

Background: BDNF promotes axonal outgrowth, whereas the underlying molecular mechanism has remained unclear.
Results: LIMK1 interacts with TrkB, and BDNF treatment could induce LIMK1 dimerization and transactivation, which facilitate axonal elongation.
Conclusion: LIMK1-regulated actin dynamics is essential for BDNF-induced axonal elongation.
Significance: These results revealed a novel mechanism for the BDNF-induced axonal elongation.
Keywords: Axon, BDNF, Neurite Outgrowth, Signal Transduction, Tyrosine-Protein Kinase (Tyrosine Kinase), LIMK1, TrkB, Axonal Elongation, Dimerization
Abstract
BDNF/TrkB signaling plays critical roles in axonal outgrowth of neurons, the process of which requires the remodeling of the cytoskeleton structure, including microtubules and filamentous actin. However, the mechanism by which BDNF/TrkB signaling regulates cytoskeleton reorganization is still unclear. Here, we identified a novel interaction between LIMK1 and TrkB, which is required for the BDNF-induced axonal elongation. We demonstrated that BDNF-induced TrkB dimerization led to LIMK1 dimerization and transphosphorylation independent of TrkB kinase activity, which could further enhance the activation and stabilization of LIMK1. Moreover, activated LIMK1 translocated to the membrane fraction and phosphorylated its substrate cofilin, thus promoting actin polymerization and axonal elongation. Our findings provided evidence of a novel mechanism for the BDNF-mediated signal transduction leading to axonal elongation.
Introduction
BDNF2 belongs to the neurotrophin family known as a group of structurally related polypeptide growth factors. The functions of BDNF depend on two distinct transmembrane receptors as follows: the high affinity TrkB receptor (a member of the receptor tyrosine kinase of the Trk subfamily) and the low affinity p75 neurotrophin receptor (p75NTR) (a member of the tumor necrosis factor family) (1, 2). Among all neurotrophins and their Trk receptors, BDNF and its major receptor TrkB are widespread and enriched in the central nervous system (CNS), and the BDNF/TrkB signaling is exceedingly important for development and normal functions of the brain (1, 3, 4). BDNF binding to TrkB receptor leads to its dimerization and activation and subsequently induces the activation of several intracellular signaling cascades (2). In the adult CNS, the BDNF/TrkB signaling is involved in synaptic plasticity and long term potentiation (3, 5). BDNF/TrkB also plays critical roles in the developing neurons such as differentiation, survival, neurite outgrowth, and branching (6–8). Self-amplifying BDNF actions through BDNF/TrkB signaling promoted axonal differentiation and growth (9). Neuronal activity could elevate the levels of surface TrkB, which made BDNF easier to promote axonal outgrowth (7, 10, 11). Deletion of TrkB decreased dendritic spine and axon varicosity density (12). The cytoskeleton of newly formed neurites is composed of microtubules with a cortex of actin close to the plasma membrane (13). Although BDNF/TrkB signaling plays an important role in axonal outgrowth through actin dynamics and microtubule assembly regulation (14), the underlying molecular mechanism has remained unclear.
The LIM kinase family of serine/threonine kinases includes two highly related members, the brain-specific LIMK1 and the ubiquitously expressed LIMK2 (15, 16). The activities of LIM kinases are modulated by Rho-GTPases through their effector kinases, Rho kinase, the p21-activated kinases 1 and 4. They phosphorylate and activate LIM kinases on conserved threonine residues, Thr-508 in LIMK1 and Thr-505 in LIMK2 (17, 18). LIM kinases regulate the architecture of actin cytoskeleton by phosphorylation and inactivation of the actin depolymerization factors ADF/cofilin, thereby participating in numerous biological processes of different cell types (18–23). LIMK1 is particularly important for regulation of neuronal morphology such as axonal outgrowth. Genetic deletion of LIMK1 results in reduction of growth cones and abnormality of dendrite morphology in vivo (19), and up-regulation of LIMK1 protein levels promotes axonal extension in cultured hippocampal neurons (22).
A growing body of evidence suggests that both BDNF/TrkB- and LIMK1/cofilin-regulated actin dynamics are involved in growth cone motility, neurite extension, synaptogenesis, and long term potentiation in neurons (24–27), which leads us to hypothesize that there may exist cross-talk between BDNF/TrkB and LIMK1/cofilin pathways. Recent studies indicated that BDNF could activate Rac and p21-activated kinase, the upstream kinases of LIMK1 (26, 28). In addition, BDNF could increase LIMK1 protein levels by relieving the microRNA miR-134 inhibition of Limk1 translation (29). However, until now there has been no direct evidence that BDNF could regulate LIMK1 activity. Here, we present evidence that LIMK1 could directly bind to TrkB, and BDNF treatment could induce LIMK1 activation and stabilization, which facilitate axonal outgrowth in hippocampal neurons.
EXPERIMENTAL PROCEDURES
Reagents and Antibodies
Human recombinant BDNF was purchased from PeproTech (Rocky Hill, NJ). K252a was from Calbiochem. Cycloheximide (CHX) was purchased from Sigma. The restriction enzymes were purchased from Fermentas (Hanover, MD). Biotinylated Tat-JMBox4 peptide was synthesized and purified by GL Biochem (Shanghai) Ltd. The Tat-JMBox4 peptide containing a 9-amino acid sequence (VIENPQYFG), which mimics the LIMK1-binding site in TrkB, was fused to cell-permeable Tat (GRRRRRRRRRRR) to interfere with the interaction between LIMK1 and TrkB. The other reagents were obtained from Sigma, unless indicated otherwise.
Sources of antibodies are as follows. Rabbit anti-phospho-cofilin (Ser-3), rabbit anti-cofilin, rabbit anti-phospho-LIMK1/2 (Thr-508/Thr-505) and rabbit anti-LIMK1 antibodies were obtained from Cell Signaling Technology (Beverly, MA). Rabbit anti-calnexin, mouse anti-FLAG (M2), rabbit anti-HA, rabbit anti-actin and mouse anti-α-tubulin antibodies were obtained from Sigma. Mouse anti-TrkB and mouse anti-LIMK1 antibodies were purchased from Pharmingen. Mouse anti-Tau-1, rabbit anti-TrkB, horseradish peroxidase (HRP)-conjugated goat anti-rabbit, or mouse IgGs were purchased from Millipore (Temecula, CA). Chicken anti-MAP2 was purchased from Abcam (Cambridge, UK). Rabbit anti-Trk (C-14) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-FLAG was from Thermo (Rockford, IL). Mouse anti-HA (HA.11) and mouse anti-neuronal class III β-tubulin (Tuj-1) were from Covance (Princeton, NJ). Rabbit anti-GFP, Alexa Fluor 594- or 488-conjugated goat or donkey anti-mouse or rabbit IgG heavy and light chains (H+L), Alexa Fluor 488-conjugated streptavidin, Alexa Fluor 594-conjugated phalloidin, and rabbit IgG were purchased from Invitrogen. Dylight 488-affinipure F(ab′)2 fragment donkey anti-chicken IgG (H+L) secondary antibody was purchased from Jackson Immunoresearch (West Grove, PA).
Plasmid Constructs
Rat FLAG-TrkB expression construct and its mutants were described previously (30). The human LIMK1 cDNA in a pENTR223.1 vector was purchased from Open Biosystems. HA-tagged (C-terminal) human LIMK1 constructs were generated as follows. The cDNA encoding the wild-type LIMK1 was amplified by PCR (forward primer 5′-GCCCAAGCTTGCCGCCACCATGAGGTTGACGCTACTTTGT-3′ and reverse primer 5′-GCTCTAGATTAGGCGTAGTCGGGCACGTCGTAAGGATAGTCGGGGACCTCAGGGTGGGC-3′). Amplified cDNAs were subcloned as HindIII-XbaI fragment into the pcDNA3.1 expression vector (Invitrogen). The C-terminal HA-tagged human LIMK1 was also subcloned into pCAGIG (Addgene Plasmid Repository) expression vector by EcoRV and NotI sites. The FLAG-T1-JMBox4 construct was generated by means of PCR. Small interfering RNAs (siRNAs) of LIMK1 were subcloned into the pSuper-EGFP vectors (OligoEngine, Seattle, WA). The target sequence for rat Limk1 genes was described previously (31, 32), 5′-GACTTGCGTAGCCTTAAGA-3′ (LIMK1 siRNA1) and 5′-GCTGGAACAATGGCTAGAA-3′ (LIMK1 siRNA2). All the constructs were confirmed by DNA sequencing.
