

Background: APOE genotype effects on Aβ accumulation were determined using new EFAD transgenic mice.
Results: In E4FAD mice, compact plaques are greater, total apoE4 is lower, less apoE4 is lipoprotein-associated, and oligomeric Aβ is higher compared with E2FAD/E3FAD, while intraneuronal Aβ is unaffected.
Conclusion: APOE4 uniquely effects Aβ accumulation.
Significance: These data provide a basis for APOE-induced AD risk.
Keywords: Alzheimer Disease, Animal Models, ApoE, Apolipoprotein Genes, Transgenic Mice, Amyloid-β, Soluble Amyloid-β species
Abstract
APOE4 is the greatest risk factor for Alzheimer disease (AD) and synergistic effects with amyloid-β peptide (Aβ) suggest interactions among apoE isoforms and different forms of Aβ accumulation. However, it remains unclear how the APOE genotype affects plaque morphology, intraneuronal Aβ, soluble Aβ42, and oligomeric Aβ (oAβ), particularly in vivo. As the introduction of human APOE significantly delays amyloid deposition in transgenic mice expressing familial AD (FAD) mutations (FAD-Tg), 5xFAD-Tg mice, which exhibit amyloid deposition by age 2 months, were crossed with apoE-targeted replacement mice to produce the new EFAD-Tg mice. Compared with 5xFAD mice, Aβ deposition was delayed by ∼4 months in the EFAD mice, allowing detection of early changes in Aβ accumulation from 2–6 months. Although plaque deposition is generally greater in E4FAD mice, E2/E3FAD mice have significantly more diffuse and E4FAD more compact plaques. As a first report in FAD-Tg mice, the APOE genotypes had no effect on intraneuronal Aβ accumulation in EFAD mice. In E4FAD mice, total apoE levels were lower and total Aβ levels higher than in E2FAD and E3FAD mice. Profiles from sequential three-step extractions (TBS, detergent, and formic acid) demonstrated that the lower level of total apoE4 is reflected only in the detergent-soluble fraction, indicating that less apoE4 is lipoprotein-associated, and perhaps less lipidated, compared with apoE2 and apoE3. Soluble Aβ42 and oAβ levels were highest in E4FAD mice, although soluble apoE2, apoE3, and apoE4 levels were comparable, suggesting that the differences in soluble Aβ42 and oAβ result from functional differences among the apoE isoforms. Thus, APOE differentially regulates multiple aspects of Aβ accumulation.
Introduction
The primary genetic risk factor for Alzheimer disease (AD)4 is APOE4, increasing the risk by ∼4- and 15-fold with a single or double allele, whereas APOE2 reduces the risk compared with APOE3. Although carriers of the APOE4 gene of apolipoprotein E (apoE) account for more than half of AD patients, the mechanism(s) by which APOE affects the pathogenesis of AD is the subject of continued inquiry (1). Plaque deposition is increased with APOE4 compared with APOE2 and APOE3 in humans and transgenic mice expressing familial AD (FAD) mutations (FAD-Tg) (2–5). However, an APOE genotype-specific effect on the accumulation of other potentially neurotoxic species of Aβ remains unclear. Research efforts to address this mechanism in vivo are hindered by the lack of: 1) tractable transgenic mouse models and 2) assays for changes in Aβ speciation and apoE solubility during the initial stages of Aβ accumulation.
Introduction of human APOE to existing FAD-Tg mice significantly delays plaque deposition, although once detected, plaque levels are generally greater with APOE4 than APOE3 (3, 6–8). For example, crossing apoE-targeted replacement mice (apoE-TR) (9) with PDAPP mice (10) delays plaque deposition from ∼10 to 18 months, although once detected, plaque levels are greater with APOE4 than APOE3 (3). To establish a tractable model, transgenic mice expressing five FAD mutations (5xFAD), which exhibit accelerated plaque deposition that is significant by 2 months (11), were crossed with apoE-TR mice to produce the EFAD mouse model. In EFAD mice, APOE genotype-specific effects on Aβ accumulation can be identified between 2 and 6 months.
Aβ pathology can refer to a number of neurotoxic forms of the peptide, making identification of “neurotoxic Aβ” unclear. Detection of different Aβ species requires complementary immunohistochemical (IHC) and biochemical approaches. By IHC, intraneuronal Aβ (12–14) and perhaps specific plaque morphologies (15, 16) are thought to contribute to Aβ pathology, although amyloid plaque burden per se may not be neurotoxic (17). Biochemical analysis has demonstrated that oligomeric Aβ (oAβ) (18–21) and soluble Aβ levels are elevated in AD brains (18), and soluble oligomeric forms of Aβ42 have been demonstrated to correlate with cognitive decline (20) and severity of disease in humans (22). The APOE genotype may affect AD risk by modulating the speciation of Aβ42, particularly oAβ levels. Possible mechanisms for this apoE isoform-specific effect include differences in Aβ clearance, degradation, and/or stabilization of oAβ (for a review see Ref. 23). Specific detection methods for oAβ are one factor limiting further understanding of apoE isoform-specific effects on oAβ levels. Thus, an ELISA for measuring oAβ levels was developed following the protocol of Xia et al. (24), enabled by the development of the new Aβ-specific antibody MOAB-2 (25).
Total apoE4 levels are lower compared with apoE2 and apoE3 in human plasma and cerebrospinal fluid for both AD patients (26–31) and age-matched controls (32), brain homogenates from AD patients (3, 33), brain homogenates from apoE-TR mice (3, 28, 34), and brain homogenates from apoE-TR/PDAPP-Tg mice (3). However, it is not known whether biochemical methods for sequential extraction differentially affect apoE isoform levels (3, 35). Traditionally, a non-ionic detergent is required to release apoE from lipoprotein particles (i.e. TBS containing 1% Triton X-100 (TBSX)) without inducing the formation of new micelles, as can occur with ionic detergents such as SDS (35–38, 41). To address this issue, a three-step sequential protein extraction protocol was optimized to account for the extraction conditions for lipoprotein-associated apoE, as well as insoluble protein from dense-core amyloid plaques (42).
In this study, development of the EFAD transgenic mouse model enabled identification of the effects of the APOE genotype on early types of Aβ accumulation. From 2 to 6 months, plaque deposition was generally greater in E4FAD mice, although E2FAD and E3FAD had primarily diffuse and E4FAD compact plaques, whereas intraneuronal Aβ levels were comparable among the APOE genotypes. Biochemical measurements of total apoE and Aβ levels, combined with an extraction method to isolate and identify the soluble, detergent-soluble, and insoluble levels of apoE, Aβ, and oAβ, revealed mechanistic interplay between the apoE isoforms and these defined species of Aβ. Although total apoE4 levels were lower than those of apoE2 and apoE3, as demonstrated previously, of particular importance is the novel finding that the decreased levels of total apoE4 occurred only in the TBSX extraction fraction, demonstrating that less apoE4 is lipoprotein-associated and perhaps that apoE4 is less lipidated compared with apoE2 and apoE3. In addition, the levels of soluble Aβ42 and oAβ were higher with apoE4.
EXPERIMENTAL PROCEDURES
EFAD Mice Development
Experiments follow the University of Illinois at Chicago Institutional Animal Care and Use Committee protocols. All breeding and colony maintenance was conducted at Taconic Laboratories. 5xFAD mice co-express five FAD mutations (APP K670N/M671L + I716V + V717I and PS1 M146L + L286V) under the control of the neuron-specific mouse Thy-1 promoter (11). The 5xFAD line of mice, Tg6799, that produce the highest amount of Aβ and are heterozygous for the 5xFAD genes were provided by Dr. R. Vassar (Northwestern University). In apoE-TR mice, the coding region of the human APOE gene replaces that of the mouse APOE gene. Homozygous apoE-TR mice were purchased from Taconic in collaboration with Dr. P. Sullivan (Duke University) (9, 43). Details on the production, genotyping, and genetic background of these mice are described in the sources cited above.
To establish colonies of EFAD mouse lines (E2FAD, E3FAD, and E4FAD), 5xFAD mice were bred to homozygous APOE2-, APOE3-, and APOE4-TR mice by Taconic Laboratories. Briefly, male APOE-TR+/+ mice on a C57/B6 background were bred with female 5xFAD+/− mice on a C57B//B6xSJL background. The resulting female mouse-APOE/APOE-TR/5xFAD+/− mice were backcrossed with male APOE-TR mice to generate APOE-TR+/+/5xFAD+/− (EFAD) mice. In this study male EFAD mice were utilized. Female mice were excluded from this initial study for consistency, as apoE isoform-specific interaction with Aβ are known to be influenced by gender (for review (44)). In EFAD mice, the levels of full-length APP were equivalent among the APOE genotypes (supplemental Fig. 1). This suggests that any observed APOE genotype-specific differences in Aβ accumulation are not mediated by altered APP expression or processing. These results are consistent with results from the PDAPP/apoE-TR mice (3) and J20/APOEfKI (45) where no significant differences in APP levels were observed with the APOE genotype.
