

Background: Cdkal1 is a type 2 diabetes (T2D) susceptibility gene responsible for tRNALys modification.
Results: CDKAL1 is a tail-anchored protein inserted in the ER via the TRC40/Get3 pathway. Its down-regulation affects the expression of some insulin granule proteins and the ER stress marker CHOP10.
Conclusion: CDKAL1 participates in translation of insulin granule proteins and ER stress.
Significance: Characterizing CDKAL1 contributes to T2D research.
Keywords: Diabetes, Endoplasmic Reticulum(ER), Insulin Secretion, Intracellular Trafficking, Protein Translocation, CDKAL1, T2D Susceptibility Gene, Tail-anchored Protein
Abstract
Genome-wide association studies have led to the identification of numerous susceptibility genes for type 2 diabetes. Among them is Cdkal1, which is associated with reduced β-cell function and insulin release. Recently, CDKAL1 has been shown to be a methylthiotransferase that modifies tRNALys to enhance translational fidelity of transcripts, including the one encoding proinsulin. Here, we report that out of several CDKAL1 isoforms deposited in public databases, only isoform 1, which migrates as a 61-kDa protein by SDS-PAGE, is expressed in human islets and pancreatic insulinoma INS-1 and MIN6 cells. We show that CDKAL1 is a novel member of the tail-anchored protein family and exploits the TCR40/Get3-assisted pathway for insertion of its C-terminal transmembrane domain into the endoplasmic reticulum. Using endo-β-N-acetylglucosaminidase H and peptide:N-glycosidase F sensitivity assays on CDKAL1 constructs carrying an N-glycosylation site within the luminal domain, we further established that CDKAL1 is an endoplasmic reticulum-resident protein. Moreover, we observed that silencing CDKAL1 in INS-1 cells reduces the expression of secretory granule proteins prochromogranin A and proICA512/ICA512-TMF, in addition to proinsulin and insulin. This correlated with reduced glucose-stimulated insulin secretion. Taken together, our findings provide new insight into the role of CDKAL1 in insulin-producing cells and help to understand its involvement in the pathogenesis of diabetes.
Introduction
Diabetes mellitus is a chronic disorder characterized by high blood glucose levels due to insufficient secretion of insulin by pancreatic β-cells in relation to metabolic needs. Its most common form, type 2 diabetes (T2D),3 results from peripheral insulin resistance and β-cell dysfunction due to the unfavorable interaction of genes with environmental factors. Recent genome-wide association studies have led to the identification of >60 T2D susceptibility genes, including Cdkal1 (Cdk5 regulatory subunit-associated protein 1-like 1) (1). Carriers of the Cdkal1 rs7754840 allele, in particular, display diminished glucose-stimulated insulin secretion and impaired proinsulin to insulin conversion (2, 3). This gene was named after its close paralogue CDK5RAP1 and, similarly to it, was speculated to inhibit Cdk5 (4) and thereby affect exocytosis of synaptic vesicles (5) and insulin granules (6–9). This hypothesis was confuted, however, as CDKAL1 was repeatedly shown neither to interact with Cdk5 nor inhibit its activity (10, 11). Meanwhile, independent work led to the discovery that Bacillus subtilis yqeV is a methylthiotransferase responsible for the modification of tRNAs (12). This modification was shown to improve codon recognition and accuracy of reading frame maintenance (13), thus ensuring fidelity during protein synthesis. Remarkably, CDKAL1, which displays sequence similarity with YqeV, was able to rescue this enzymatic activity in a yqeV strain. Wei et al. (11) elegantly demonstrated that CDKAL1 is indeed a methylthiotransferase that specifically modifies tRNALys in mammals. The lack of CDKAL1 caused misreading of the Lys codon, and amino acids were erroneously incorporated during translation, resulting in aberrant proteolytic processing of proinsulin and a probable increase in its degradation.
Here, we show that down-regulation of CDKAL1 in insulinoma cells perturbs levels of not only insulin but also chromogranin A and islet cell autoantigen 512 (ICA512/IA-2), two other components of insulin secretory granules. We additionally show that CDKAL1 is a new tail-anchored (TA) protein that exploits the TRC40/Get3 pathway for its insertion in the ER membrane. Finally, we report that CDKAL1 mRNA is up-regulated upon induction of the unfolded protein response, further suggesting a role for CDKAL1 in the regulation of the secretory pathway.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
Rat insulinoma INS-1 cells were grown and electroporated as described (14). Human pancreatic islets isolated from an organ donor with written consent for research use were kindly provided by Barbara Ludwig and Stefan Bornstein (Medical Clinic III, University Clinic, Dresden University of Technology).
