (29, 39, 40, 55), an observation in accord with involvement of NF-κB in IL-36γ regulation as detected herein. In contrast, IFNγ as single stimulus fails to mediate IL-36γ in keratinocytes (38, 55). This incapability of IFNγ to induce IL-36γ in this cell type reflects the incapability of IFNγ to efficiently activate NF-κB and moreover the universal lack of T-bet in keratinocytes. Proinflammatory activation of keratinocytes by IL-36γ indicates its functional role in dermal inflammation (52). In fact, IL-36γ is up-regulated in the lesional skin of psoriatic patients (29, 38, 39) and in murine models of this disease (38, 39). A crucial role of IL-36R ligands in dermal pathology is furthermore suggested by psoriasis-like symptoms observed in skin-specific IL-36α transgenic mice (56). Notably, a deficiency of the IL-36Ra results in severe generalized pustular psoriasis in humans (53).
Induction of genes different from IFNγ by T-bet is of particular interest under conditions where both parameters display discordant pathophysiological functions. As already alluded to, this applies exemplarily to murine CIA, a common in vivo model for rheumatoid arthritis where IFNγ shows some protective (18), but T-bet pathogenic action (16). Notably, T-bet especially in DC is crucial for the pathogenesis of CIA (16). Data presented herein thus point to IL-36γ as T-bet-dependent/DC-derived pathogenic mediator of experimental arthritis. In fact, IL-36γ is expressed in joints of mice undergoing CIA, and as detected by exposure of cells to IL-36β, IL-36R activation up-regulates inflammatory parameters in human synoviocytes and articular chondrocytes, respectively (57). To investigate CIA along with IL-36γ deficiency is thus an important matter of future investigation.
Detailed reports on IL-36γ production by myeloid cells are missing, although one initial study in passing mentioned expression of the cytokine by LPS-stimulated monocytes (31). Moreover, one most recent report demonstrates up-regulation of IL-36γ mRNA by TLR7 ligation in cultured murine bone marrow-derived DC. However, likely due to short term activation for 6 h, induction remained modest, and molecular mechanisms activating the IL-36γ promoter were not investigated (58). Interestingly, expression of IL-36γ is detectable in mononuclear infiltrates of lesional psoriatic skin and actually correlates with that of IFNγ (38). It is tempting to speculate in this context that early IL-36γ derived from macrophages/DC may have the potential to efficiently perpetuate local production of inflammatory parameters by autocrine action on DC (59, 60) and paracrine action on adjacent cells such as CD4+ T cells and keratinocytes (53, 60). In fact, herein we confirm proinflammatory action of IL-36γ on primary human keratinocytes. Specifically, was mature IL-36γ, but not pro-IL-36γ, able to induce itself and beyond that crucial parameters of cutaneous inflammation such as TNFα, CCL20 (61), S100A7 (62), and iNOS (63). Data altogether reflect the inherent potential of IL-36γ to initiate a broad keratinocyte-driven inflammatory cascade. Induction of IL-36γ by T-bet in the partly overlapping macrophage/DC cell compartment as described herein likewise functionally relates IL-36γ to initiation of Th1-like immune responses. In fact, administration of the IL-36R ligand IL-36β to mice is associated with enhanced ex vivo Th1 activity (60) and with differentiation of naïve CD4+ T cells toward Th1 by IL-36R ligands in vitro (64). IL-36R ligands, including IL-36γ, moreover amplify DC maturation (59, 60). Current data thus altogether imply that IL-36γ derived from the macrophages/DC compartment is part of a self-amplifying loop (52) in which NF-κB, activated by IL-1/TNFα or TLR ligands (55), and T-bet, potentially activated by early innate IFNγ (65), may contribute to rapid implementation of strong immunoactivation either to install efficient host defense or as crucial component of tissue-destructive inflammatory processes.
This work was supported by the August Scheidel-Stiftung, Frankfurt, Germany (to M. B.).
Microarray data are available at the NCBI GEO accession number GSE37595.