Tissue Harvesting
Two-, 4-, and 6-month-old mice were anesthetized with sodium pentobarbital (50 mg/kg) and perfused transcardially with ice-cold PBS containing protease inhibitors (Calbiochem, set 3). Directly following perfusion, brains were removed and dissected at the midline. Left hemi-brains from mice at each age were drop-fixed in 4% paraformaldehyde for 48 h followed by storage at 4 °C in PBS plus 0.05% sodium azide (NaN3) until use. Right hemi-brains were dissected on ice into cortex, hippocampus, and cerebellum, immediately snap-frozen in liquid nitrogen, and stored at −80 °C until use.
Immunohistochemistry for Aβ
Paraformaldehyde-fixed left hemi-brains were incubated in two sequential 30% sucrose solutions (in TBS) for 24 h each, frozen on dry ice, and cut sagittally at 35-μm thickness on a sliding microtome; then sections were stored in cryoprotectant at −20 °C. Immediately prior to staining, tissue sections were washed in TBS (three times for 10 min each), incubated in 88% formic acid (FA) (8 min), permeabilized with 0.25% Triton X-100 in TBS (TBST; three times for 10 min each), and blocked with 5% bovine serum albumin (BSA) in TBST for 1 h. Free-floating sections were subsequently incubated with an anti-Aβ antibody, MOAΒ-2 (mouse IgG2b, 1:1000 dilution of 0.5 mg/ml stock (25)), and an anti-NeuN antibody (mouse IgG1, 1:1000 dilution, Chemicon) diluted in TBST containing 2% BSA overnight on an oscillatory rotator. Next, sections were washed in TBST (six times for 10 min each), incubated with Alexa fluorophore-conjugated isotype-specific secondary antibodies diluted 1:200 in TBST containing 2% BSA for 1 h, washed in TBST (three times for 10 min each), washed in TBS (three times for 10 min each), and mounted on glass coverslips with ProLong Gold antifade mounting medium containing DAPI (Invitrogen). Images were captured on a Zeiss Axio Imager M1 under identical capture settings at ×20 or ×63 magnification.
Protein Extractions
Serial extractions of brain tissue were performed essentially as described (42). Briefly, frozen tissue from dissected brains was homogenized in 15 volumes (w/v) of TBS. Samples were centrifuged (100,000 × g, 1 h at 4 °C) and the TBS-soluble fraction was aliquoted prior to freezing in liquid nitrogen and stored at −80 °C. The pellet was washed in TBS, resuspended in 15 volumes of TBS buffer containing 1% TBSX and mixed gently by rotation at 4 °C for 30 min. Samples were centrifuged (100,000 × g, 1 h at 4 °C), and the TBSX-soluble fraction was aliquoted and frozen as described for TBS. The pellet was washed with TBSX, resuspended in 70% FA to 150 mg/ml based on pellet weight, and mixed by rotation at room temperature for 2 h with occasional vortexing. Samples were centrifuged (100,000 × g, 1 h at 4 °C), and the FA-soluble fraction was neutralized (with 20 volumes of 1 m Tris base), aliquoted, and frozen at −80 °C. Total protein content in the TBS and TBSX extractions was determined via colorimetric micro-BCA assay per the manufacturer's instructions (Thermo Scientific). Because of interference of Tris and FA with the BCA assay, total protein in FA extractions was determined via a Quick Start Bradford protein micro-assay.
ELISAs
ApoE and Aβ42
ApoE levels were determined as described previously (42), using apoE2, -E3, and -E4 standards (Calbiochem) (0–100 ng/well). Briefly, apoE was measured using anti-apoE (WuE4) capture antibody, anti-apoE detection antibody (Calbiochem), and an HRP-conjugated secondary antibody. Initially Aβ42 levels were measured in hippocampal extractions from 2-, 4-, and 6-month-old mice (Fig. 1) using Wako ELISA kits as described previously (42). All subsequent Aβ42 ELISAs used HJ7.4 as the capture antibody, biotinylated antibody HJ5.1 as the detection antibody, and streptavidin-poly-HRP-conjugated secondary antibody (provided by Dr. D. Holtzman, Washington University) (2). For apoE and Aβ42 ELISAs, the protein/peptide standards were reconstituted in TBS, TBSX, or FA at concentrations equivalent to those in the assayed samples, as appropriate. All data were normalized to the amount of total protein in each extraction sample. For Fig. 5, total Aβ or apoE levels in each extraction fraction (i.e. TBS + TBSX + FA) were divided by the total protein in each fraction.
FIGURE 1.

Human APOE genotype-specific delay of Aβ accumulation in 5xFAD mice: EFAD transgenic mice. A, representative images of sagittal brain sections of 2-, 4-, and 6-month-old 5xFAD and EFAD mice immunostained for Aβ (red) and NeuN (green), ×20 magnification (scale bar = 500 μm). B, total Aβ42 levels in the hippocampus of 6-month-old 5xFAD and EFAD mice measured by ELISA. Data are expressed as mean ± S.E. and were analyzed by one-way ANOVA followed by Tukey's multiple comparison post hoc analysis. *, p < 0.05 versus m-apoE; #, p < 0.05 versus apoE2 and apoE3.
FIGURE 5.

The lower levels of total apoE4 are the result of a decrease only in the TBSX extraction fraction. Extraction profiles of apoE (A and Aβ42 (B) using a three-step sequential protein extraction (TBS, TBSX, and FA) in the hippocampus of 2-, 4-, and 6-month-old EFAD mice measured by ELISA. Data are expressed as mean ± S.E. and were analyzed by two-way ANOVA followed by Bonferroni's multiple comparison post hoc analysis. *, p < 0.05 versus 2 or 4 months; #, p < 0.05 versus apoE2 and apoE3.
Oligomeric Aβ
oAβ levels were determined based on a modified protocol by Xia et al. (24) using the Aβ antibody MOAB-2 (25). MOAB-2 was developed in the LaDu laboratory and is a high-affinity antibody specific for Aβ that does not detect APP. For the oAβ ELISA, MOAB-2 was used as a capture antibody and biotinylated MOAB-2 as a detection antibody based on a previous protocol (46). This ELISA pairing detects synthetic oAβ preparations but not monomeric Aβ using preparations of oAβ optimized for the original protocol (data not shown) (46). These oAβ preparations were used as a standard for measuring soluble oAβ levels in EFAD mice. In addition, the MOAB-2-based oAβ42 ELISA detects oligomeric but not monomeric Aβ42 using synthetic preparations characterized previously by our laboratory (data not shown) (47, 48).
Thioflavin S Staining for Plaques
Thio-S plaque staining and quantification was conducted by an investigator blinded to the APOE genotype using every ninth tissue section for six consecutive sections, beginning with the lateral-most section in the region of interest (ROI). Sections were washed in TBS (six times for 5 min each), mounted on glass coverslips, allowed to dry, rehydrated in Milli-Q water for 2 min, and stained in 0.1% Thio-S (dissolved in 50% EtOH plus 50% 1× PBS) for 5 min in the dark. The tissue was destained in 80% EtOH (twice for 5 min each) in the dark and mounted with VectaShield fluorescence mounting medium. The quantification of plaque deposition was performed as described (49). Briefly, images were captured on a NanoZoomer slide scanner (Hamamatsu Photonics), exported with NDP viewer software (Hamamatsu Photonics), and converted to 8-bit grayscale using ACDSee Pro 2 software (ACD Systems). The converted images were thresholded to highlight plaques and to diminish background signal (all images were thresholded at the same value). The objects identified after thresholding were inspected individually to confirm each object as a plaque. The subiculum and frontal cortex in each image were then outlined and analyzed with the “analyze particles” function in NIH ImageJ software. Plaques were evaluated for total number and percentage of area covered (total Thio-S immunoreactivity/ROI). Because plaques smaller than 5.5 μm were not readily distinguishable from background Thio-S fluorescence, limits were set in the ImageJ software such that all plaques greater than 5.5 μm were included, and plaques smaller than 5.5 μm were excluded.
Plaque Morphology
Preliminary analysis using tissue from EFAD mice indicated that plaques were of three major classifications, consistent with previous descriptions (16, 50): 1) diffuse = no center and weak Thio-S staining with a wispy morphology; 2) dense core with halos = an obvious center that stains brightly with Thio-S and a surrounding halo of weakly stained fibrils; and 3) compact = core plaques that stain very brightly with Thio-S, appear almost spherical, are generally smaller than plaques with halos, and have no obvious halo of fibrils. The quantification of plaque morphology was carried out using every 18th section of subiculum for three consecutive sections as described, using an NIH ImageJ Plugin for a counting application. The total numbers of each plaque type were expressed as a percentage of total plaques counted for each mouse.