Cell Cultures and Transfections
HEK293 and PC12 cells were cultured as described previously (33). Briefly, HEK293 cells were cultured in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% fetal bovine serum (FBS), 2 mm l-glutamine, and 100 units/ml penicillin/streptomycin. PC12 cells were grown in DMEM containing 5% FBS, 10% horse serum, 2 mm l-glutamine, and 100 units/ml penicillin/streptomycin. Dissociated cells were centrifuged and resuspended in electroporation buffer, mixed with 2–4 μg of plasmid, and electroporated (Amaxa Biosystems) using the fixed program (A-023 for HEK293 and U-029 for PC12). Electroporated cells were transferred to poly-d-lysine (Sigma)-coated dishes. 48–72 h after transfection, lysates were collected and analyzed by immunoprecipitation and immunoblotting.
Dissociated hippocampal neurons from embryonic day 18 (E18) Sprague-Dawley rat brains were prepared as described previously (30). In brief, embryos were removed at E18 and then placed into ice-cold Hanks' balanced salt solution without Ca2+ and Mg2+. Following cleaning of meninges, the hippocampi were collected, digested with 0.05% trypsin/EDTA for 10 min at 37 °C, and dissociated with a fire-polished Pasteur pipette in a 1:1 mixture of DMEM/F-12 (Invitrogen) with 10% FBS. Neurons were collected after centrifugation at 80 × g for 8 min. Neurons were then transfected by electroporation using the fixed program (O-003) and plated onto a culture dish coated with 0.1 mg/ml poly-d-lysine. Neurons were cultured in Neurobasal medium (Invitrogen) plus 2% B27 (Invitrogen), 0.5 mm l-glutamine (Invitrogen), and 100 units/ml penicillin/streptomycin (Sigma) at 37 °C with 5% CO2. In some experiments, neurons cultured for 2 days in vitro (DIV2) were transfected using Lipofectamine 2000 (Invitrogen) in Opti-MEM (Invitrogen) following the manufacturer's instructions. 4 h after transfection, the Opti-MEM was changed to Neurobasal medium with or without 50 ng/ml BDNF. 20 h later, neurons were fixed for immunofluorescence staining. The lengths of axon (Tau-1-immunolabeled) and dendrite (MAP2-immunolabeled) were measured at DIV3 and DIV5, respectively.
BDNF Stimulation and Western Blot Analysis
Hippocampal neurons (DIV3) were serum-starved for 10 h and stimulated with BDNF (50 ng/ml) for 20 min. For the inhibitor treatment, serum-starved cultures were pretreated with K252a (100 nm) or Tat-JMBox4 (1 μm) in Neurobasal medium for 30 min at 37 °C and 5% CO2 and stimulated with BDNF (50 ng/ml) in the presence of the inhibitors. For the half-life experiments, neurons were pretreated with CHX (20 μg/ml) and Tat-JMBox4 (1 μm) at 37 °C. 30 min later, neurons were stimulated with BDNF (50 ng/ml) for the indicated times. Following stimulation, neurons were lysed with TNE lysis buffer (10 mm Tris at pH 7.4, 150 mm NaCl, 1 mm EDTA, and 1% Nonidet P-40) containing protease and phosphatase inhibitors (Roche Applied Science). Cell debris was removed by centrifugation at 16,000 × g for 15 min. Protein extracts were boiled with SDS sample buffer (Invitrogen). For the LIMK1 homo-dimerization experiment, protein extracts were boiled with SDS sample buffer without dithiothreitol (DTT). Western blot analysis was carried out according to the protocol described previously (20). In brief, equal volumes of samples were loaded and separated by SDS-PAGE and transferred to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad) in a Tris-glycine buffer containing 20% methanol. The PVDF membranes were blocked for 1 h in TBS containing 5% BSA or 5% nonfat milk and then immunoblotted with the respective primary antibodies in TBS containing 3% BSA or 3% nonfat milk overnight at 4 °C, washed three times in TBS containing 0.1% Tween 20, and followed by incubation with the corresponding HRP-conjugated secondary antibody (1:5000) for 1 h at room temperature. After washing three times in TBS with 0.1% Tween 20, the Western blot signals were detected using the ECL detection kit (Millipore, Temecula, CA) and quantified by Quantity One (Bio-Rad).
Cell Fractionation
Cytoplasmic and membrane-associated proteins were isolated from primary hippocampal neurons using the ProteoJETTM membrane protein extraction kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's instructions (34). In brief, plated neurons in a 6-cm culture plate (5 × 106 cells) were stimulated with BDNF (50 ng/ml) for 30 min; then the culture medium was removed, and the cells were rinsed once with 4 ml of ice-cold Cell Wash Solution. 1.5 ml of ice-cold Cell Permeabilization Buffer was added into the plate and incubated for 10 min at 4 °C with gentle rocking. All liquid from permeabilized neurons was collected as cytoplasmic fraction. Then the permeabilized neurons were incubated in 1 ml of ice-cold Membrane Protein Extraction Buffer for 30 min at 4 °C with constant shaking. The mixture was collected and centrifuged at 16,000 × g for 15 min at 4 °C; the supernatant was transferred into a new tube as a membrane protein fraction. To determine protein localization and extraction efficiency, fractions were subjected to SDS-PAGE and immunoblotted with primary antibody corresponding to either LIMK1, calnexin (membrane protein), or tubulin (cytosol protein).
Co-immunoprecipitation and Immunoblotting
48 h after electroporation with the indicated constructs, HEK293 cells were lysed in 500 μl of TNE lysis buffer containing protease and phosphatase inhibitors and were centrifuged at 16,000 × g for 15 min. The supernatants were transferred to a new tube, and 60 μl of cell lysates were directly denatured by SDS sample buffer. The rest were incubated with rabbit anti-FLAG antibody or rabbit anti-HA antibody for 2 h at 4 °C and then mixed with protein A-Sepharose (Sigma) overnight at 4 °C. After washing three times, the immunoprecipitates were eluted, boiled in SDS sample buffer, then separated on SDS-PAGE, and immunoblotted with the indicated antibodies.
For endogenous interaction, rat brain or cultured neurons were lysed in TNE lysis buffer with protease and phosphatase inhibitors and centrifuged at 16,000 × g for 15 min at 4 °C. Equal volumes of lysates were incubated with rabbit anti-TrkB (1:200) antibody or rabbit IgG (1:200), respectively. The immunocomplex was precipitated by protein A-Sepharose beads. The bound proteins were denatured and analyzed by immunoblotting with mouse anti-LIMK1 and mouse anti-TrkB antibodies.
Immunofluorescence Staining
Immunofluorescence experiments were carried out as described previously (20). Neurons were fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.4% Triton X-100 for 30 min, and blocked with 10% goat serum or donkey serum for 1 h at room temperature. After incubation with the primary antibodies overnight at 4 °C, neurons were washed three times with PBS and incubated with their respective subtype-specific fluorescent secondary antibodies for 1 h at room temperature. Fluorescent images were obtained by a Zeiss LSM780 microscope (Microstructural Platform of Shandong University), and quantification of axonal and dendritic length and fluorescence intensity of growth cones was performed using MetaMorph software (Universal Imaging Corporation, West Chester, PA).
Statistical Analysis
Data were assessed by Student's t test or analysis of variance (one-way ANOVA), followed by post hoc tests. All values in the figures represent means ± S.E. Differences were considered significant at p < 0.05.