- T-bet
- T-box expressed in T cells
- CIA
- collagen-induced arthritis
- DC
- dendritic cell
- IKK
- IB kinase
- NF-B
- nuclear factor-B
- PMA
- phorbol-12-myristate-13-acetate
- TLR
- Toll-like receptor
- nt
- nucleotide
- ANOVA
- analysis of variance
- iNOS
- inducible NOS.
REFERENCES
- 1. Lazarevic V., Glimcher L. H. (2011) T-bet in disease. Nat. Immunol. 12, 597–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Powell N., Canavan J. B., MacDonald T. T., Lord G. M. (2010) Transcriptional regulation of the mucosal immune system mediated by T-bet. Mucosal. Immunol. 3, 567–577 [DOI] [PubMed] [Google Scholar]
- 3. Szabo S. J., Kim S. T., Costa G. L., Zhang X., Fathman C. G., Glimcher L. H. (2000) A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100, 655–669 [DOI] [PubMed] [Google Scholar]
- 4. Ravindran R., Foley J., Stoklasek T., Glimcher L. H., McSorley S. J. (2005) Expression of T-bet by CD4 T cells is essential for resistance to Salmonella infection. J. Immunol. 175, 4603–4610 [DOI] [PubMed] [Google Scholar]
- 5. Sullivan B. M., Jobe O., Lazarevic V., Vasquez K., Bronson R., Glimcher L. H., Kramnik I. (2005) Increased susceptibility of mice lacking T-bet to infection with Mycobacterium tuberculosis correlates with increased IL-10 and decreased IFN-γ production. J. Immunol. 175, 4593–4602 [DOI] [PubMed] [Google Scholar]
- 6. Lighvani A. A., Frucht D. M., Jankovic D., Yamane H., Aliberti J., Hissong B. D., Nguyen B. V., Gadina M., Sher A., Paul W. E., O'Shea J. J. (2001) T-bet is rapidly induced by interferon-γ in lymphoid and myeloid cells. Proc. Natl. Acad. Sci. U.S.A. 98, 15137–15142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lugo-Villarino G., Maldonado-Lopez R., Possemato R., Penaranda C., Glimcher L. H. (2003) T-bet is required for optimal production of IFN-γ and antigen-specific T cell activation by dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 100, 7749–7754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lugo-Villarino G., Ito S., Klinman D. M., Glimcher L. H. (2005) The adjuvant activity of CpG DNA requires T-bet expression in dendritic cells. Proc. Natl. Acad. Sci. U.S.A. 102, 13248–13253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lipscomb M. W., Chen L., Taylor J. L., Goldbach C., Watkins S. C., Kalinski P., Butterfield L. H., Wesa A. K., Storkus W. J. (2009) Ectopic T-bet expression licenses dendritic cells for IL-12-independent priming of type 1 T cells in vitro. J. Immunol. 183, 7250–7258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rutitzky L. I., Smith P. M., Stadecker M. J. (2009) T-bet protects against exacerbation of schistosome egg-induced immunopathology by regulating Th17-mediated inflammation. Eur. J. Immunol. 39, 2470–2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lazarevic V., Chen X., Shim J. H., Hwang E. S., Jang E., Bolm A. N., Oukka M., Kuchroo V. K., Glimcher L. H. (2011) T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORγt. Nat. Immunol. 12, 96–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lexberg M. H., Taubner A., Albrecht I., Lepenies I., Richter A., Kamradt T., Radbruch A., Chang H. D. (2010) IFN-γ and IL-12 synergize to convert in vivo generated Th17 into Th1/Th17 cells. Eur. J. Immunol. 40, 3017–3027 [DOI] [PubMed] [Google Scholar]