Intraneuronal Aβ Counts
The number of neurons containing intraneuronal Aβ was determined using unbiased stereology (51) and counted by an investigator blinded to APOE genotype using sections immunostained with MOAB-2 (for Aβ) and NeuN. Briefly, every ninth tissue section for six consecutive sections was counted beginning with the lateral-most section in which the ROI was first identified. The ROI (subiculum, frontal cortex, or CA3) was outlined at ×10 magnification using the NeuN fluorescence channel. An optical fractionator design was then used for systematic random sampling of the entire ROI, to make unbiased estimates of total neuronal counts using a three-dimensional optical dissector counting probe. The parameters assume an average mounted tissue thickness of 25 μm (to account for tissue shrinkage) with 5-μm guard zones, a counting frame size of 200 × 200 μm, and an SRS grid size of 300 × 300 μm. Neurons were counted by moving through the entire depth of tissue in both the Aβ and NeuN fluorescence channels at each count site, to ensure the accuracy of intraneuronal Aβ counts (to disqualify extracellular Aβ and/or nonspecific signal). The number of Aβ-containing neurons, calculated by the stereology software based on the above parameters and the measured volume of each ROI, was expressed as the number of neurons/mm3 of tissue.
Western Blots
Total protein (17.5 μg) from TBSX-extracted EFAD cortex was run on 12% bis-Tris SDS-polyacrylamide gels and transferred to an 0.2-μm PVDF membrane followed by overnight incubation in primary antibodies against N-terminal APP (1:2500, Invitrogen) or β-actin as a loading control (1:10,000, Invitrogen). Membranes were washed, incubated in HRP-conjugated secondary antibody raised against mouse IgG, developed with super ECL reagent (Pierce), and exposed using Kodak ImageStation MI software.
Statistical Analyses
Data were analyzed by one-way analysis of variance (ANOVA) followed by Tukey's post hoc analysis (Figs. 1B and 6) or by two-way ANOVA followed by Bonferroni's post hoc analysis (all other figures) using GraphPad Prism version 4 for Macintosh. p < 0.05 was considered significant.
FIGURE 6.
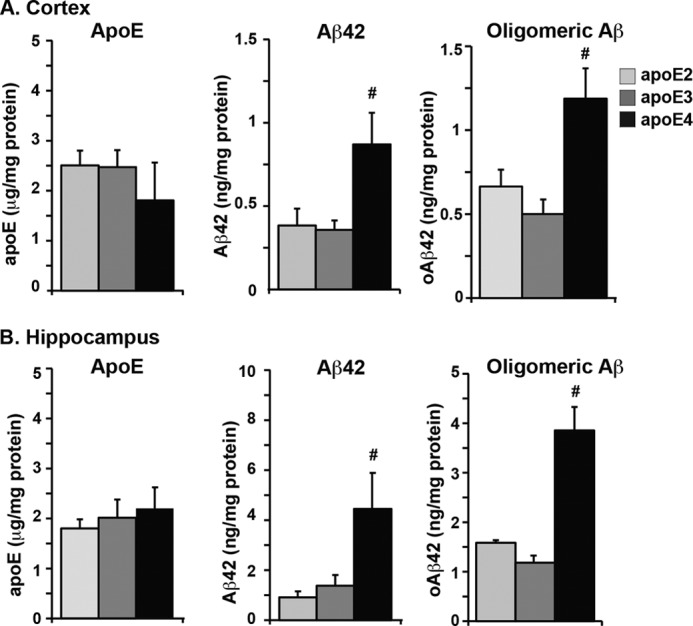
Soluble Aβ42 and soluble oligomeric Aβ levels are higher in E4FAD mice compared with E2FAD and E3FAD mice. ApoE, Aβ42, and oAβ in the TBS/soluble extraction fraction of the cortex (A) and hippocampus (B in 6-month-old EFAD mice measured by ELISA. Data are expressed as mean ± S.E. and were analyzed by one-way ANOVA followed by Tukey's multiple comparison post hoc analysis. #, p < 0.05 versus apoE2 and apoE3.
RESULTS
Human APOE Genotype-specific Delay of Aβ Accumulation in 5xFAD Mice: EFAD Transgenic Mice
EFAD mice were developed by crossing 5xFAD mice with the three strains of human apoE-TR mice, resulting in E2FAD, E3FAD, and E4FAD mice. Given the abundant Aβ pathology in 5xFAD mice at 2 months (11), total Aβ deposition was examined in 2-, 4-, and 6-month-old EFAD mice by IHC using the monoclonal anti-Aβ antibody MOAΒ-2 (Fig. 1A), a new anti-Aβ antibody that does not recognize full-length or soluble APP fragments (25). The overall regional pattern of Aβ accumulation in the brain was similar among the 5xFAD and EFAD mice, with no APOE genotype-specific changes. Aβ accumulation appeared first in the subiculum of the hippocampal formation and in the deep layers of the frontal cortex, spreading to the outer layers of the cortex and CA1 region of the hippocampus. Consistent with previous reports (11), 5xFAD mice (expressing endogenous mouse apoE) showed extracellular Aβ deposition as early as 2 months, which increased with age from 2 to 6 months (Fig. 1A). Compared with 5xFAD mice, Aβ deposition was delayed ∼4 months in the EFAD mice, with accumulation earliest in E4FAD > E3FAD = E2FAD. Aβ accumulation appeared earlier and was greater in the subiculum compared with the frontal cortex, allowing for regional, as well as temporal, comparisons of the effects of the APOE genotype on the progression of Aβ accumulation.
To confirm the difference in total Aβ accumulation observed by IHC between 5xFAD and EFAD mice, and among the APOE genotypes in the EFAD mice, Aβ42 levels were measured in brain homogenates at 6 months by an Aβ42-specific ELISA (Fig. 1B). Consistent with the Aβ accumulation detected by IHC, the levels of total Aβ42 were higher in 5xFAD compared with EFAD mice. Aβ42 levels were highest in E4FAD compared with E2FAD and E3FAD mice. EFAD mice demonstrated APOE genotype-dependent changes in Aβ accumulation that are significant in mice aged 2 to 6 months, thus providing a tractable model.
Plaque Deposition Is Greatest in E4FAD, an Effect Modulated by Brain Region and APOE Genotype-specific Plaque Morphology
Although amyloid plaques may not correlate directly with a cognitive decline in AD patients (52), they remain a key determinant in the diagnosis of AD. Importantly, APOE4 is associated with a higher plaque burden than APOE2 and APOE3 (53). Therefore, the effect of APOE genotype on amyloid plaque burden in the EFAD mice was determined using Thio-S, a stain specific for parallel β-sheet structure (Fig. 2). Representative images from 2-, 4-, and 6-month-old EFAD mice demonstrate that Thio-S staining is initiated at 4 months in the subiculum and deep layers of the frontal cortex (Fig. 2A) and is increased by 6 months for all APOE genotypes, a pattern consistent with IHC for total Aβ (Fig. 1A). These data were quantified to determine the number and percentage of area covered by plaques (frontal cortex (Fig. 2B) and subiculum (Fig. 2C)), measures that produced equivalent differences among the APOE genotypes. Overall, plaque deposition was greatest in E4FAD mice compared with E2FAD and E3FAD mice and was significant at 4 months in the frontal cortex and subiculum and at 6 months in the frontal cortex.
FIGURE 2.

Plaque deposition is greatest in E4FAD, an effect modulated by the brain region and APOE genotype-specific plaque morphology. A, representative images of sagittal brain sections from 2-, 4-, and 6-month-old EFAD mice stained with Thio-S (green), ×20 magnification (scale bar = 500 μm). Quantification of the number of plaques and percentage of area covered by plaques in the frontal cortex (B) and subiculum (C). D, plaque morphology. Shown are representative images of the primary types of plaques in EFAD brain, ×63 magnification: diffuse, dense-cored with halos, and compact (scale bar = 20 μm). E, quantification of plaque morphology in the subiculum. Shown is the percentage of each plaque type in 4- and 6-month-old EFAD mice. Data are expressed as mean ± S.E. and were analyzed by two-way ANOVA followed by Bonferroni's multiple comparison post hoc analysis. *, p < 0.05 versus 2 or 4 months; #, p < 0.05 versus apoE2 and apoE3; ‡, p < 0.05 versus apoE3.
It is interesting to note that at 6 months, in the subiculum, plaque levels in both E2FAD and E4FAD mice were significantly higher than in E3FAD mice (Fig. 2C). Recent reports from the “oldest of the old” studies demonstrate significant plaque burden in the absence of cognitive deficits in APOE2 subjects (54, 55). Importantly, in a case study of an APOE2/2 subject, plaque morphology was described as fleecy/diffuse compared with the compact/dense core morphology of APOE4 AD patients (15). Based on this potential difference in plaque morphology between the E2FAD and E4FAD mice, plaques in the subiculum (at 4 and 6 months) were classified and quantified according to the following scale: 1) diffuse = no obvious dense core; 2) dense-cored, with halos = an obvious center with a halo of fibrils; 3) compact = dense-cored plaques that appear spherical (Fig. 2D). The majority of the plaques in EFAD mice were either diffuse or compact, and this distribution in plaque morphology did not change with age (Fig. 2E). In E2FAD and E3FAD mice the majority of plaques were diffuse, accounting for more than 50% of total plaques, whereas diffuse plaques were significantly lower in E4FAD mice (e.g. 30% at 6 months). The reverse pattern was observed with compact plaques; ∼20% of the plaques in E2FAD and E3FAD mice were compact, whereas 40–50% of plaques in E4FAD mice were compact. In addition, from 4 to 6 months, the proportion of diffuse plaques decreased and that of compact plaques increased only in the E4FAD mice (Fig. 2E). This trend suggests that APOE2 and APOE3 may maintain plaques in a diffuse morphology, whereas APOE4 facilitates compact plaque formation, the plaque morphology traditionally associated with AD pathology (15).