RESULTS
Identification of LIMK1 as a Novel TrkB-interacting Protein
In our yeast two-hybrid screening, we used the intracellular domain of TrkB (amino acids 454–821) as a bait to screen a human brain cDNA library. Among the 98 positive clones, three clones, which encoded LIMK1, were isolated. To corroborate the interplay between LIMK1 and TrkB, HA-tagged LIMK1 was co-transfected with FLAG-tagged TrkB in HEK293 cells, and a co-immunoprecipitation experiment was carried out to examine whether TrkB could form a complex with LIMK1. FLAG-TrkB was clearly detected by HA-LIMK1 immunoprecipitates, and conversely HA-LIMK1 was observed in FLAG-TrkB immunoprecipitates, which suggested that LIMK1 and TrkB could associate with each other in physical conditions (Fig. 1, A and B). To exclude the possibility that this interaction was an artifact because of protein overexpression, the endogenous TrkB/LIMK1 co-immunoprecipitation assay was performed using rat brain lysates. We found that LIMK1 was co-immunoprecipitated with TrkB by specific TrkB antibody but not by a preimmune IgG under the endogenous level (Fig. 1C). We also examined the endogenous interaction between the two proteins using hippocampal neuron cultures. We found that BDNF stimulation could enhance the interaction between LIMK1 and TrkB (Fig. 4D). Furthermore, we performed immunostaining experiments to compare the subcellular distribution of LIMK1 and TrkB in the primary cultured hippocampal neurons (DIV3). Punctate staining of LIMK1 was widely distributed and principally aggregated in the perinuclear region and growth cones and partially co-localized with TrkB (Fig. 1D). Because of the expression of p75NTR and TrkB isoform 1 (T1) in hippocampal neurons, we also examined the interaction between the overexpressed HA-LIMK1 and FLAG-p75NTR or FLAG-T1. We found that they could not form a complex with LIMK1 (data not shown). These results suggest that LIMK1 has the specific ability to form a complex with TrkB in hippocampal neurons.
FIGURE 1.

Identification of LIMK1 as a novel TrkB-interacting protein. A and B, LIMK1 associates with the TrkB receptor. Lysates from HEK293 cells transfected with HA-LIMK1 and/or FLAG-TrkB constructs were immunoprecipitated (IP) with anti-HA or anti-FLAG antibody. Then Western blot analysis was performed to detect immunoprecipitated proteins. IB, immunoblot. C, endogenous LIMK1 interacts with TrkB receptor. Rat brain lysates were subjected to immunoprecipitation with polyclonal rabbit anti-TrkB antibody or normal rabbit IgG followed by immunoblotting with mouse anti-TrkB and mouse anti-LIMK1 antibodies, respectively. D, LIMK1 co-localizes with the TrkB receptor in hippocampal neurons. The hippocampal pyramidal neurons transfected with HA-LIMK1 were immunostained with mouse anti-LIMK1 (red) and rabbit anti-Trk (C-14) (green) antibodies. Lower panels showed enlarged images of framed regions. The arrows indicate LIMK1 and TrkB co-localization. Scale bar, 20 μm.
FIGURE 4.

TrkB/LIMK1 interaction is required for BDNF-induced LIMK1 homo-dimerization, phosphorylation, and redistribution. A, Tat-JMBox4 peptide can efficiently transduce into cultured hippocampal neurons. Hippocampal neurons were treated with 1 μm biotinylated Tat-JMBox4 peptide for 30 min, then cells were fixed and immunostained with mouse anti-Tuj1 followed by incubation with Alexa Fluor 594-conjugated goat anti-mouse antibody and Alexa Fluor 488-conjugated streptavidin. Scale bar, 40 μm. B, Tat-JMBox4 peptide disrupts the TrkB/LIMK1 interaction in overexpressed HEK293 cells. HEK293 cells co-transfected with the indicated constructs were lysed 30 min after Tat-JMBox4 treatment (1 μm), and immunoprecipitation (IP) experiments were carried out as described in Fig. 1A. IB, immunoblot. C, quantification of the TrkB/LIMK1 association ratio (n = 3, **, p < 0.01; Student's t test). Con, control. D, Tat-JMBox4 peptide blocks BDNF-enhanced TrkB/LIMK1 interaction. Hippocampal neurons were pretreated with 1 μm Tat-JMBox4 peptide for 30 min followed by 50 ng/ml BDNF treatment for 30 min, and then cell lysates were collected and immunoprecipitated with polyclonal rabbit anti-TrkB antibody or normal rabbit IgG followed by immunoblotting with mouse anti-TrkB and mouse anti-LIMK1 antibodies, respectively. E, Tat-JMBox4 peptide blocks BDNF-induced LIMK1 homo-dimerization. Hippocampal neurons were treated as described previously in D, and then cells were lysed and examined by SDS-PAGE in the absence of DTT. F, Tat-JMBox4 peptide inhibits BDNF-induced phosphorylation of LIMK1 (Thr-508) and cofilin (Ser-3). Cultured hippocampal neurons were pretreated as described in Fig. 4D, and protein extracts were immunoblotted with the indicated antibodies. G, quantification of LIMK1 and cofilin phosphorylation (n = 3, *, p < 0.05; **, p < 0.01 versus control group; #, p < 0.05 versus BDNF-treated group; one-way ANOVA). H, Tat-JMBox4 peptide blocks BDNF-induced translocation of LIMK1. Serum-starved neurons were treated as described previously, and protein extracts were separated into cytosol and membrane fractions. I, quantification of LIMK1 levels in membrane and cytosol fractions (n = 3, *, p < 0.05 versus control group; #, p < 0.05 versus BDNF-treated group; one-way ANOVA).
BDNF Induces LIMK1 Homo-dimerization and Redistribution
TrkB, which is a receptor tyrosine kinase, could be activated by BDNF stimulation and transduce several signaling pathways (2). Given that LIMK1 could associate with TrkB, we considered whether the activity of LIMK1 could be modulated by BDNF stimulation in developing neurons, because both of them are essential for the development of CNS as described above. We used specific antibodies to detect p-LIMK1 (Thr-508), which is an active form of LIMK1, and total LIMK1. After 10 h of serum starvation, cultured neurons were stimulated with BDNF (50 ng/ml) for 30 min. Cell lysates were then collected for SDS-PAGE. Western blot analysis showed that p-LIMK1/LIMK1 was significantly increased after BDNF stimulation. Intriguingly, although there was a declining trend, the effect could not be totally blocked by pretreatment of K252a, which is a Trk tyrosine kinase inhibitor (Fig. 2, A and B). These data suggested that BDNF-induced LIMK1 phosphorylation (Thr-508) was independent of TrkB tyrosine kinase activity. Hence, we considered the following question. “What is the mechanism to interpret BDNF-induced phosphorylation of LIMK1?” BDNF and other neurotrophins binding to their receptors invoke the formation of noncovalently associated receptor dimers. The dimerization of receptors induces autophosphorylation in the kinase domain of the Trk receptors, followed by the activation of various signaling pathways (35). Coincidentally, the activity and stability of LIMK1 is also regulated by homo-dimerization (36). LIMK1 homo-dimerization could induce its transphosphorylation and thus increase LIMK1 activity. Hence, we tried to test whether BDNF could promote LIMK1 homo-dimerization. For this experiment, neurons were treated with or without BDNF (50 ng/ml) for 30 min, and cell extracts were subjected to SDS-PAGE in the absence or presence of dithiothreitol, which was used to reduce disulfide bonds and thus inhibit dimer formation, and then immunoblotted with anti-LIMK1 antibody. Indeed, we observed that BDNF stimulation led to increased dimerization of endogenous LIMK1 in hippocampal neurons, the effect of which was not inhibited by K252a (Fig. 2C). Other studies have reported that LIMK1 activation resulted in LIMK1 redistribution from the cytosol to the plasma membrane (37, 38). We then examined the subcellular localization of LIMK1 before and after BDNF treatment. Neuronal lysates were separated into membrane and cytosol fractions to assess the distribution of LIMK1. We used calnexin and α-tubulin as membrane and cytosolic protein markers, respectively, to examine the cross-contamination and extraction efficiency as described previously (34). Notably, we detected a significant increase in membrane-associated LIMK1 levels after BDNF stimulation. In the cytosol fraction, LIMK1 levels were decreased by BDNF stimulation (Fig. 2, D and E). Taken together, these data suggested that BDNF stimuli promoted LIMK1 homo-dimerization and subcellular redistribution and thus increased LIMK1 activity, the effect of which was independent of TrkB kinase activity.