Cloning of Rat/Human CDKAL1 cDNA
RNA was isolated from INS-1 cells and human pancreatic islets with an RNeasy mini kit (Qiagen); reverse transcription of 2 μg of RNA was performed with Superscript II reverse transcriptase (Invitrogen) using oligo(dT). cDNA was then amplified with primers annealing to 5′/3′-UTRs, based on human Ref Seq NM_017774.3 and rat Ref Seq XM_341524.3, which has in the meantime been discontinued (rat 5′-UTR, 5′-tttcctgacagtggcttgtg-3′; rat 3′-UTR, 5′-tgagaactggtgctgttgct-3′; human 5′-UTR, 5′-tagactaattgcagataattaagag-3′; human 3′-UTR, 5′-ctttatagatgtttccattagttg-3′). PCR products were cloned into the pCRII vector by TA cloning (Invitrogen) and verified by DNA sequencing. The sequence of rat CDKAL1 cDNA was scanned against the database, and no other genes with significant relatedness were identified.
Plasmids
All constructs used here were made by standard recombinant DNA techniques and verified by sequencing. Full-length human CDKAL1 cDNA (IMAGE clone 40117357) was purchased from Open Biosystems. Its open reading frame was amplified by PCR, flanked with HindIII and XbaI restriction sites, and subcloned in-frame with three HA epitopes at its C terminus, as described (15). pGEM4-hCDKAL1-Nglyc was generated by amplifying human CDKAL1 with the following primers: fw-KpnI 5′-ctcggtaccatgccttctgcatcctg-3′ and rev-BspEI 5′-tttgtcaaggtctataattccggaat-3′. The KpnI-BspEI-digested PCR product replaced the cytochrome b5 coding sequence into pGEM4-N-glyc-b5 (16). pEG-CDKAL1-Nglyc was obtained by replacing the enhanced GFP coding sequence in pEGFPN1 (Invitrogen) with the PCR-amplified hCDKAL1-Nglyc containing HindIII-NotI restriction sites at the 5′- and 3′-ends. mVenus-17 has been described (17). Sec61β-Nglyc plasmid was a kind gift from S. High.
Cell Extract and Western Blotting
INS-1 cells were washed with ice-cold PBS and extracted in lysis buffer (10 mm Tris-HCl, pH 8.0, 140 mm NaCl, 1% Triton X-100, 1 mm EDTA, 1% inhibitor mixture (Sigma)) at 4 °C. Protein measurements and immunoblotting were performed as described (14) using the following antibodies: polyclonal rabbit anti-CDKAL1#1 (GTX110020, GeneTex); anti-CDKAL1#2 (HPA014374, Sigma); goat anti-CDKAL1#3 (EB08288, Everest Biotech); rabbit anti-CDKAL1#4 (ab74020, Abcam); monoclonal mouse anti-ICA512 (19); anti-GAPDH (Novus Biologicals); anti-γ-tubulin (Sigma); polyclonal rabbit anti-TRC40 (20); anti-CHOP10 (Santa Cruz Biotechnology); anti-CGA (BD Biosciences); anti-PC1/3, anti-PC2, and anti-carboxypeptidase H (Chemicon International); anti-transferrin receptor (Zymed Laboratories Inc.); anti-ribophorin I from Gert Kreibich (New York University School of Medicine, New York) (21); and anti-Sec61α from R. Zimmermann (22). To detect endogenous proinsulin/insulin, cell extracts were resolved on 15% Tris/Tricine gels with nonreducing loading buffer. Immunoblotting was performed with monoclonal mouse anti-insulin (Sigma). Chemiluminescence was developed and quantified as described (14, 23).