Intraneuronal Aβ Levels Are Comparable among the APOE Genotypes
Intraneuronal Aβ has been observed prior to extracellular plaques in human AD tissue and in mouse models and has been linked to neurotoxicity (56, 57). As described previously, MOAB-2 is an anti-Aβ antibody that specifically detects Aβ but not APP. MOAB-2 demonstrates strong intraneuronal immunoreactivity in 5xFAD and 3xTg mouse brain tissue that precedes extracellular Aβ deposition (25). To accurately measure the extent of intracellular Aβ accumulation without the confounding contribution of extracellular Aβ deposition, the total number of Aβ-containing neurons was counted in the frontal cortex (see Fig. 3A for a representative image and Fig. 3B for quantification) and subiculum (Fig. 3C) at 2 and 4 months and in the CA3 region of the hippocampus (Fig. 3D) at 4 and 6 months using IHC with MOAB-2 and unbiased stereological methods. The number of Aβ-containing neurons increased significantly with time in all three brain regions (Fig. 3, B–D). However, there were no significant APOE genotype-specific differences in the total number of Aβ-containing neurons in any region analyzed.
FIGURE 3.

Intraneuronal Aβ levels are comparable among the APOE genotypes. A, representative images of cortex in sagittal brain sections from 2- and 4-month-old EFAD mice immunostained for Aβ (red) and NeuN (green), ×20 magnification (scale bar = 20 μm). Shown are total numbers of Aβ-containing neurons in the frontal cortex (B) at 2 and 4 months; in the subiculum (C) at 2 and 4 months; and in the CA3 region (D) at 4 and 6 months in EFAD mice counted via unbiased stereology. Data are expressed as mean ± S.E. and were analyzed by two-way ANOVA followed by Bonferroni's multiple comparison post hoc analysis. *, p < 0.05 versus 2 or 4 months.
Total apoE Levels Are Lower and Total Aβ42 Levels Are Higher in E4FAD Mice Compared with E2FAD and E3FAD Mice
Total apoE (Fig. 4A) and Aβ42 (Fig. 4B) levels in 2-, 4-, and 6-month-old EFAD mice were measured by ELISAs in the cerebellum (a brain region resistant to Aβ pathology) as well as in the cortex and hippocampus (regions susceptible to Aβ pathology). For IHC, the subiculum was specifically analyzed, as it was the region of the hippocampus with the greatest Aβ accumulation. However, for biochemical measurements, it was necessary to homogenize the entire hippocampus, thus “diluting” the subiculum-specific Aβ42 accumulation, although hippocampal Aβ levels remained significantly higher than in the cortex, as observed with IHC. In general, across age and brain regions, total apoE4 levels were lower than apoE2 and apoE3 (Fig. 4A). Specifically, apoE4 levels are significantly lower than apoE2 and apoE3 in the cerebellum at 4 months, in the cortex at 2, 4, and 6 months, and in the hippocampus at 2 and 4 months of age (for specific comparisons, see Fig. 4A). Previous studies in humans and mice report lower apoE4 levels compared with apoE3, although little data are published that compare apoE4 with apoE2 levels (3, 26, 33–35, 58, 59). Interestingly, total apoE levels in the EFAD mice were lowest in the cerebellum, significantly higher in the cortex, and highest in the hippocampus (Fig. 4A, dashed lines mean the data are collapsed by genotype and age within a region; p < 0.001). In addition, apoE levels did not change in response to age, and thus there was no APOE genotype-specific response to increasing Aβ accumulation (Fig. 4B).
FIGURE 4.

Total apoE levels are lower and total Aβ42 levels are higher in E4FAD mice compared with E2FAD and E3FAD mice. Shown are total levels of apoE (A) and Aβ42 (B) in the cerebellum, hippocampus, and cortex of 2-, 4-, and 6-month-old EFAD mice measured by ELISA (n.m. = not measured). The dashed lines mark the mean of apoE within the CB < CX < H (p < 0.001). Data are expressed as mean ± S.E. and were analyzed by two-way ANOVA followed by Bonferroni's multiple comparison post hoc analysis. *, p < 0.05 versus 2 or 4 months; #, p < 0.05 versus apoE2 and apoE3; ‡, p < 0.05 versus apoE3.
In the cortex and hippocampus, total Aβ42 levels increased with age in an APOE genotype-specific manner (Fig. 4B). In the cortex, Aβ42 levels were generally low at 2 months but increased significantly by 4 months such that E4FAD > E3FAD = E2FAD. It is interesting to note that at 6 months, the levels of Aβ42 in E2FAD and E4FAD mice were both significantly higher than in E3FAD mice. In the hippocampus, Aβ42 levels were low at 2 months but increased significantly by 4 and 6 months such that E4FAD > E3FAD = E2FAD. The Aβ42 levels in the cerebellum were below detection level until 6 months, when the values were low and equivalent among the genotypes (Fig. 4B). As observed for apoE, Aβ42 levels were lowest in the cerebellum, higher in the cortex, and highest in the hippocampus (Fig. 4B, p < 0.001; note the y axis values change for the cerebellum and cortex versus the hippocampus). These data in the EFAD mice from 2 to 6 months for total Aβ42 levels are consistent with Aβ immunoreactivity (Fig. 1A) and Thio-S staining (Fig. 2), demonstrating the earliest accumulation of Aβ in the subiculum followed by the deep layers of the frontal cortex.
In summary, total apoE and Aβ42 levels follow a similar pattern of brain region-dependent accumulation that is initiated in the subiculum and the deep layers of the frontal cortex. Total apoE levels did not change in response to age (2–6 months), although apoE4 levels were generally lower than apoE2 and apoE3 at each age and in each brain region. Total Aβ42 levels increased with age in disease-susceptible regions and were generally higher with apoE4 compared with apoE2 and apoE3. Thus, APOE4 promotes Aβ accumulation in the hippocampus (subiculum) and cortex as demonstrated by IHC (Figs. 1A), Thio-S staining (Fig. 2A), and biochemical analysis (Fig. 4).
The Lower Levels of Total apoE4 Are the Result of a Decrease Only in the TBSX Extraction Fraction
Previously, a three-step extraction protocol was optimized for the detection of apoE and Aβ in the presence of increasing amyloid deposition, focusing particularly on the full extraction of insoluble apoE and Aβ42 with FA, as plaques in the 5xFAD mice are primarily compact/dense-cored (42). Thus, this protocol is a sequential protein extraction in TBS (“soluble”), TBSX (“detergent-soluble”), and FA (“insoluble”). Fig. 5 depicts the extraction profiles for apoE (Fig. 5A) and Aβ42 (Fig. 5B) from the hippocampus of 2-, 4-, and 6-month-old EFAD mice, as this is the region with the earliest signs of Aβ accumulation (Figs. 1, 2, and 4B). In the TBS-soluble fraction, apoE levels did not differ with age or by APOE genotype (Fig. 5A). ApoE2 and apoE3 were extracted primarily to the TBSX fraction, whereas apoE4 levels were significantly lower in this fraction at 2, 4, and 6 months. In the FA fraction, apoE levels increased from 4 to 6 months with all APOE genotypes, consistent with the increase in amyloid plaques (53). The lower total apoE4 levels compared with total apoE2 and apoE3 levels (Fig. 4A) were reflected primarily in the TBSX fraction of the extraction profile (Fig. 5A). Further, the distribution of apoE across the extraction profiles reveals that the proportion of apoE4 in the TBSX fraction is specifically lower in the brain regions that are susceptible to Aβ pathology (cortex and hippocampus) and not in the region resistant to Aβ pathology (cerebellum; data not shown). Thus, using an entirely different approach from previous reports, these data provide evidence that the levels of lipoprotein-associated apoE4 are lower than those of apoE2 and apoE3.
In AD patients, soluble Aβ levels in the brain correlate with disease severity (52). In EFAD mice, Aβ42 increased with age in the TBS-soluble fractions from 2 to 6 months (Fig. 5B). At 4 and 6 months, TBS-soluble Aβ42 levels were significantly higher in E4FAD mice compared with E2FAD and E3FAD mice. Indeed, at 6 months, Aβ levels in the E4FAD mice were at least 4-fold higher than in E2FAD or E3FAD mice. In the TBSX fractions, Aβ42 did not change significantly with age or APOE genotype. FA-extracted Aβ42 levels increased with age for all APOE genotypes and were significantly higher in E4FAD mice at 4 and 6 months compared with E2FAD and E3FAD mice (Fig. 5B). At 6 months, the Aβ42 levels in the FA fraction of the E4FAD mice were at least 4-fold higher than in E2FAD or E3FAD mice, comparable to the difference in the Aβ42 levels in the TBS fraction. The high levels of Aβ42 in the FA fraction of the hippocampus of E4FAD mice, as well as the increased Aβ immunoreactivity (Fig. 1A) and Thio-S staining (Fig. 2) in the subiculum of the E4FAD mice, is consistent with total Aβ42 levels (Fig. 4B).