FIGURE 2.

BDNF induces LIMK1 phosphorylation, homo-dimerization, and redistribution. A, BDNF stimuli increases phosphorylation (Thr-508) of LIMK1 in a TrkB tyrosine kinase activity-independent manner. Hippocampal neurons (DIV3) were serum-starved overnight and treated with vehicle or BDNF (50 ng/ml) for 30 min. K252a was pretreated to inhibit Trk kinase activity. Lysates were collected for SDS-PAGE and immunoblotted with the indicated antibodies. B, quantitation of LIMK1 phosphorylation (n = 4, *, p < 0.05; **, p < 0.01 versus control group; one-way ANOVA). Con, control; NS, not significant. C, BDNF induces LIMK1 homo-dimerization independent of TrkB receptor tyrosine kinase activity. Hippocampal neurons were treated as in A, and protein extracts were boiled with SDS sample buffer with or without dithiothreitol (DTT) followed by immunoblotting with anti-LIMK1 antibody. D, BDNF stimuli induces LIMK1 redistribution. Hippocampal neurons were serum-starved and stimulated with vehicle or BDNF (50 ng/ml) for 30 min, and then cytoplasmic and membrane fractions were isolated and immunoblotted with anti-LIMK1, anti-calnexin, or anti-tubulin antibodies. E, quantification of LIMK1 protein levels in membrane and cytosol respectively (n = 3, *, p < 0.05; **, p < 0.01; Student's t test).
Juxtamembrane Domain in TrkB Is Responsible for Its Interaction with LIMK1
To examine whether disrupting the interaction between TrkB and LIMK1 could block BDNF-induced LIMK1 dimerization and transactivation, first we need to identify the key domain in TrkB that mediates its binding with LIMK1. TrkB receptor consists of the following five regions: extracellular, transmembrane, juxtamembrane (JM), tyrosine kinase (TK), and C-terminal (CT). To examine the potential region in TrkB responsible for the interaction with LIMK1, various domains (C terminus, tyrosine kinase, and juxtamembrane) were gradually deleted from the full-length of TrkB (TrkB-FL) (Fig. 3A). Co-immunoprecipitation experiments showed that TrkB ΔCT and TrkB ΔTK but not TrkB-JM0 (deletion of the entire TrkB intracellular domain) could be pulled down by HA-LIMK1 (Fig. 3, B and C), suggesting that JM region in TrkB is necessary for its interaction with LIMK1. To further clarify the specific sequence in the JM domain responsible for this interaction, the 80-amino acid JM domain was divided into five parts as described previously (30) (Fig. 3D). We found TrkB-JM5 (TrkB ΔTK) and TrkB-JM4 but not TrkB-JM3, TrkB-JM2, TrkB-JM1, and TrkB-JM0 could be pulled down by HA-LIMK1 (Fig. 3, E and F), suggesting that the 9-amino acid sequence between JM3 and JM4 (VIENPQYFG named as JMBox4) is necessary for the TrkB/LIMK1 interaction. The truncated TrkB isoform 1 (T1) is highly expressed in the adult CNS containing a short cytoplasmic region lacking kinase domain (39). We found T1 could not associate with LIMK1 (Fig. 3G). Nevertheless, the chimeric receptor with JMBox4 transplanted into T1 (T1-JMBox4) could be immunoprecipitated by HA-LIMK1 (Fig. 3G), which indicated that the 9-amino acid sequence of JMBox4 in TrkB is not only necessary but is also sufficient for TrkB/LIMK1 interaction. As the JMBox4 sequence in TrkB has high similarity with the amino acid sequence in TrkA or TrkC (Fig. 3I), we therefore examined whether the other Trks can also interact with LIMK1. Not surprisingly, we found that LIMK1 can indeed associate with TrkA or TrkC (Fig. 3H).
FIGURE 3.

Juxtamembrane region in TrkB is responsible for its interaction with LIMK1. A, schematic presentation of FLAG-TrkB and its deletion constructs. ΔCT indicates deletion of the C-terminal region of TrkB; ΔTK indicates deletion of both tyrosine kinase and the C-terminal regions of TrkB; JM0 indicates deletion of the whole intracellular region of TrkB. B and C, lack of TrkB JM region disrupts TrkB/LIMK1 interaction. Lysates of HEK293 cells transfected with FLAG-TrkB deletion mutants and HA-LIMK1 were immunoprecipitated (IP) with rabbit anti-HA antibody, followed by immunoblotting (IB) with mouse anti-FLAG or anti-HA antibodies, respectively. D, schematic representation of the deletion mutants of TrkB constructs containing juxtamembrane domain. EC, extracellular; TM, transmembrane. E and F, 9 amino acids between JM3 and JM4 (JMBox4) are required for TrkB/LIMK1 interaction. HEK293 cells were transiently transfected with the indicated constructs. Protein extracts were immunoprecipitated with rabbit anti-FLAG antibodies and immunoblotted with mouse anti-FLAG or mouse anti-HA antibodies. G, JMBox4 is sufficient for the binding between TrkB and LIMK1. Chimera FLAG-T1-JMBox4 was generated by PCR. HEK293 cells were transfected with the indicated constructs. Protein extracts were immunoprecipitated by rabbit anti-FLAG antibodies and immunoblotted with mouse anti-FLAG or mouse anti-HA antibodies. H, all three Trk receptors can associate with LIMK1. Lysates from HEK293 cells co-transfected with the indicated constructs were immunoprecipitated with rabbit anti-FLAG antibody, followed by immunoblotting with mouse anti-HA and mouse anti-FLAG antibodies. I, multiple sequence alignment on the JMBox4 region of three Trk receptors is performed by Clustal Omega.
TrkB/LIMK1 Interaction Is Necessary for BDNF-induced LIMK1 Homo-dimerization and Increased LIMK1 Activity
We next want to determine whether the BDNF-induced LIMK1 homo-dimerization and redistribution are mediated by the interaction between TrkB and LIMK1. Based on our previous TrkB/LIMK1 interaction domain mapping study, we used a synthesized peptide consisting of the 9-amino acid sequence of JMBox4 in TrkB fused to the membrane permeability domain of human immunodeficiency virus-1 (HIV)-Tat (40, 41). First, we examined whether the synthesized Tat fusion peptide could successfully transduce into cells. We treated cultured neurons with 1 μm of biotinylated Tat-JMBox4 fusion peptide. 30 min later, cells were fixed and stained with Alexa Fluor 488-conjugated streptavidin. We observed that the Tat fusion peptide successfully transduced into ∼100% of cells (Fig. 4A). Next, we tested whether the Tat-JMBox4 peptide could disrupt the TrkB and LIMK1 interaction by competitively binding with LIMK1. Tat-JMBox4 peptide (1 μm) was added to HEK293 cells, that transiently overexpressed FLAG-TrkB and HA-LIMK1. Immunoprecipitation experiments were carried out 30 min later. We found that the amount of FLAG-TrkB immunoprecipitated by HA-LIMK1 was decreased to 14.8 ± 7.3% when Tat-JMBox4 was present (Fig. 4, B and C), suggesting that TrkB/LIMK1 association could be blocked by the treatment of Tat-JMBox4. We next tested whether the Tat-JMBox4 peptide could block endogenous interaction between TrkB and LIMK1. Indeed, we found that Tat-JMBox4 significantly blocked BDNF enhanced TrkB/LIMK1 interaction (Fig. 4D). Therefore, we could use Tat-JMBox4 as a blocker for TrkB/LIMK1 interaction. We found that BDNF-increased LIMK1 homo-dimerization was totally blocked by Tat-JMBox4 pretreatment (Fig. 4E). BDNF-induced phosphorylation of LIMK1 (Thr-508) was also significantly decreased by pretreatment with Tat-JMBox4 (Fig. 4, F and G). More interestingly, BDNF-induced phosphorylation of cofilin, the downstream effector of LIMK1, was also decreased by Tat-JMBox4 treatment as well (Fig. 4, F and G), suggesting that BDNF-induced LIMK1 phosphorylation depends on TrkB/LIMK1 interaction and LIMK1 dimerization. Furthermore, we found that BDNF-induced translocation of LIMK1 to membrane fraction was also blocked by the Tat-JMBox4 peptide (Fig. 4, H and I). Together, these observations suggest that TrkB/LIMK1 interaction is essential for BDNF stimulus-induced LIMK1 homo-dimerization and transactivation, which contributes to BDNF-increased phosphorylation of LIMK1 and its downstream effector cofilin.