- 13. Kriegova E., Fillerova R., Tomankova T., Hutyrova B., Mrazek F., Tichy T., Kolek V., du Bois R. M., Petrek M. (2011) T-helper cell type-1 transcription factor T-bet is up-regulated in pulmonary sarcoidosis. Eur. Respir. J. 38, 1136–1144 [DOI] [PubMed] [Google Scholar]
- 14. Martinez F. O., Gordon S., Locati M., Mantovani A. (2006) Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization. New molecules and patterns of gene expression. J. Immunol. 177, 7303–7311 [DOI] [PubMed] [Google Scholar]
- 15. Bettelli E., Sullivan B., Szabo S. J., Sobel R. A., Glimcher L. H., Kuchroo V. K. (2004) Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J. Exp. Med. 200, 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang J., Fathman J. W., Lugo-Villarino G., Scimone L., von Andrian U., Dorfman D. M., Glimcher L. H. (2006) Transcription factor T-bet regulates inflammatory arthritis through its function in dendritic cells. J. Clin. Invest. 116, 414–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Krakowski M., Owens T. (1996) Interferon-γ confers resistance to experimental allergic encephalomyelitis. Eur. J. Immunol. 26, 1641–1646 [DOI] [PubMed] [Google Scholar]
- 18. Manoury-Schwartz B., Chiocchia G., Bessis N., Abehsira-Amar O., Batteux F., Muller S., Huang S., Boissier M. C., Fournier C. (1997) High susceptibility to collagen-induced arthritis in mice lacking IFN-γ receptors. J. Immunol. 158, 5501–5506 [PubMed] [Google Scholar]
- 19. Mühl H., Pfeilschifter J. (2003) Anti-inflammatory properties of proinflammatory interferon-γ. Int. Immunopharmacol. 3, 1247–1255 [DOI] [PubMed] [Google Scholar]
- 20. Bachmann M., Dragoi C., Poleganov M. A., Pfeilschifter J., Mühl H. (2007) Interleukin-18 directly activates T-bet expression and function via p38 mitogen-activated protein kinase and nuclear factor-κB in acute myeloid leukemia-derived predendritic KG-1 cells. Mol. Cancer Ther. 6, 723–731 [DOI] [PubMed] [Google Scholar]
- 21. Dinarello C. A., Novick D., Puren A. J., Fantuzzi G., Shapiro L., Mühl H., Yoon D. Y., Reznikov L. L., Kim S. H., Rubinstein M. (1998) Overview of interleukin-18. More than an interferon-γ inducing factor. J. Leukoc. Biol. 63, 658–664 [PubMed] [Google Scholar]
- 22. Mühl H., Pfeilschifter J. (2004) Interleukin-18 bioactivity. A novel target for immunopharmacological anti-inflammatory intervention. Eur. J. Pharmacol. 500, 63–71 [DOI] [PubMed] [Google Scholar]
- 23. St Louis D. C., Woodcock J. B., Franzoso G., Blair P. J., Carlson L. M., Murillo M., Wells M. R., Williams A. J., Smoot D. S., Kaushal S., Grimes J. L., Harlan D. M., Chute J. P., June C. H., Siebenlist U., Lee K. P. (1999) Evidence for distinct intracellular signaling pathways in CD34+ progenitor to dendritic cell differentiation from a human cell line model. J. Immunol. 162, 3237–3248 [PubMed] [Google Scholar]
- 24. Dinarello C., Arend W., Sims J., Smith D., Blumberg H., O'Neill L., Goldbach-Mansky R., Pizarro T., Hoffman H., Bufler P., Nold M., Ghezzi P., Mantovani A., Garlanda C., Boraschi D., Rubartelli A., Netea M., van der Meer J., Joosten L., Mandrup-Poulsen T., Donath M., Lewis E., Pfeilschifter J., Martin M., Kracht M., Muehl H., Novick D., Lukic M., Conti B., Solinger A., Kelk P., Peyman K., van de Veerdonk F., Gabel C. (2010) IL-1 family nomenclature. Nat. Immunol. 11, 973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bachmann M., Paulukat J., Pfeilschifter J., Mühl H. (2009) Molecular mechanisms of IL-18BP regulation in DLD-1 cells. Pivotal direct action of the STAT1/GAS axis on the promoter level. J. Cell Mol. Med. 13, 1987–1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Iwamoto S., Iwai S., Tsujiyama K., Kurahashi C., Takeshita K., Naoe M., Masunaga A., Ogawa Y., Oguchi K., Miyazaki A. (2007) TNF-α drives human CD14+ monocytes to differentiate into CD70+ dendritic cells evoking Th1 and Th17 responses. J. Immunol. 179, 1449–1457 [DOI] [PubMed] [Google Scholar]