Soluble Aβ42 and Soluble Oligomeric Aβ Levels Are Higher in E4FAD Mice Compared with E2FAD and E3FAD Mice
Soluble Aβ42 and soluble oAβ are increased in AD patients and are considered important for AD progression (52). However, the effect of APOE genotype on these species remains unclear. Therefore, soluble apoE, Aβ42, and oAβ were compared in 6-month-old EFAD mice (Fig. 6). Soluble apoE levels were comparable in the cortex (Fig. 6A) and hippocampus (Fig. 6B) and did not vary significantly by APOE genotype. Therefore, any differences observed in Aβ speciation are likely mediated by differences in the function rather than the amount of each apoE isoform. Soluble Aβ42 levels were higher in the hippocampus than the cortex in EFAD mice (Fig. 6), consistent with early Aβ accumulation in this region. Further, in both the cortex and the hippocampus, soluble Aβ42 levels were 2-fold and 3-fold higher, respectively, in E4FAD mice compared with E2FAD and E3FAD mice. An ELISA for measuring oAβ was developed using MOAB-2, based on the protocol developed by Xia et al. (Ref. 24, see details under “Methods”). As with soluble Aβ42, oAβ levels were higher in the hippocampus than the cortex. In E4FAD mice, oAβ levels were ∼2-fold higher in the cortex and 3-fold higher in the hippocampus compared with those in E2FAD and E3FAD mice. Thus, Aβ accumulates preferentially as soluble and oligomeric forms in the presence of APOE4, and comparing cortex to hippocampus, these soluble species of Aβ appear to increase with the accumulation of total Aβ. Overall, these data demonstrate that APOE genotype modulates Aβ speciation, an effect likely mediated by functional differences among the apoE isoforms.
DISCUSSION
In currently available apoE/FAD-Tg mouse models, expression of human APOE significantly delays plaque deposition (for review Ref. 44). To generate a more tractable model to investigate the apoE isoform effect on Aβ accumulation, 5xFAD mice (11) were crossed with apoE-TR mice (9), producing the EFAD mouse model. In 5xFAD mice, neurons produce primarily human Aβ42 and show early and aggressive Aβ42 deposition, with significant intraneuronal Aβ accumulation at 6 weeks, significant plaque deposition at 2 months, and subtle changes in synaptic markers at 9 months. ApoE-TR mice are perhaps the most biologically relevant transgenic mouse model for apoE, as human apoE is expressed primarily by glia at physiologically regulated levels. In EFAD mice, Aβ deposition is delayed ∼4 months compared with 5xFAD mice, creating a window from 2 to 6 months during which comparisons of multiple forms of Aβ accumulation can be measured, although this is still early in the overall process of Aβ accumulation in either the hippocampus or the cortex. Compared with E2FAD and E3FAD mice, E4FAD mice consistently demonstrated accelerated Aβ accumulation, greater total levels of Aβ42, and selective increases in soluble Aβ42 and oAβ levels. Interestingly, rather than simply delaying Aβ accumulation relative to E3FAD mice, E2FAD mice displayed Aβ accumulation similar to E3FAD. The development of these mice will allow future studies to further probe specific mechanisms for the observed changes among the apoE isoforms, as well as apoE isoform-specific changes in Aβ accumulation.
Autosomal dominant mutations that increase either total Aβ or specifically Aβ42 cause FAD. However, the identity of the specific assembly(s) of Aβ42 that causes the eventual neurotoxicity characteristic of AD remains unclear. Although the total plaque burden does not correlate with the degree of dementia or neurodegenerative pathology in humans (52), plaque staging has identified particular plaque morphologies that likely contribute to neurotoxicity more than others (15, 16). Overall, E4FAD mice display the highest plaque load, as expected based on previous studies in humans and transgenic mice (for review see Ref. 53). The exception is that the plaque burden in E2FAD mice was equivalent to E4FAD mice in the subiculum at 6 months; the subiculum is the most affected region in the oldest mice studied. However, analysis of plaque morphology revealed that whereas the majority of the APOE4 plaques are compact, the majority of the APOE2 plaques are diffuse. This APOE2-induced deposition of diffuse plaques is novel in a mouse model and consistent with the previous work by Dr. C. Kawas and co-workers (54, 55) who report that increased diffuse plaques are associated with APOE2 in cognitively normal subjects compared with compact plaques in APOE4 AD patients from the oldest of the old study.
The structural properties and composition of different plaque types may correspond to functional differences. A renewed interest in the pathological role of plaques has suggested that oAβ species accumulate at the periphery of dense-cored plaques (60, 61) and that compact plaques enhance Aβ toxicity indirectly by creating a platform on which toxic oAβ species can accumulate or stabilize. This effect would be enhanced with APOE4, as it is associated with a larger proportion of compact plaques. Diffuse plaques may prevent Aβ-induced toxicity by depriving soluble oAβ of a stable foundation. Determining the effect of APOE genotype on the origins, structural characteristics, and neurotoxicity of these different plaque types in EFAD mice is a crucial next step.
In addition to plaque morphology, recent evidence indicates that intraneuronal Aβ accumulation may be an important proximal neurotoxic event in AD pathogenesis (for review see Ref. 56, 57). Indeed, intraneuronal Aβ has been correlated with cognitive deficits in FAD-Tg mice. However, even the existence of Aβ deposits within neurons has been challenged recently by Winton et al. (62). These authors purport that it is actually intraneuronal APP being detected by antibodies thought to be specific for Aβ. MOAB-2 is an anti-Aβ antibody that does not detect APP (25). MOAB-2 demonstrates strong intraneuronal immunoreactivity in 5xFAD and 3xTg mouse brain tissue that precedes extracellular Aβ deposition. The current study represents one of the first reports of the effects of APOE genotype on intraneuronal Aβ in an FAD-Tg mouse model. Using MOAB-2, the accumulation of intraneuronal Aβ was comparable between the isoforms in a specific region at a given age, but accumulation increased significantly with age and varied with the AD susceptibility of the brain region. These data are in apparent contrast with data from a single study in humans that reports an increase in intraneuronal Aβ with one or two alleles of APOE4 (63). However, this human study measured Aβ40, whereas the EFAD mice express almost exclusively Aβ42, which is one possible explanation for the discrepancy. To our knowledge, no studies have yet reported the effect of APOE genotype on intraneuronal Aβ accumulation in an apoE/FAD-Tg mouse.
In E4FAD mice, total apoE levels in the brain are lower compared with E2FAD and E3FAD, as observed previously in human and apoE Tg mouse models (35–41). With TBS, TBSX, and FA extraction, apoE2 and apoE3 from the EFAD mouse tissue extract primarily to the TBSX fraction, consistent with the extraction of apoE-containing lipoproteins from plasma. However, the lower total apoE4 levels are reflected only in their significant reduction in the TBSX fraction, as the levels of apoE2, apoE3, and apoE4 are comparable in the TBS- and FA-extracted fractions. Further, this apoE4-specific distribution appears to occur in the hippocampus and cortex, which are AD-susceptible brain regions, and not the cerebellum, a disease-resistant region. Because the TBSX fraction contains the apoE extracted specifically from lipoprotein particles, potential interpretations of these data are that less apoE4 is lipoprotein-associated and/or apoE4-containing lipoproteins are less lipidated. Although a substantial amount of research has been devoted to understanding the functional differences between lipidated and non-lipidated apoE, including its ability to bind Aβ (64–69) or apoE receptors (69–72), surprisingly little is known about the effect of the apoE isoform on the lipidation state of CNS-relevant lipoproteins. Although the general dogma in the field is that apoE4 is less lipidated than apoE3, the number of publications that compare the lipidation state of apoE4 particles to apoE3 particles in vivo is severely limited. Relevant in vitro data include, for example, the results that glia-mediated degradation of apoE4 is increased and cholesterol release is reduced in primary glial cultures from apoE-TR mice expressing apoE4 compared with glial cells expressing apoE3 (35, 73). Thus, the extraction profiles for the apoE isoforms, as presented in this article, will provide critical, novel information for interpreting results from AD therapeutics that target apoE levels and/or its level of lipidation (74–77).
The neurotoxicity of soluble oAβ is of interest because of the apparent correlation of oAβ levels with disease severity; soluble oligomers correlate with cognitive decline (Mini Mental State Examination (MMSE) scores) and tangle stage (20). In FAD-Tg mice, soluble oAβ suppresses synaptic function, reduces synaptic plasticity, and impairs learning and memory (52). Prior work from our laboratory demonstrates that oAβ-induced deficits in synaptic plasticity (78) and neuronal viability (79) are enhanced in the presence of APOE4 compared with APOE2 and APOE3. The current work illustrates that soluble Aβ42 and oAβ levels are increased in the TBS-soluble fractions of the disease-vulnerable cortex and hippocampus in the E4FAD mice, and the preferential accumulation of Aβ42 results in an increase in both oAβ and plaques. As discussed above, the APOE4-specific increase in compact plaque formation may provide a platform for the assembly of oAβ.