BDNF Stimuli Retards LIMK1 Degradation through TrkB/LIMK1 Interaction
There is evidence that BDNF increases LIMK1 expression levels in synaptoneurosomes by relieving the miR-134 inhibition of Limk1 translation (29). To shed light on the long term effect of BDNF on LIMK1, we tried to examine the alteration in LIMK1 levels after long periods of BDNF treatment. Indeed, we observed markedly increased LIMK1 protein levels after 5–20 h BDNF (50 ng/ml) treatment, although LIMK1 protein levels were unchanged in unstimulated group (Fig. 5, A and B). The increased LIMK1 protein levels upon BDNF treatment could be due to either increased Limk1 translation as described above or attenuated LIMK1 protein degradation. A previous study demonstrated that LIMK1 was a stable protein with a half-life of ∼20 h, and LIMK1 homodimer formation and transphosphorylation could facilitate the stabilization of LIMK1 and attenuate LIMK1 degradation (36). To determine whether BDNF has an influence on LIMK1 stability, we analyzed the degradation rate of the LIMK1 protein in primary cultured hippocampal neurons in the presence or absence of BDNF. Serum-starved neurons were pretreated with CHX (20 μg/ml) to block de novo protein synthesis and then incubated with BDNF (50 ng/ml) for various times (1, 5, 15, and 20 h). Cell lysates were collected at the indicated time points to detect LIMK1 protein levels by Western blot analysis. We found that endogenous LIMK1 was gradually degraded with a half-life of ∼20 h when CHX was present, which was compatible with a previous study (Fig. 5, C and D) (36). However, when treated with BDNF, the half-life of LIMK1 protein was prolonged to >20 h (Fig. 5, C and D). The effect of BDNF on LIMK1 degradation was blocked by Tat-JMBox4 treatment (1 μm, applied before and continuing throughout BDNF treatment as the same as CHX), indicating that BDNF-promoted LIMK1 stability was dependent on TrkB/LIMK1 interaction (Fig. 5, C and D). Taken together, we conclude that BDNF cannot only increase LIMK1 protein synthesis but also retard LIMK1 degradation by promoting LIMK1 homodimer formation.
FIGURE 5.

BDNF stimuli retards LIMK1 degradation through TrkB/LIMK1 interaction. A, BDNF stimuli increased LIMK1 protein levels. Cultured hippocampal neurons were serum-starved and treated with vehicle or 50 ng/ml BDNF for the indicated times, and then cells were lysed and Western blot analysis was performed. Con, control. B, quantification of LIMK1 levels (n = 3, *, p < 0.05 versus 0 h; Student's t test). C, retarded LIMK1 degradation by BDNF treatment can be blocked by Tat-JMBox4 peptide. Serum-starved hippocampal neurons were treated with CHX (20 μg/ml) and/or Tat-JMBox4 (1 μm) 30 min before BDNF (50 ng/ml) administration. After the indicated times, neurons were lysed for Western blot analysis. D, quantification of LIMK1 levels (n = 3, *, p < 0.05; **, p < 0.01 versus 0 h; one-way ANOVA).
TrkB/LIMK1 Interaction Is Required for BDNF-induced Axonal Growth
Previous studies reported that up-regulation of LIMK1 levels in primary hippocampal neurons promoted axonal growth, although down-regulation of LIMK1 had the opposite effect (20, 22). BDNF stimulation also resulted in increased axonal length as described previously (42). We hypothesized that the effect of BDNF on LIMK1 would be involved in the BDNF-regulated axonal elongation. To test this hypothesis, we first tested the interference efficiency of LIMK1 siRNAs and showed that they markedly reduced LIMK1 levels in cultured PC12 cells (Fig. 6A). Next, we examined the effect of BDNF on axonal length of the cultured hippocampal neurons that were transfected with HA-LIMK1 or LIMK1 siRNA, respectively. We found that up-regulation of LIMK1 levels increased axonal length whereas siRNA knocking down LIMK1 levels decreased axonal length, the results of which are in conformity with previous studies (20, 22), and BDNF treatment could significantly promote axonal elongation. Neurons overexpressing LIMK1 still showed responsiveness to BDNF treatment and displayed significantly longer axon in the presence of BDNF, whereas neurons transfected with LIMK1 siRNA1 or LIMK1 siRNA2 lost their responsiveness to BDNF stimuli in axonal elongation (Fig. 6, B and C), indicating that the effect of BDNF on axonal outgrowth was LIMK1-dependent. To further investigate whether the TrkB/LIMK1 interaction was required for BDNF-induced axonal outgrowth, we treated primary hippocampal neurons with the Tat-JMBox4 peptide to interfere with the association between TrkB and LIMK1. We found that the BDNF-induced axonal extension was blocked by Tat-JMBox4 treatment (Fig. 6D). Furthermore, we found BDNF-promoted dendritic outgrowth could also be blocked by the Tat-JMBox4 peptide (data not shown). Next, we carried out quantitative immunofluorescence staining on hippocampal growth cones using anti-p-LIMK1 (Thr-508) and anti-p-cofilin (Ser-3) antibodies to determine whether LIMK1 mediates BDNF-induced axonal elongation through controlling cofilin activity that is regulated by the phosphorylation of its Ser-3 residue. We found that BDNF stimulation increased p-LIMK1, p-cofilin, and filamentous actin (F-actin) levels in hippocampal growth cones, and the effects were blocked by the Tat-JMBox4 peptide (Fig. 6, E and F). These data suggested that BDNF-enhanced LIMK1 activity could phosphorylate and inactivate cofilin leading to actin polymerization and axonal elongation. Together, these data revealed that BDNF-facilitated axonal growth is dependent on LIMK1 function and that TrkB/LIMK1 interaction plays an important role in this process.
FIGURE 6.

TrkB/LIMK1 interaction is required for BDNF-induced axonal growth. A, LIMK1 siRNA efficiently decreased the expression of endogenous LIMK1. PC12 cells were transiently transfected with LIMK1 siRNA. 72 h after transfection, cells were lysed and immunoblotted with anti-LIMK1 and anti-actin antibodies. B, LIMK1 is required for BDNF-induced axonal elongation. Cultured hippocampal neurons were transfected with HA-LIMK1 or LIMK1 siRNA as described under “Experimental Precedures.” 20 h after BDNF (50 ng/ml) treatment, neurons were fixed and immunostained with rabbit anti-GFP antibodies. Scale bar, 20 μm. C, quantification of axonal length (n = 56–72, **, p < 0.01; Student's t test, #, p < 0.05; ##, p < 0.01; one-way ANOVA). NS, not significant. D, Tat-JMBox4 peptide can block BDNF-induced axonal elongation. Hippocampal neurons were pretreated with Tat-JMBox4 peptide 30 min before BDNF administration. 20 h after BDNF treatment, cells were fixed and immunostained with mouse anti-Tau-1 (red) and chicken anti-MAP2 (green) antibodies. The length of Tau-1-positive neurite was measured as axonal length at DIV3. Representative merged images are shown, and quantification of axonal length is shown below (n = 63–77, **, p < 0.01 versus control group; ##, p < 0.01 versus BDNF-treated group, one-way ANOVA). Scale bar, 20 μm. E, Tat-JMBox4 peptide can block BDNF increased p-LIMK1 and p-cofilin levels and actin polymerization in growth cones. Serum-starved neurons were treated with Tat-JMBox4 peptide (1 μm) and/or BDNF (50 ng/ml for 30 min) followed by fixation and immunostaining. Scale bar, 10 μm. F, quantification of fluorescence intensity of p-LIMK1, p-cofilin, and F-actin (n = 25–38, *, p < 0.05; **, p < 0.01 versus control group; one-way ANOVA).