- 27. Sallusto F., Lanzavecchia A. (1994) Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and down-regulated by tumor necrosis factor α. J. Exp. Med. 179, 1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Paulukat J., Bosmann M., Nold M., Garkisch S., Kämpfer H., Frank S., Raedle J., Zeuzem S., Pfeilschifter J., Mühl H. (2001) Expression and release of IL-18 binding protein in response to IFN-γ. J. Immunol. 167, 7038–7043 [DOI] [PubMed] [Google Scholar]
- 29. Debets R., Timans J. C., Homey B., Zurawski S., Sana T. R., Lo S., Wagner J., Edwards G., Clifford T., Menon S., Bazan J. F., Kastelein R. A. (2001) Two novel IL-1 family members, IL-1δ and IL-1 ϵ, function as an antagonist and agonist of NF-κB activation through the orphan IL-1 receptor-related protein 2. J. Immunol. 167, 1440–1446 [DOI] [PubMed] [Google Scholar]
- 30. Dinarello C. A. (2011) Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kumar S., McDonnell P. C., Lehr R., Tierney L., Tzimas M. N., Griswold D. E., Capper E. A., Tal-Singer R., Wells G. I., Doyle M. L., Young P. R. (2000) Identification and initial characterization of four novel members of the interleukin-1 family. J. Biol. Chem. 275, 10308–10314 [DOI] [PubMed] [Google Scholar]
- 32. Cho J. Y., Grigura V., Murphy T. L., Murphy K. (2003) Identification of cooperative monomeric Brachyury sites conferring T-bet responsiveness to the proximal IFN-γ promoter. Int. Immunol. 15, 1149–1160 [DOI] [PubMed] [Google Scholar]
- 33. Carreira S., Dexter T. J., Yavuzer U., Easty D. J., Goding C. R. (1998) Brachyury-related transcription factor Tbx2 and repression of the melanocyte-specific TRP-1 promoter. Mol. Cell Biol. 18, 5099–5108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kispert A., Hermann B. G. (1993) The Brachyury gene encodes a novel DNA-binding protein. EMBO J. 12, 4898–4899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tada M., Smith J. C. (2001) T-targets. Clues to understanding the functions of T-box proteins. Dev. Growth Differ. 43, 1–11 [DOI] [PubMed] [Google Scholar]
- 36. Edwards A. D., Diebold S. S., Slack E. M., Tomizawa H., Hemmi H., Kaisho T., Akira S., Reis e Sousa C. (2003) Toll-like receptor expression in murine DC subsets. Lack of TLR7 expression by CD8 α+ DC correlates with unresponsiveness to imidazoquinolines. Eur. J. Immunol. 33, 827–833 [DOI] [PubMed] [Google Scholar]
- 37. Benoit M., Desnues B., Mege J. L. (2008) Macrophage polarization in bacterial infections. J. Immunol. 181, 3733–3739 [DOI] [PubMed] [Google Scholar]
- 38. Carrier Y., Ma H. L., Ramon H. E., Napierata L., Small C., O'Toole M., Young D. A., Fouser L. A., Nickerson-Nutter C., Collins M., Dunussi-Joannopoulos K., Medley Q. G. (2011) Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo. Implications in psoriasis pathogenesis. J. Invest. Dermatol. 131, 2428–2437 [DOI] [PubMed] [Google Scholar]