The apoE isoform-specific effects on soluble species of Aβ continues to be the focus of extensive research efforts, and a number of potential molecular mechanisms have been proposed. These include effects on: Aβ oligomerization (80, 81), Aβ clearance though multiple mechanisms including intracellular uptake and degradation by glia (82–85) and neurons (86, 87), clearance across the blood-brain barrier (88, 89), extracellular enzymatic degradation (64), ISF-mediated clearance (90), and perivascular drainage (40) in addition to the potential interplay between plaque morphology and apoE and oAβ levels (for example, see Ref. 39). Further, whether these effects are mediated by direct binding of Aβ to apoE (68) or by apoE-specific pathways and receptors add a further layer of complexity (76).
Modulation of the levels of apoE is an attractive target for AD therapy. Indeed, recent evidence has demonstrated that bexarotene (an RXR agonist) increases apoE levels and decreases soluble Aβ in mice within hours and significantly reduces insoluble Aβ after 3 days, although plaque levels at 3 months remained unchanged (76). However, application of these data to AD patients is difficult, as this initial study used transgenic mice producing human Aβ but expressing mouse apoE. Therefore, it is important to demonstrate drug efficacy on Aβ pathology, and in particular Aβ speciation, in FAD-Tg mice that express the human apoE isoforms prior to clinical trial initiation. Although drug intervention trials have not yet been performed on the EFAD mice, these mice are a model of early Aβ pathology, express the human apoE isoforms, demonstrate apoE isoform-specific effects on early Aβ speciation, and are thus an ideal model for efficient evaluation of drug therapies targeting apoE. In addition, initial therapeutic testing in a transgenic mouse expressing the human apoE isoforms is critical, as APOE4 carriers can exhibit a differential response to certain therapeutic interventions, complicating the interpretation of drug trials, which can lead to costly failure after extensive phase 3 clinical trials (53).
In conclusion, EFAD mice represent a tractable new mouse model that allowed the identification of the APOE genotype effects on the earliest accumulations of Aβ (from 2 to 6 months). Although APOE genotype-specific differences were observed in plaque deposition and morphology, intraneuronal Aβ deposition was not APOE genotype-specific. Compared with APOE2 and APOE3, APOE4 promoted higher levels of total Aβ42, as well as soluble Aβ42 and oAβ. In addition, the levels of apoE4 were lower and less apoE4 appeared to be lipoprotein-associated compared with apoE2 or apoE3. Thus, a number of apoE isoform-specific mechanisms were identified in vivo that likely contribute to the effect of the APOE genotype on AD risk. EFAD mice can be used for further study of these mechanisms. These mice may also provide a model for the efficient evaluation of both AD drug prevention and treatment paradigms. The risk imparted by APOE4 extends to other cerebral insults with amyloid deposition, including traumatic brain injury, cerebral hemorrhage, and stroke, suggesting that EFAD mice may also provide a model for assessing additional therapeutic strategies.
This work was supported, in whole or in part, by National Institutes of Health Grants P01AG030128 (through the NIA) (to M. J. L.), P01AG030128-03S1 (through the NIA) (to E. N. H.), and 5T32-AG036697 (to K. L. Y.); Alzheimer's Association Grant ZEN-08-899000 (to M. J. L.); and a University of Illinois at Chicago Center for Clinical and Translational Science Grant UL1RR029879 (to M. J. L.).

This article contains supplemental Fig. 1.
- AD
- Alzheimer disease
- Aβ
- amyloid-β
- apoE
- apolipoprotein E
- APP
- amyloid precursor protein
- FA
- formic acid
- FAD
- familial AD
- FAD-Tg
- transgenic mice expressing APP and/or PS1 with FAD mutations
- apoE-TR
- apoE-targeted replacement mice
- 5xFAD
- mice expressing five FAD mutations
- EFAD
- 5xFAD mice crossed with apoE-TR mice
- oAβ
- oligomeric Aβ
- ROI
- region of interest
- Thio-S
- thioflavin S
- bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- ANOVA
- analysis of variance
- IHC
- immunohistochemical or immunohistochemistry.
REFERENCES
- 1. Bu G. (2009) Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim J., Jiang H., Park S., Eltorai A. E., Stewart F. R., Yoon H., Basak J. M., Finn M. B., Holtzman D. M. (2011) Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-β amyloidosis. J. Neurosci. 31, 18007–18012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bales K. R., Liu F., Wu S., Lin S., Koger D., DeLong C., Hansen J. C., Sullivan P. M., Paul S. M. (2009) Human APOE isoform-dependent effects on brain β-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Drzezga A., Grimmer T., Henriksen G., Mühlau M., Perneczky R., Miederer I., Praus C., Sorg C., Wohlschläger A., Riemenschneider M., Wester H. J., Foerstl H., Schwaiger M., Kurz A. (2009) Effect of APOE genotype on amyloid plaque load and gray matter volume in Alzheimer disease. Neurology 72, 1487–1494 [DOI] [PubMed] [Google Scholar]
- 5. Grimmer T., Tholen S., Yousefi B. H., Alexopoulos P., Förschler A., Förstl H., Henriksen G., Klunk W. E., Mathis C. A., Perneczky R., Sorg C., Kurz A., Drzezga A. (2010) Progression of cerebral amyloid load is associated with the apolipoprotein E ϵ4 genotype in Alzheimer's disease. Biol. Psychiatry 68, 879–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buttini M., Yu G. Q., Shockley K., Huang Y., Jones B., Masliah E., Mallory M., Yeo T., Longo F. M., Mucke L. (2002) Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid β peptides but not on plaque formation. J. Neurosci. 22, 10539–10548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Holtzman D. M., Bales K. R., Tenkova T., Fagan A. M., Parsadanian M., Sartorius L. J., Mackey B., Olney J., McKeel D., Wozniak D., Paul S. M. (2000) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 97, 2892–2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fryer J. D., Simmons K., Parsadanian M., Bales K. R., Paul S. M., Sullivan P. M., Holtzman D. M. (2005) Human apolipoprotein E4 alters the amyloid-β 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J. Neurosci. 25, 2803–2810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sullivan P. M., Mezdour H., Aratani Y., Knouff C., Najib J., Reddick R. L., Quarfordt S. H., Maeda N. (1997) Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem. 272, 17972–17980 [DOI] [PubMed] [Google Scholar]
- 10. Games D., Adams D., Alessandrini R., Barbour R., Berthelette P., Blackwell C., Carr T., Clemens J., Donaldson T., Gillespie F., Guido T., Hagopian S., Johnson-Wood K., Khan K., Lee M., Leibowitz P., Lieberburg I., Little S., Masliah E., McConlogue L., Montoya-Zavala, Mucke L., Paganini L., Penniman E., Power M., Schenk D., Seubert P., Snyder B., Soriano F., Tan H., Vitale J., Wadsworth S., Wolozin B., Zhao J. (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 373, 523–527 [DOI] [PubMed] [Google Scholar]
- 11. Oakley H., Cole S. L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., Berry R., Vassar R. (2006) Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fernández-Vizarra P., Fernández A. P., Castro-Blanco S., Serrano J., Bentura M. L., Martínez-Murillo R., Martínez A., Rodrigo J. (2004) Intra- and extracellular Aβ and PHF in clinically evaluated cases of Alzheimer's disease. Histol. Histopathol. 19, 823–844 [DOI] [PubMed] [Google Scholar]
- 13. D'Andrea M. R., Nagele R. G., Wang H. Y., Lee D. H. (2002) Consistent immunohistochemical detection of intracellular β-amyloid42 in pyramidal neurons of Alzheimer's disease entorhinal cortex. Neurosci. Lett. 333, 163–166 [DOI] [PubMed] [Google Scholar]
- 14. D'Andrea M. R., Nagele R. G., Wang H. Y., Peterson P. A., Lee D. H. (2001) Evidence that neurones accumulating amyloid can undergo lysis to form amyloid plaques in Alzheimer's disease. Histopathology 38, 120–134 [DOI] [PubMed] [Google Scholar]
- 15. Thal D. R., Griffin W. S., Braak H. (2008) Parenchymal and vascular Aβ-deposition and its effects on the degeneration of neurons and cognition in Alzheimer's disease. J. Cell. Mol. Med. 12, 1848–1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thal D. R., Capetillo-Zarate E., Del Tredici K., Braak H. (2006) The development of amyloid β protein deposits in the aged brain. Sci. Aging Knowledge Environ. 2006, re1. [DOI] [PubMed] [Google Scholar]
- 17. Haass C., Selkoe D. J. (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 8, 101–112 [DOI] [PubMed] [Google Scholar]
- 18. Kuo Y. M., Emmerling M. R., Vigo-Pelfrey C., Kasunic T. C., Kirkpatrick J. B., Murdoch G. H., Ball M. J., Roher A. E. (1996) Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 271, 4077–4081 [DOI] [PubMed] [Google Scholar]