DISCUSSION
In this study, we have found a novel interaction between LIMK1 and TrkB and identified a 9-amino acid sequence in the TrkB JM domain, which is necessary and sufficient for TrkB/LIMK1 association. Furthermore, we demonstrated that BDNF stimulation could induce LIMK1 dimerization and transphosphorylation leading to activation and stabilization of LIMK1, the effect of which was dependent on the association between TrkB and LIMK1. Finally, we showed that LIMK1 and TrkB/LIMK1 interactions were required for BDNF-enhanced actin polymerization and axonal elongation.
Our studies provide three new insights into the molecular mechanisms underlying BDNF-enhanced LIMK1 activation and axonal elongation. To our knowledge, this is the first time to show the novel interaction between LIMK1 and TrkB. Previous studies have demonstrated that TrkB knock-out mice had a reduction of dendritic spine and axon varicosity density and showed impairments in hippocampus-dependent learning (12, 43, 44). Intriguingly, LIMK1 knock-out mice had similar phenotypes such as abnormal morphology of axon and dendrites and deficits in hippocampus-dependent learning (19). Therefore, we hypothesized that there might exist cross-talk between BDNF/TrkB and LIMK1 signaling pathways. Here, we demonstrated for the first time that TrkB could directly interact with LIMK1 both at exogenous and endogenous levels. T1 and p75NTR do not have the ability to form a complex with LIMK1, which indicated that LIMK1 associated with TrkB specifically. Moreover, we found that the nine amino acids (VIENPQYFG) in TrkB juxtamembrane region play a crucial role in linking TrkB to LIMK1 as the short sequence was necessary and sufficient for the association between TrkB and LIMK1.
Second, we found a novel mechanism underlying BDNF-induced LIMK1 activation, which was independent of TrkB kinase activity. A previous study reported that BDNF, actions dependent on TrkB tyrosine kinase activity, phosphorylated and activated Tiam1, which resulted in activation of Rac1, one of the upstream kinases of LIMK1 (28). Intriguingly, we found that BDNF-induced phosphorylation of LIMK1 (Thr-508) could not be blocked by K252a, although there was a declining trend. It suggested that BDNF induced phosphorylation of LIMK1, which, to a large extent, did not rely on TrkB tyrosine kinase activity. A previous study has revealed that LIMK1 can autophosphorylate on serine, tyrosine, and threonine residues in vitro (15). Moreover, LIMK1 homo-dimerization leads to further activation and stabilization of LIMK1 by transphosphorylation (36). In our work, we found that BDNF stimulation led to LIMK1 homo-dimerization, and this effect can be inhibited by Tat-JMBox4, a synthesized cell-penetrating peptide used to interfere with the association between TrkB and LIMK1. We therefore postulated that the BDNF could induce LIMK1 homodimer formation and transphosphorylation through TrkB/LIMK1 interaction, the effect of which was independent of TrkB tyrosine kinase activity. Indeed, we found that BDNF-increased p-LIMK1 (Thr-508) levels were blocked by Tat-JMBox4 peptide. LIMK1 is predominantly expressed in the cytoplasm of neurons (45). Other studies showed that activated LIMK1 translocated to the plasma membrane (37, 38). In agreement with previous studies, we found that BDNF stimulation reduced cytoplasmic LIMK1 but increased membrane-associated LIMK1 levels. Activated LIMK1 translocation to the plasma membrane might play an important role in cytoskeleton remodeling by phosphorylation of cofilin beneath the plasma membrane.
Moreover, we found that BDNF not only had the short-term effect on LIMK1 phosphorylation but also had the long term effect on LIMK1 protein degradation. LIMK1 is a quite stable protein with a half-life of ∼20 h, and LIMK1 homo-dimerization and transphosphorylation could further extend its half-life (36). We found BDNF treatment could attenuate LIMK1 degradation and thus prolong the half-life of LIMK1, and the effect was blocked by the Tat-JMBox4 peptide. Our data suggested that BDNF induced LIMK1 homo-dimerization, and further activation played a vital role in impeding LIMK1 degradation. A Previous study indicated that exposure of neurons to BDNF could increase LIMK1 protein synthesis and spine growth by relieving miR-134 inhibition of Limk1 translation (29). Therefore, BDNF could increase LIMK1 levels by synergetic effects on both the LIMK1 synthesis and degradation. Taken together, although most of BDNF effects are mediated by TrkB kinase activity, we found that BDNF-enhanced LIMK1 activation relied on dimerization rather than kinase activity of the TrkB receptor.
Third, we showed that TrkB/LIMK1 interaction was required for BDNF-enhanced actin polymerization and axonal elongation. In neurons, LIMK1 plays important roles in the regulation of growth cone motility and axon development (46). Overexpression of LIMK1 accelerates axon formation and elongation in cultured hippocampal neurons, although down-regulation of LIMK1 has the opposite effect (22). Overexpression of the LIM domain of LIMK1, a dominant negative form to inhibit endogenous LIMK1, impeded NGF or Ras-induced neurite extension in PC12 cells (47). However, other research groups demonstrated that both up-regulation and down-regulation of LIMK1 significantly suppressed growth cone motility and NGF-induced neurite extension in PC12 cells and chick dorsal root ganglion neurons (31, 46). Actually, the discrepancy results may be due to the different expressing duration of LIMK1. There is evidence that short term post-transfection of wild-type LIMK1 (<24 h) promotes axon outgrowth, whereas long term up-regulation of LIMK1 (>24 h) leads to growth cone collapse and axon retraction in primary hippocampal neurons (20). Thus, a proper duration of activated LIMK1 is critical to modulate cofilin activity for growth cone motility and neurite extension. Hence, we examined the axonal length 20 h after Lipofectamine transfection of wild-type LIMK1 in DIV2 neurons. Consistent with previous results, we found that overexpression of LIMK1 increased axonal length, and siRNA knockdown of LIMK1 decreased axonal length.
Previous works have proved that BDNF facilitates axon elongation through BDNF/TrkB signaling (42, 48, 49). In this study, we found that BDNF had no effect on axonal elongation when LIMK1 was knocked down by siRNA, and BDNF stimulation further enhanced axonal growth in primary neurons transfected with wild-type LIMK1, suggesting that BDNF could induce axonal elongation in a manner dependent on LIMK1 activity. Moreover, we observed that BDNF-induced axonal extension could be blocked by Tat-JMBox4, indicating that BDNF promoting axonal extension relied on TrkB/LIMK1 interaction. A previous study had demonstrated that BDNF/TrkB signaling could activate Rac in cultured cortical neurons (28) and increase F-actin polymerization via promoting phosphorylation of p21-activated kinase and cofilin in rat hippocampal slices (26). However, there was evidence that BDNF could modulate RhoA activity and decrease p-cofilin via p75NTR in retinal growth cones (50). The contradictory results may be due to the different cell types that express high levels of TrkB or p75NTR, respectively. In our study, we found that BDNF-enhanced LIMK1 activity could phosphorylate cofilin and promote actin polymerization that leads to axonal elongation.
In conclusion, our study revealed that TrkB interacted with LIMK1 via its JMBox4 region. Furthermore, BDNF-induced LIMK1 dimerization and phosphorylation depended on TrkB/LIMK1 interaction but not TrkB tyrosine kinase activity. Finally, we found that BDNF-induced LIMK1 dimerization and transactivation played an essential role in BDNF-enhanced actin polymerization and axonal elongation (Fig. 7). These findings provide insights into the mechanistic link between LIMK1-regulated actin dynamic and BDNF-induced axonal elongation.