In Vitro Transcription/Translation and Protease Protection Assay
In vitro translation of rat CDKAL1 in pCRII was performed with the TnT-T7 Quick-coupled Transcription/Translation System (Promega) according to the manufacturer's protocol. Reactions performed in the presence of [35S]methionine were analyzed by SDS-PAGE and phosphorimaging (BAS 1800II phosphorimager (Fuji)); reactions performed with nonradioactive methionine were resolved by SDS-PAGE and analyzed by immunoblot. Transcription and translation of hCDKAL1-Nglyc, b5-Nglyc, preprolactin, and Sec61β-Nglyc constructs, translocation reactions with rough microsomes (RM), or liposomes prepared by extrusion, digestions with proteinase K, immunoprecipitation with anti-opsin monoclonal antibody (from P. Hargrave (see Ref. 24)), and SDS-PAGE analysis were performed as described previously (18, 23, 25). TRC-40 depletion from rabbit reticulocyte lysate was done as described (20). Partial removal of TRC40 and any peripherally associated protein from RM was achieved by incubation of 50 μl of RM for 15 min on ice in a 0.5-ml total volume of stripping buffer (250 mm sucrose, 50 mm triethanolamine, 50 mm EDTA, 1 m KCl, 5 mm DTT). The sample was then layered on top of a discontinuous gradient composed of 0.1 ml of 0.6 m and 0.1 ml of 0.4 m sucrose both containing 5 mm DTT and 50 mm triethanolamine. After centrifugation at 4 °C for 1 h at 65,000 rpm in a Beckman TLA 100.1 rotor, the RM pellet was washed once with membrane buffer (250 mm sorbitol, 50 mm Hepes-KOH, pH 7.2, 70 mm KOAc, 5 mm potassium-EGTA, 2.5 mm MgAc, 4 mm DTT) and resuspended in 50 μl of the same buffer. Sec61α-depleted proteoliposomes were generated from a detergent extract of rat microsomal membrane proteins using 0.5% deoxy-Big CHAP (Calbiochem) in membrane buffer; after 30 min of incubation on ice, the extract was centrifuged at 4 °C for 1 h at 60,000 rpm in a Beckman TLA100.1 rotor. The soluble fraction was then reconstituted into proteoliposomes by detergent removal as described (23).
Alkaline Sucrose Gradient
INS-1 cells transfected with CDKAL1-HA3 were homogenized in HB buffer (250 mm sucrose, 4 mm Hepes, pH 7.4, 1% protease inhibitor mixture (Sigma)) with a ball-bearing cell cracker, and cell extracts were centrifuged for 10 min at 1000 × g to recover the post-nuclear supernatant. The latter was separated in a high speed supernatant and a high speed pellet (HSP) by centrifugation at 4 °C for 30 min at 150,000 × g in a Beckman TLA100.1 rotor. The HSP was resuspended in HB buffer (half of the starting homogenate volume), treated with an equal volume of 0.2 m Na2CO3 at pH 11.5 for 30 min on ice, and then brought to 1.5 m sucrose, 0.1 m Na2CO3 in a final volume of 0.5 ml. The sample was layered under a discontinuous sucrose gradient composed of 2 ml of 1.2 m and 1.5 ml of 0.25 m sucrose, both containing 0.1 m Na2CO3. After centrifugation overnight (40,000 rpm, 4 °C, in Beckman MLS-50 rotor), 1-ml fractions were collected from the top, TCA-precipitated, and analyzed by SDS-PAGE.
Insulin and Proinsulin Quantitation
INS-1 cells were treated as described previously (26). Briefly, cells were preincubated at 37 °C for 1 h in resting buffer (with 0 mm glucose) and then for an additional 2 h in resting or stimulation buffer (25 mm glucose, 5 mm KCl). The medium was collected, and the cells were extracted as described (14). Insulin content in the cells and in the medium was measured with the sensitive rat insulin RIA kit (Linco Research). Proinsulin content was measured with a rat ELISA kit (Mercodia AB, Uppsala, Sweden). Values were normalized to those of total and secreted insulin and proinsulin under control conditions in resting buffer.
RNA Interference
INS-1 cells were seeded or transfected with CDKAL1-HA3 on day 1. On day 2, cells were transfected with a mixture of siRNA oligos (30 nm each for CDKAL1, 25 nm each for CHOP10, and equivalent amounts for control Firefly luciferase siRNA) in OptiMEM with DharmaFECT4 (Dharmacon). On day 4, cells were either processed for insulin RIA or harvested and processed for RT-quantitative PCR and immunoblot. Sequences of the siRNA oligos (Riboxx GmbH, Radebeul, Germany) are provided in supplemental Table S1.
Real Time PCR
Total RNA isolation and retrotranscription was described earlier. Quantitative PCR was performed using Platinum SYBR Green quantitative PCR mixture (Invitrogen) with an Mx4000 Multiplex quantitative real time PCR system (Stratagene). The variation of CHOP10 and CDKAL1 transcripts after thapsigargin treatment relative to the untreated control was calculated using the ΔΔCt method (27), with β-actin as the reference gene. Primer sequences are provided in supplemental Table S2.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and immunostained overnight at 4 °C with mouse monoclonal anti-HA (Sigma), followed by 1 h of incubation with goat anti-mouse Alexa488-conjugated IgGs (Molecular Probes). ER staining in HeLa cells was achieved by incubation with 50 μg/ml concanavalin A-AlexaFluor594 (Molecular Probes). In INS-1 cells, the ER was visualized by co-transfection of ER-localized mVenus-17. Nuclei were counterstained with DAPI (Sigma). Cells were imaged with a Zeiss confocal microscope (LSM510 Meta) using a 63× Plan Apochromat lens (1.4 NA). Single confocal sections are shown, which were prepared with Photoshop software (Adobe).