- 19. Selkoe D. J. (2011) Resolving controversies on the path to Alzheimer's therapeutics. Nat. Med. 17, 1060–1065 [DOI] [PubMed] [Google Scholar]
- 20. Tomic J. L., Pensalfini A., Head E., Glabe C. G. (2009) Soluble fibrillar oligomer levels are elevated in Alzheimer's disease brain and correlate with cognitive dysfunction. Neurobiol. Dis. 35, 352–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jin M., Shepardson N., Yang T., Chen G., Walsh D., Selkoe D. J. (2011) Soluble amyloid β-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc. Natl. Acad. Sci. U.S.A. 108, 5819–5824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McLean C. A., Cherny R. A., Fraser F. W., Fuller S. J., Smith M. J., Beyreuther K., Bush A. I., Masters C. L. (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann. Neurol. 46, 860–866 [DOI] [PubMed] [Google Scholar]
- 23. Huang Y., Mucke L. (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148, 1204–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xia W., Yang T., Shankar G., Smith I. M., Shen Y., Walsh D. M., Selkoe D. J. (2009) A specific enzyme-linked immunosorbent assay for measuring β-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch. Neurol. 66, 190–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Youmans K. L., Tai L. M., Kanekiyo T., Stine W. B., Jr., Michon S. C., Nwabuisi-Heath E., Manelli A. M., Fu Y., Riordan S., Eimer W. A., Binder L., Bu G., Yu C., Hartley D. M., LaDu M. J. (2012) Intraneuronal Aβ detection in 5xFAD mice by a new Aβ-specific antibody. Mol. Neurodegener. 7, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Poirier J. (2005) Apolipoprotein E, cholesterol transport and synthesis in sporadic Alzheimer's disease. Neurobiol. Aging 26, 355–361 [DOI] [PubMed] [Google Scholar]
- 27. Poirier J. (2008) Apolipoprotein E represents a potent gene-based therapeutic target for the treatment of sporadic Alzheimer's disease. Alzheimers Dement. 4, S91–S97 [DOI] [PubMed] [Google Scholar]
- 28. Ramaswamy G., Xu Q., Huang Y., Weisgraber K. H. (2005) Effect of domain interaction on apolipoprotein E levels in mouse brain. J. Neurosci. 25, 10658–10663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Raffai R. L., Dong L. M., Farese R. V., Jr., Weisgraber K. H. (2001) Introduction of human apolipoprotein E4 “domain interaction” into mouse apolipoprotein E. Proc. Natl. Acad. Sci. U.S.A. 98, 11587–11591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bertrand P., Poirier J., Oda T., Finch C. E., Pasinetti G. M. (1995) Association of apolipoprotein E genotype with brain levels of apolipoprotein E and apolipoprotein J (clusterin) in Alzheimer disease. Brain Res. Mol. Brain Res. 33, 174–178 [DOI] [PubMed] [Google Scholar]
- 31. Glöckner F., Meske V., Ohm T. G. (2002) Genotype-related differences of hippocampal apolipoprotein E levels only in early stages of neuropathological changes in Alzheimer's disease. Neuroscience 114, 1103–1114 [DOI] [PubMed] [Google Scholar]
- 32. Cruchaga C., Kauwe J. S., Nowotny P., Bales K., Pickering E. H., Mayo K., Bertelsen S., Hinrichs A., The Alzheimer's Disease Neuroimaging Initiative, Fagan A. M., Holtzman D. M., Morris J. C., Goate A. M. (2012) Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer's disease. Hum. Mol. Genet. 21, 4558–4571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Beffert U., Cohn J. S., Petit-Turcotte C., Tremblay M., Aumont N., Ramassamy C., Davignon J., Poirier J. (1999) Apolipoprotein E and β-amyloid levels in the hippocampus and frontal cortex of Alzheimer's disease subjects are disease-related and apolipoprotein E genotype-dependent. Brain Res. 843, 87–94 [DOI] [PubMed] [Google Scholar]
- 34. Sullivan P. M., Han B., Liu F., Mace B. E., Ervin J. F., Wu S., Koger D., Paul S., Bales K. R. (2011) Reduced levels of human apoE4 protein in an animal model of cognitive impairment. Neurobiol. Aging 32, 791–801 [DOI] [PubMed] [Google Scholar]
- 35. Riddell D. R., Zhou H., Atchison K., Warwick H. K., Atkinson P. J., Jefferson J., Xu L., Aschmies S., Kirksey Y., Hu Y., Wagner E., Parratt A., Xu J., Li Z., Zaleska M. M., Jacobsen J. S., Pangalos M. N., Reinhart P. H. (2008) Impact of apolipoprotein E (ApoE) polymorphism on brain ApoE levels. J. Neurosci. 28, 11445–11453 18987181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cushley R. J., Okon M. (2002) NMR studies of lipoprotein structure. Annu. Rev. Biophys. Biomol. Struct. 31, 177–206 [DOI] [PubMed] [Google Scholar]
- 37. Gangabadage C. S., Zdunek J., Tessari M., Nilsson S., Olivecrona G., Wijmenga S. S. (2008) Structure and dynamics of human apolipoprotein CIII. J. Biol. Chem. 283, 17416–17427 [DOI] [PubMed] [Google Scholar]
- 38. Krul E. S., Cole T. G. (1996) Quantitation of apolipoprotein E. Methods Enzymol. 263, 170–187 [DOI] [PubMed] [Google Scholar]
- 39. Jones P. B., Adams K. W., Rozkalne A., Spires-Jones T. L., Hshieh T. T., Hashimoto T., von Armin C. A., Mielke M., Bacskai B. J., Hyman B. T. (2011) Apolipoprotein E: isoform specific differences in tertiary structure and interaction with amyloid-β in human Alzheimer brain. PLoS One 6, e14586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hawkes C. A., Sullivan P. M., Hands S., Weller R. O., Nicoll J. A., Carare R. O. (2012) Disruption of arterial perivascular drainage of amyloid-β from the brains of mice expressing the human APOE ϵ4 allele. PLoS One 7, e41636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang N., Weng W., Breslow J. L., Tall A. R. (1996) Scavenger receptor BI (SR-BI) is up-regulated in adrenal gland in apolipoprotein A-I and hepatic lipase knock-out mice as a response to depletion of cholesterol stores. In vivo evidence that SR-BI is a functional high density lipoprotein receptor under feedback control. J. Biol. Chem. 271, 21001–21004 [DOI] [PubMed] [Google Scholar]
- 42. Youmans K. L., Leung S., Zhang J., Maus E., Baysac K., Bu G., Vassar R., Yu C., LaDu M. J. (2011) Amyloid-β42 alters apolipoprotein E solubility in brains of mice with five familial AD mutations. J. Neurosci. Methods 196, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sullivan P. M., Mezdour H., Quarfordt S. H., Maeda N. (1998) Type III hyperlipoproteinemia and spontaneous atherosclerosis in mice resulting from gene replacement of mouse Apoe with human Apoe*2. J. Clin. Invest. 102, 130–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tai L. M., Youmans K. L., Jungbauer L., Yu C., LaDu M. J. (2011) Introducing human APOE into Aβ transgenic mouse models. Int. J. Alzheimers Dis. 2011, 810981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bien-Ly N., Gillespie A. K., Walker D., Yoon S. Y., Huang Y. (2012) Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci. 32, 4803–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moore B. D., Rangachari V., Tay W. M., Milkovic N. M., Rosenberry T. L. (2009) Biophysical analyses of synthetic amyloid-β(1–42) aggregates before and after covalent cross-linking. Implications for deducing the structure of endogenous amyloid-β oligomers. Biochemistry 48, 11796–11806 [DOI] [PubMed] [Google Scholar]
- 47. Dahlgren K. N., Manelli A. M., Stine W. B., Jr., Baker L. K., Krafft G. A., LaDu M. J. (2002) Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 277, 32046–32053 [DOI] [PubMed] [Google Scholar]
- 48. Stine W. B., Jr., Dahlgren K. N., Krafft G. A., LaDu M. J. (2003) In vitro characterization of conditions for amyloid-β peptide oligomerization and fibrillogenesis. J. Biol. Chem. 278, 11612–11622 [DOI] [PubMed] [Google Scholar]
- 49. Kim D., Tsai L. H. (2009) Bridging physiology and pathology in AD. Cell 137, 997–1000 [DOI] [PubMed] [Google Scholar]
- 50. Fiala J. C. (2007) Mechanisms of amyloid plaque pathogenesis. Acta Neuropathol. 114, 551–571 [DOI] [PubMed] [Google Scholar]
- 51. West M. J., Slomianka L., Gundersen H. J. (1991) Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat. Rec. 231, 482–497 [DOI] [PubMed] [Google Scholar]
- 52. Larson M. E., Lesné S. E. (2012) Soluble Aβ oligomer production and toxicity. J. Neurochem. 120, Suppl. 1, 125–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Verghese P. B., Castellano J. M., Holtzman D. M. (2011) Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol. 10, 241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Berlau D. J., Corrada M. M., Head E., Kawas C. H. (2009) APOE ϵ2 is associated with intact cognition but increased Alzheimer pathology in the oldest old. Neurology 72, 829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Berlau D. J., Kahle-Wrobleski K., Head E., Goodus M., Kim R., Kawas C. (2007) Dissociation of neuropathologic findings and cognition: case report of an apolipoprotein E ϵ2/ϵ2 genotype. Arch. Neurol. 64, 1193–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gouras G. K., Tampellini D., Takahashi R. H., Capetillo-Zarate E. (2010) Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer's disease. Acta Neuropathol. 119, 523–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bayer T. A., Wirths O. (2010) Intracellular accumulation of amyloid-β: a predictor for synaptic dysfunction and neuron loss in Alzheimer's disease. Front. Aging Neurosci. 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sullivan P. M., Mace B. E., Maeda N., Schmechel D. E. (2004) Marked regional differences of brain human apolipoprotein E expression in targeted replacement mice. Neuroscience 124, 725–733 [DOI] [PubMed] [Google Scholar]