- 39. Johnston A., Xing X., Guzman A. M., Riblett M., Loyd C. M., Ward N. L., Wohn C., Prens E. P., Wang F., Maier L. E., Kang S., Voorhees J. J., Elder J. T., Gudjonsson J. E. (2011) IL-1F5, -F6, -F8, and -F9. A novel IL-1 family signaling system that is active in psoriasis and promotes keratinocyte antimicrobial peptide expression. J. Immunol. 186, 2613–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Muhr P., Zeitvogel J., Heitland I., Werfel T., Wittmann M. (2011) Expression of interleukin (IL)-1 family members upon stimulation with IL-17 differs in keratinocytes derived from patients with psoriasis and healthy donors. Br. J. Dermatol. 165, 189–193 [DOI] [PubMed] [Google Scholar]
- 41. Beima K. M., Miazgowicz M. M., Lewis M. D., Yan P. S., Huang T. H., Weinmann A. S. (2006) T-bet binding to newly identified target gene promoters is cell type-independent but results in variable context-dependent functional effects. J. Biol. Chem. 281, 11992–12000 [DOI] [PubMed] [Google Scholar]
- 42. Jenner R. G., Townsend M. J., Jackson I., Sun K., Bouwman R. D., Young R. A., Glimcher L. H., Lord G. M. (2009) The transcription factors T-bet and GATA-3 control alternative pathways of T-cell differentiation through a shared set of target genes. Proc. Natl. Acad. Sci. U.S.A. 106, 17876–17881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matsuda J. L., George T. C., Hagman J., Gapin L. (2007) Temporal dissection of T-bet functions. J. Immunol. 178, 3457–3465 [DOI] [PubMed] [Google Scholar]
- 44. Lewis D. A., Hengeltraub S. F., Gao F. C., Leivant M. A., Spandau D. F. (2006) Aberrant NF-κB activity in HaCaT cells alters their response to UVB signaling. J. Invest. Dermatol. 126, 1885–1892 [DOI] [PubMed] [Google Scholar]
- 45. Stewart J. J., Fischbeck J. A., Chen X., Stargell L. A. (2006) Non-optimal TATA elements exhibit diverse mechanistic consequences. J. Biol. Chem. 281, 22665–22673 [DOI] [PubMed] [Google Scholar]
- 46. Rice K. L., Heidari M., Taplin R. H., Kees U. R., Greene W. K. (2009) Transcriptional regulation of the human ALDH1A1 promoter by the oncogenic homeoprotein TLX1/HOX11. Hematol. Rev. 1, e13 [Google Scholar]
- 47. Dinarello C. A. (1996) Biologic basis for interleukin-1 in disease. Blood 87, 2095–2147 [PubMed] [Google Scholar]
- 48. Lind H., Haugen A., Zienolddiny S. (2007) Differential binding of proteins to the IL1B-31 T/C polymorphism in lung epithelial cells. Cytokine 38, 43–48 [DOI] [PubMed] [Google Scholar]
- 49. Taylor S. L., Renshaw B. R., Garka K. E., Smith D. E., Sims J. E. (2002) Genomic organization of the interleukin-1 locus. Genomics 79, 726–733 [DOI] [PubMed] [Google Scholar]
- 50. Towne J. E., Garka K. E., Renshaw B. R., Virca G. D., Sims J. E. (2004) Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to NF-κB and MAPKs. J. Biol. Chem. 279, 13677–13688 [DOI] [PubMed] [Google Scholar]
- 51. Towne J. E., Renshaw B. R., Douangpanya J., Lipsky B. P., Shen M., Gabel C. A., Sims J. E. (2011) Interleukin-36 (IL-36) ligands require processing for full agonist (IL-36α, IL-36β, and IL-36γ) or antagonist (IL-36Ra) activity. J. Biol. Chem. 286, 42594–42602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Towne J. E., Sims J. E. (2012) IL-36 in psoriasis. Curr. Opin. Pharmacol. 12, 486–490 [DOI] [PubMed] [Google Scholar]