FIGURE 7.
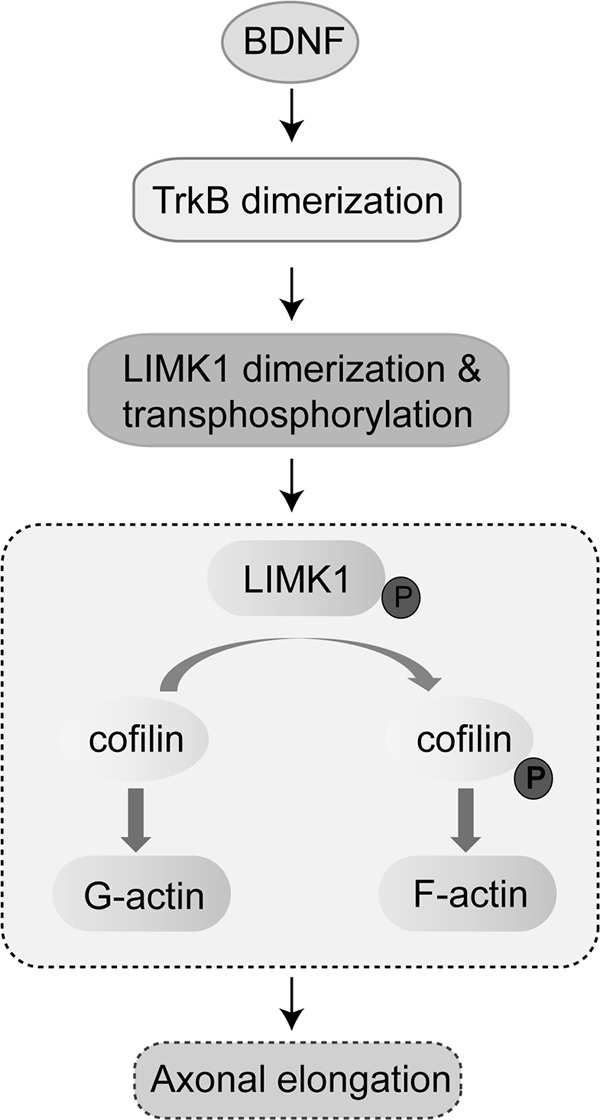
Schematic illustrating a novel mechanism for BDNF-induced axonal elongation. LIMK1 is a novel TrkB interaction molecule, and BDNF stimulation leads to LIMK1 activation and stabilization by homo-dimerization and transphosphorylation. Activated LIMK1 phosphorylates cofilin that results in polymerization of actin and promotion of axonal elongation.
This work was supported by National Natural Science Foundation of China Grants 31071254 and 31130026, National 973 Basic Research Program of China Grants 2012CB911000 and 2009CB941403, State Program of National Natural Science Foundation of China for Innovative Research Group Grant 81021001, and the Independent Innovation Foundation of Shandong University.
- BDNF
- brain-derived neurotrophic factor
- ANOVA
- analysis of variance
- CHX
- cycloheximide
- DIV
- days in vitro
- JM
- juxtamembrane
- TK
- tyrosine kinase
- CT
- C-terminal.
REFERENCES
- 1. Bartkowska K., Turlejski K., Djavadian R. L. (2010) Neurotrophins and their receptors in early development of the mammalian nervous system. Acta Neurobiol. Exp. 70, 454–467 [DOI] [PubMed] [Google Scholar]
- 2. Chao M. V. (2003) Neurotrophins and their receptors. A convergence point for many signaling pathways. Nat. Rev. Neurosci. 4, 299–309 [DOI] [PubMed] [Google Scholar]
- 3. Cunha C., Brambilla R., Thomas K. L. (2010) A simple role for BDNF in learning and memory? Front. Mol. Neurosci. 3, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Santos A. R., Comprido D., Duarte C. B. (2010) Regulation of local translation at the synapse by BDNF. Prog. Neurobiol. 92, 505–516 [DOI] [PubMed] [Google Scholar]
- 5. Purcell A. L., Carew T. J. (2003) Tyrosine kinases, synaptic plasticity, and memory. Insights from vertebrates and invertebrates. Trends Neurosci. 26, 625–630 [DOI] [PubMed] [Google Scholar]
- 6. Danzer S. C., Crooks K. R., Lo D. C., McNamara J. O. (2002) Increased expression of brain-derived neurotrophic factor induces formation of basal dendrites and axonal branching in dentate granule cells in hippocampal explant cultures. J. Neurosci. 22, 9754–9763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Goldberg J. L. (2003) How does an axon grow? Genes Dev. 17, 941–958 [DOI] [PubMed] [Google Scholar]
- 8. Labelle C., Leclerc N. (2000) Exogenous BDNF, NT-3, and NT-4 differentially regulate neurite outgrowth in cultured hippocampal neurons. Brain Res. Dev. Brain Res. 123, 1–11 [DOI] [PubMed] [Google Scholar]
- 9. Cheng P. L., Song A. H., Wong Y. H., Wang S., Zhang X., Poo M. M. (2011) Self-amplifying autocrine actions of BDNF in axon development. Proc. Natl. Acad. Sci. U.S.A. 108, 18430–18435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goldberg J. L., Espinosa J. S., Xu Y., Davidson N., Kovacs G. T., Barres B. A. (2002) Retinal ganglion cells do not extend axons by default. Promotion by neurotrophic signaling and electrical activity. Neuron 33, 689–702 [DOI] [PubMed] [Google Scholar]
- 11. Zhao L., Sheng A. L., Huang S. H., Yin Y. X., Chen B., Li X. Z., Zhang Y., Chen Z. Y. (2009) Mechanism underlying activity-dependent insertion of TrkB into the neuronal surface. J. Cell Sci. 122, 3123–3136 [DOI] [PubMed] [Google Scholar]
- 12. Luikart B. W., Nef S., Virmani T., Lush M. E., Liu Y., Kavalali E. T., Parada L. F. (2005) TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J. Neurosci. 25, 3774–3786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Govek E. E., Newey S. E., Van Aelst L. (2005) The role of the Rho GTPases in neuronal development. Genes Dev. 19, 1–49 [DOI] [PubMed] [Google Scholar]
- 14. Namekata K., Harada C., Taya C., Guo X., Kimura H., Parada L. F., Harada T. (2010) Dock3 induces axonal outgrowth by stimulating membrane recruitment of the WAVE complex. Proc. Natl. Acad. Sci. U.S.A. 107, 7586–7591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pröschel C., Blouin M. J., Gutowski N. J., Ludwig R., Noble M. (1995) Limk1 is predominantly expressed in neural tissues and phosphorylates serine, threonine, and tyrosine residues in vitro. Oncogene 11, 1271–1281 [PubMed] [Google Scholar]
- 16. Takahashi H., Koshimizu U., Nakamura T. (1998) A novel transcript encoding truncated LIM kinase 2 is specifically expressed in male germ cells undergoing meiosis. Biochem. Biophys. Res. Commun. 249, 138–145 [DOI] [PubMed] [Google Scholar]
- 17. Scott R. W., Olson M. F. (2007) LIM kinases. Function, regulation, and association with human disease. J. Mol. Med. 85, 555–568 [DOI] [PubMed] [Google Scholar]
- 18. Bernard O. (2007) Lim kinases, regulators of actin dynamics. Int. J. Biochem. Cell Biol. 39, 1071–1076 [DOI] [PubMed] [Google Scholar]
- 19. Meng Y., Zhang Y., Tregoubov V., Janus C., Cruz L., Jackson M., Lu W. Y., MacDonald J. F., Wang J. Y., Falls D. L., Jia Z. (2002) Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35, 121–133 [DOI] [PubMed] [Google Scholar]
- 20. Rosso S., Bollati F., Bisbal M., Peretti D., Sumi T., Nakamura T., Quiroga S., Ferreira A., Cáceres A. (2004) LIMK1 regulates Golgi dynamics, traffic of Golgi-derived vesicles, and process extension in primary cultured neurons. Mol. Biol. Cell 15, 3433–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bagheri-Yarmand R., Mazumdar A., Sahin A. A., Kumar R. (2006) LIM kinase 1 increases tumor metastasis of human breast cancer cells via regulation of the urokinase-type plasminogen activator system. Int. J. Cancer 118, 2703–2710 [DOI] [PubMed] [Google Scholar]
- 22. Tursun B., Schlüter A., Peters M. A., Viehweger B., Ostendorff H. P., Soosairajah J., Drung A., Bossenz M., Johnsen S. A., Schweizer M., Bernard O., Bach I. (2005) The ubiquitin ligase Rnf6 regulates local LIM kinase 1 levels in axonal growth cones. Genes Dev. 19, 2307–2319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nishimura Y., Yoshioka K., Bernard O., Bereczky B., Itoh K. (2006) A role of LIM kinase 1/cofilin pathway in regulating endocytic trafficking of EGF receptor in human breast cancer cells. Histochem. Cell Biol. 126, 627–638 [DOI] [PubMed] [Google Scholar]