- 59. Vitek M. P., Brown C. M., Colton C. A. (2009) APOE genotype-specific differences in the innate immune response. Neurobiol. Aging 30, 1350–1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Koffie R. M., Meyer-Luehmann M., Hashimoto T., Adams K. W., Mielke M. L., Garcia-Alloza M., Micheva K. D., Smith S. J., Kim M. L., Lee V. M., Hyman B. T., Spires-Jones T. L. (2009) Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc. Natl. Acad. Sci. U.S.A. 106, 4012–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Spires-Jones T. L., de Calignon A., Meyer-Luehmann M., Bacskai B. J., Hyman B. T. (2011) Monitoring protein aggregation and toxicity in Alzheimer's disease mouse models using in vivo imaging. Methods 53, 201–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Winton M. J., Lee E. B., Sun E., Wong M. M., Leight S., Zhang B., Trojanowski J. Q., Lee V. M. (2011) Intraneuronal APP, not free Aβ peptides in 3xTg-AD mice: implications for Tau versus Aβ-mediated Alzheimer neurodegeneration. J. Neurosci. 31, 7691–7699 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63. Christensen D. Z., Schneider-Axmann T., Lucassen P. J., Bayer T. A., Wirths O. (2010) Accumulation of intraneuronal Aβ correlates with ApoE4 genotype. Acta Neuropathol. 119, 555–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jiang Q., Lee C. Y., Mandrekar S., Wilkinson B., Cramer P., Zelcer N., Mann K., Lamb B., Willson T. M., Collins J. L., Richardson J. C., Smith J. D., Comery T. A., Riddell D., Holtzman D. M., Tontonoz P., Landreth G. E. (2008) ApoE promotes the proteolytic degradation of Aβ. Neuron 58, 681–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hirsch-Reinshagen V., Maia L. F., Burgess B. L., Blain J. F., Naus K. E., McIsaac S. A., Parkinson P. F., Chan J. Y., Tansley G. H., Hayden M. R., Poirier J., Van Nostrand W., Wellington C. L. (2005) The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer's disease. J. Biol. Chem. 280, 43243–43256 [DOI] [PubMed] [Google Scholar]
- 66. LaDu M. J., Falduto M. T., Manelli A. M., Reardon C. A., Getz G. S., Frail D. E. (1994) Isoform-specific binding of apolipoprotein E to β-amyloid. J. Biol. Chem. 269, 23403–23406 [PubMed] [Google Scholar]
- 67. LaDu M. J., Lukens J. R., Reardon C. A., Getz G. S. (1997) Association of human, rat, and rabbit apolipoprotein E with β-amyloid. J. Neurosci. Res. 49, 9–18 [PubMed] [Google Scholar]
- 68. LaDu M. J., Munson G. W., Jungbauer L., Getz G. S., Reardon C. A., Tai L. M., Yu C. (2012) Preferential interactions between ApoE-containing lipoproteins and Aβ revealed by a detection method that combines size exclusion chromatography with non-reducing gel-shift. Biochim. Biophys. Acta 1821, 295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. LaDu M. J., Stine W. B., Jr., Narita M., Getz G. S., Reardon C. A., Bu G. (2006) Self-assembly of HEK cell-secreted ApoE particles resembles ApoE enrichment of lipoproteins as a ligand for the LDL receptor-related protein. Biochemistry 45, 381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Manelli A. M., Stine W. B., Van Eldik L. J., LaDu M. J. (2004) ApoE and Aβ1–42 interactions: effects of isoform and conformation on structure and function. J. Mol. Neurosci. 23, 235–246 [DOI] [PubMed] [Google Scholar]
- 71. Holtzman D. M., Herz J., Bu G. (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2, a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hauser P. S., Narayanaswami V., Ryan R. O. (2011) Apolipoprotein E: from lipid transport to neurobiology. Prog. Lipid Res. 50, 62–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gong J. S., Kobayashi M., Hayashi H., Zou K., Sawamura N., Fujita S. C., Yanagisawa K., Michikawa M. (2002) Apolipoprotein E (ApoE) isoform-dependent lipid release from astrocytes prepared from human ApoE3 and ApoE4 knock-in mice. J. Biol. Chem. 277, 29919–29926 [DOI] [PubMed] [Google Scholar]
- 74. Wang H., Durham L., Dawson H., Song P., Warner D. S., Sullivan P. M., Vitek M. P., Laskowitz D. T. (2007) An apolipoprotein E-based therapeutic improves outcome and reduces Alzheimer's disease pathology following closed head injury: evidence of pharmacogenomic interaction. Neuroscience 144, 1324–1333 [DOI] [PubMed] [Google Scholar]
- 75. Sadowski M. J., Pankiewicz J., Scholtzova H., Mehta P. D., Prelli F., Quartermain D., Wisniewski T. (2006) Blocking the apolipoprotein E/amyloid-β interaction as a potential therapeutic approach for Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 103, 18787–18792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Cramer P. E., Cirrito J. R., Wesson D. W., Lee C. Y., Karlo J. C., Zinn A. E., Casali B. T., Restivo J. L., Goebel W. D., James M. J., Brunden K. R., Wilson D. A., Landreth G. E. (2012) ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mahley R. W., Weisgraber K. H., Huang Y. (2006) Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 103, 5644–5651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Trommer B. L., Shah C., Yun S. H., Gamkrelidze G., Pasternak E. S., Stine W. B., Manelli A., Sullivan P., Pasternak J. F., LaDu M. J. (2005) ApoE isoform-specific effects on LTP: blockade by oligomeric amyloid-β1–42. Neurobiol. Dis. 18, 75–82 [DOI] [PubMed] [Google Scholar]
- 79. Manelli A. M., Bulfinch L. C., Sullivan P. M., LaDu M. J. (2007) Aβ42 neurotoxicity in primary co-cultures: effect of apoE isoform and Aβ conformation. Neurobiol. Aging 28, 1139–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cerf E., Gustot A., Goormaghtigh E., Ruysschaert J. M., Raussens V. (2011) High ability of apolipoprotein E4 to stabilize amyloid-β peptide oligomers, the pathological entities responsible for Alzheimer's disease. FASEB J. 25, 1585–1595 [DOI] [PubMed] [Google Scholar]
- 81. Petrlova J., Hong H. S., Bricarello D. A., Harishchandra G., Lorigan G. A., Jin L. W., Voss J. C. (2011) A differential association of Apolipoprotein E isoforms with the amyloid-β oligomer in solution. Proteins 79, 402–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Koistinaho M., Lin S., Wu X., Esterman M., Koger D., Hanson J., Higgs R., Liu F., Malkani S., Bales K. R., Paul S. M. (2004) Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 10, 719–726 [DOI] [PubMed] [Google Scholar]
- 83. Mandrekar S., Jiang Q., Lee C. Y., Koenigsknecht-Talboo J., Holtzman D. M., Landreth G. E. (2009) Microglia mediate the clearance of soluble Aβ through fluid phase macropinocytosis. J. Neurosci. 29, 4252–4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Basak J. M., Verghese P. B., Yoon H., Kim J., Holtzman D. M. (2012) Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J. Biol. Chem. 287, 13959–13971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Thal D. R. (2012) The role of astrocytes in amyloid β-protein toxicity and clearance. Exp. Neurol. 236, 1–5 [DOI] [PubMed] [Google Scholar]
- 86. Vekrellis K., Ye Z., Qiu W. Q., Walsh D., Hartley D., Chesneau V., Rosner M. R., Selkoe D. J. (2000) Neurons regulate extracellular levels of amyloid β-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 20, 1657–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Wirths O., Bayer T. A. (2012) Intraneuronal Aβ accumulation and neurodegeneration: lessons from transgenic models. Life Sci. 91, 1148–1152 [DOI] [PubMed] [Google Scholar]
- 88. Deane R., Sagare A., Hamm K., Parisi M., Lane S., Finn M. B., Holtzman D. M., Zlokovic B. V. (2008) apoE isoform-specific disruption of amyloid β peptide clearance from mouse brain. J. Clin. Invest. 118, 4002–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bachmeier C., Beaulieu-Abdelahad D., Crawford F., Mullan M., Paris D. (2012) Stimulation of the retinoid X receptor facilitates β-amyloid clearance across the blood-brain barrier. J. Mol. Neurosci., in press [DOI] [PubMed] [Google Scholar]
- 90. Castellano J. M., Kim J., Stewart F. R., Jiang H., DeMattos R. B., Patterson B. W., Fagan A. M., Morris J. C., Mawuenyega K. G., Cruchaga C., Goate A. M., Bales K. R., Paul S. M., Bateman R. J., Holtzman D. M. (2011) Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3, 89ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]