- 53. Marrakchi S., Guigue P., Renshaw B. R., Puel A., Pei X. Y., Fraitag S., Zribi J., Bal E., Cluzeau C., Chrabieh M., Towne J. E., Douangpanya J., Pons C., Mansour S., Serre V., Makni H., Mahfoudh N., Fakhfakh F., Bodemer C., Feingold J., Hadj-Rabia S., Favre M., Genin E., Sahbatou M., Munnich A., Casanova J. L., Sims J. E., Turki H., Bachelez H., Smahi A. (2011) Interleukin-36 receptor antagonist deficiency and generalized pustular psoriasis. N. Engl. J. Med. 365, 620–628 [DOI] [PubMed] [Google Scholar]
- 54. van de Veerdonk F. L., Stoeckman A. K., Wu G., Boeckermann A. N., Azam T., Netea M. G., Joosten L. A., van der Meer J. W., Hao R., Kalabokis V., Dinarello C. A. (2012) IL-38 binds to the IL-36 receptor and has biological effects on immune cells similar to IL-36 receptor antagonist. Proc. Natl. Acad. Sci. U.S.A. 109, 3001–3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lian L. H., Milora K. A., Manupipatpong K. K., Jensen L. E. (2012) The double-stranded RNA analogue polyinosinic-polycytidylic acid induces keratinocyte pyroptosis and release of IL-36γ. J. Invest. Dermatol. 132, 1346–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blumberg H., Dinh H., Trueblood E. S., Pretorius J., Kugler D., Weng N., Kanaly S. T., Towne J. E., Willis C. R., Kuechle M. K., Sims J. E., Peschon J. J. (2007) Opposing activities of two novel members of the IL-1 ligand family regulate skin inflammation. J. Exp. Med. 204, 2603–2614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Magne D., Palmer G., Barton J. L., Mézin F., Talabot-Ayer D., Bas S., Duffy T., Noger M., Guerne P. A., Nicklin M. J., Gabay C. (2006) The new IL-1 family member IL-1F8 stimulates production of inflammatory mediators by synovial fibroblasts and articular chondrocytes. Arthritis Res. Ther. 8, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tortola L., Rosenwald E., Abel B., Blumberg H., Schäfer M., Coyle A. J., Renauld J. C., Werner S., Kisielow J., Kopf M. (2012) Psoriasiform dermatitis is driven by IL-36-mediated DC-keratinocyte crosstalk. J. Clin. Invest., 122, 3965–3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mutamba S., Allison A., Mahida Y., Barrow P., Foster N. (2012) Expression of IL-1Rrp2 by human myelomonocytic cells is unique to DCs and facilitates DC maturation by IL-1F8 and IL-1F9. Eur. J. Immunol. 42, 607–617 [DOI] [PubMed] [Google Scholar]
- 60. Vigne S., Palmer G., Lamacchia C., Martin P., Talabot-Ayer D., Rodriguez E., Ronchi F., Sallusto F., Dinh H., Sims J. E., Gabay C. (2011) IL-36R ligands are potent regulators of dendritic and T cells. Blood 118, 5813–5823 [DOI] [PubMed] [Google Scholar]
- 61. Schön M. P., Boehncke W. H. (2005) Psoriasis. N. Engl. J. Med. 352, 1899–1912 [DOI] [PubMed] [Google Scholar]
- 62. Eckert R. L., Broome A. M., Ruse M., Robinson N., Ryan D., Lee K. (2004) S100 proteins in the epidermis. J. Invest. Dermatol. 123, 23–33 [DOI] [PubMed] [Google Scholar]
- 63. Mühl H., Bachmann M., Pfeilschifter J. (2011) Inducible NO synthase and antibacterial host defence in times of Th17/Th22/T22 immunity. Cell Microbiol. 13, 340–348 [DOI] [PubMed] [Google Scholar]
- 64. Vigne S., Palmer G., Martin P., Lamacchia C., Strebel D., Rodriguez E., Olleros M. L., Vesin D., Garcia I., Ronchi F., Sallusto F., Sims J. E., Gabay C. (2012) IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood 120, 3478–3487 [DOI] [PubMed] [Google Scholar]
- 65. Wehner R., Dietze K., Bachmann M., Schmitz M. (2011) The bidirectional cross-talk between human dendritic cells and natural killer cells. J. Innate Immun. 3, 258–263 [DOI] [PubMed] [Google Scholar]