- 24. Tahirovic S., Bradke F. (2009) Neuronal polarity. Cold Spring Harbor Perspect. Biol. 1, a001644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shen K., Cowan C. W. (2010) Guidance molecules in synapse formation and plasticity. Cold Spring Harbor Perspect. Biol. 2, a001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rex C. S., Lin C. Y., Kramár E. A., Chen L. Y., Gall C. M., Lynch G. (2007) Brain-derived neurotrophic factor promotes long term potentiation-related cytoskeletal changes in adult hippocampus. J. Neurosci. 27, 3017–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lu B. (2003) BDNF and activity-dependent synaptic modulation. Learn. Mem. 10, 86–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miyamoto Y., Yamauchi J., Tanoue A., Wu C., Mobley W. C. (2006) TrkB binds and tyrosine phosphorylates Tiam1, leading to activation of Rac1 and induction of changes in cellular morphology. Proc. Natl. Acad. Sci. U.S.A. 103, 10444–10449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schratt G. M., Tuebing F., Nigh E. A., Kane C. G., Sabatini M. E., Kiebler M., Greenberg M. E. (2006) A brain-specific microRNA regulates dendritic spine development. Nature 439, 283–289 [DOI] [PubMed] [Google Scholar]
- 30. Huang S. H., Duan S., Sun T., Wang J., Zhao L., Geng Z., Yan J., Sun H. J., Chen Z. Y. (2011) JIP3 mediates TrkB axonal anterograde transport and enhances BDNF signaling by directly bridging TrkB with kinesin-1. J. Neurosci. 31, 10602–10614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Endo M., Ohashi K., Mizuno K. (2007) LIM kinase and slingshot are critical for neurite extension. J. Biol. Chem. 282, 13692–13702 [DOI] [PubMed] [Google Scholar]
- 32. Horita Y., Ohashi K., Mukai M., Inoue M., Mizuno K. (2008) Suppression of the invasive capacity of rat ascites hepatoma cells by knockdown of Slingshot or LIM kinase. J. Biol. Chem. 283, 6013–6021 [DOI] [PubMed] [Google Scholar]
- 33. Yin Y. X., Sun Z. P., Huang S. H., Zhao L., Geng Z., Chen Z. Y. (2010) RanBPM contributes to TrkB signaling and regulates brain-derived neurotrophic factor-induced neuronal morphogenesis and survival. J. Neurochem. 114, 110–121 [DOI] [PubMed] [Google Scholar]
- 34. Claus C., Chey S., Heinrich S., Reins M., Richardt B., Pinkert S., Fechner H., Gaunitz F., Schäfer I., Seibel P., Liebert U. G. (2011) Involvement of p32 and microtubules in alteration of mitochondrial functions by rubella virus. J. Virol. 85, 3881–3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schecterson L. C., Bothwell M. (2010) Neurotrophin receptors. Old friends with new partners. Dev. Neurobiol. 70, 332–338 [DOI] [PubMed] [Google Scholar]
- 36. Li R., Soosairajah J., Harari D., Citri A., Price J., Ng H. L., Morton C. J., Parker M. W., Yarden Y., Bernard O. (2006) Hsp90 increases LIM kinase activity by promoting its homo-dimerization. FASEB J. 20, 1218–1220 [DOI] [PubMed] [Google Scholar]
- 37. Matsui S., Matsumoto S., Adachi R., Kusui K., Hirayama A., Watanabe H., Ohashi K., Mizuno K., Yamaguchi T., Kasahara T., Suzuki K. (2002) LIM kinase 1 modulates opsonized zymosan-triggered activation of macrophage-like U937 cells. Possible involvement of phosphorylation of cofilin and reorganization of actin cytoskeleton. J. Biol. Chem. 277, 544–549 [DOI] [PubMed] [Google Scholar]
- 38. Buchan A. M., Lin C. Y., Choi J., Barber D. L. (2002) Somatostatin, acting at receptor subtype 1, inhibits Rho activity, the assembly of actin stress fibers, and cell migration. J. Biol. Chem. 277, 28431–28438 [DOI] [PubMed] [Google Scholar]
- 39. Ohira K., Hayashi M. (2009) A new aspect of the TrkB signaling pathway in neural plasticity. Curr. Neuropharmacol. 7, 276–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Toda S., Shen H. W., Peters J., Cagle S., Kalivas P. W. (2006) Cocaine increases actin cycling. Effects in the reinstatement model of drug seeking. J. Neurosci. 26, 1579–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heitz F., Morris M. C., Divita G. (2009) Twenty years of cell-penetrating peptides. From molecular mechanisms to therapeutics. Br. J. Pharmacol. 157, 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yoshimura T., Kawano Y., Arimura N., Kawabata S., Kikuchi A., Kaibuchi K. (2005) GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137–149 [DOI] [PubMed] [Google Scholar]
- 43. Tyler W. J., Alonso M., Bramham C. R., Pozzo-Miller L. D. (2002) From acquisition to consolidation. On the role of brain-derived neurotrophic factor signaling in hippocampus-dependent learning. Learn. Mem. 9, 224–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Minichiello L., Korte M., Wolfer D., Kühn R., Unsicker K., Cestari V., Rossi-Arnaud C., Lipp H. P., Bonhoeffer T., Klein R. (1999) Essential role for TrkB receptors in hippocampus-mediated learning. Neuron 24, 401–414 [DOI] [PubMed] [Google Scholar]
- 45. Bernard O., Ganiatsas S., Kannourakis G., Dringen R. (1994) Kiz-1, a protein with LIM zinc finger and kinase domains, is expressed mainly in neurons. Cell Growth Differ. 5, 1159–1171 [PubMed] [Google Scholar]
- 46. Endo M., Ohashi K., Sasaki Y., Goshima Y., Niwa R., Uemura T., Mizuno K. (2003) Control of growth cone motility and morphology by LIM kinase and Slingshot via phosphorylation and dephosphorylation of cofilin. J. Neurosci. 23, 2527–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Birkenfeld J., Betz H., Roth D. (2001) Inhibition of neurite extension by overexpression of individual domains of LIM kinase 1. J. Neurochem. 78, 924–927 [DOI] [PubMed] [Google Scholar]
- 48. Tucker K. L. (2002) Neurotrophins and the control of axonal outgrowth. Panminerva Med. 44, 325–333 [PubMed] [Google Scholar]
- 49. Cosker K. E., Shadan S., van Diepen M., Morgan C., Li M., Allen-Baume V., Hobbs C., Doherty P., Cockcroft S., Eickholt B. J. (2008) Regulation of PI3K signaling by the phosphatidylinositol transfer protein PITPα during axonal extension in hippocampal neurons. J. Cell Sci. 121, 796–803 [DOI] [PubMed] [Google Scholar]
- 50. Gehler S., Gallo G., Veien E., Letourneau P. C. (2004) p75 neurotrophin receptor signaling regulates growth cone filopodial dynamics through modulating RhoA activity. J. Neurosci. 24, 4363–4372 [DOI] [PMC free article] [PubMed] [Google Scholar]