Miscellaneous Treatments
Endo-H and PNGaseF treatments were performed on INS-1 cell extracts according to the manufacturer's protocol (New England Biolabs). Iodoacetamide (IAA) treatment was performed starting with 2 μl of in vitro translated rat CDKAL1 lysate or 10 μl of transfected INS-1 cell extract; samples were mixed with an equal volume of DTT buffer (100 mm DTT, 500 mm sucrose, 250 mm Tris-HCl, pH 8.8, 5% SDS, 0.04% bromphenol blue) and boiled at 100 °C for 3 min; after cooling down to room temperature, IAA was added to reach 300 mm, and samples were incubated at 37 °C for 45 min and then loaded on gels for SDS-PAGE.
Statistics and Graphics
Quantification of Western blots was performed with Image Gauge Version 3.45 (Fuji, Tokyo, Japan). Data were analyzed using the unpaired Student's t test. The results are presented as mean ± S.D. from at least three independent experiments. Histograms were prepared with Excel (Microsoft).
RESULTS
Insulinoma INS-1 Cells and Human Islets Express CDKAL1 Isoform 1
Four human Cdkal1 variants are deposited in Ensembl (ENSG 00000145996), but only two alternatively spliced proteins are listed. The first (ENST 00000274695) and second (ENST 00000378610) transcripts only differ in the length of their 5′-UTR and encode isoform 1, which is 579 amino acids for a predicted mass of ∼65 kDa (Fig. 1A). A third transcript (ENST 00000378624) originates isoform 2, which is only 488 amino acids (∼55 kDa) because exons 4 and 13 are skipped due to alternative splicing; additionally, compared with isoform 1, the first 95 residues are replaced by a distinct 25-residue sequence due to a downstream “ATG” start codon in a different reading frame that leads to an alternative translation of exon 3. The last transcript (ENST 00000476517) encodes a partial fragment of isoform 1 and 2. In contrast, only two shorter proteins of 97 and 133 residues are listed for rat Cdkal1 (Ensembl ENSRNOG 00000043136).
FIGURE 1.

Detection of isoform1 in insulin-producing cells. A, schematic representation of protein and exon structures of human CDKAL1 isoform1 and 2. The transcript coding isoform 2 lacks exons 4 and 13, and its translation starts from an internal ATG codon in a different reading frame than isoform 1. The different N-terminal portion in the two isoforms is highlighted either in light or dark gray. B, RT-PCR products amplified from INS-1 cells and human islet RNA with primers specific for rat and human CDKAL1 (lanes 3 and 6, respectively). For negative control reactions, no reverse transcriptase was added to the samples in lanes 1 and 4; no PCR primers were added to the samples in lanes 2 and 5. C, in vitro transcription and translation of rat CDKAL1 cDNA subcloned into pCRII. Reactions ± cDNA were subsequently analyzed by SDS-PAGE and immunoblot with anti-CDKAL1#2. The specific band corresponding to translated CDKAL1 is marked with *. A 55-kDa-reactive protein recognized by the same antibody is marked with ●.
RT-PCR for Cdkal1 mRNA from rat INS-1 cells, a widely used in vitro β-cell model, yielded only a single product of ∼2 kb (Fig. 1B, lane 3). A single Cdkal1 transcript of similar size was also amplified from human islets (Fig. 1B, lane 6). Sequence analysis and translation in silico of both products identified them as isoform 1. Specifically, the rat transcript encoded a protein of 578 residues that is 95 and 89% identical to mouse and human CDKAL1, respectively (supplemental Fig. S1). Although CDKAL1 isoform 1 has an estimated mass of 65 kDa, in vitro transcription and translation of the rat Cdkal1 cDNA generated a 61-kDa protein, as detected by immunoblotting with an anti-CDKAL1 antibody (Fig. 1C, lane 2, asterisk). Notably, the same antibody also recognized a prominent 55-kDa protein in rabbit reticulocyte lysate (Fig. 1C, dots). The size of this smaller CDKAL1-immunoreactive species was compatible with the predicted size of CDKAL1 isoform 2.
We next compared the reactivity of several commercial anti-CDKAL1 antibodies (Ab1–4) generated against different regions of human CDKAL1 (Fig. 2A) on extracts of various cell types (Fig. 2B). Ab2 was used for the immunoblot shown in Fig. 1. In all cases, extracts of INS-1 cells transfected with human CDKAL1 isoform 1 tagged at the C terminus with three HA epitopes (CDKAL1-HA3) were used as positive controls. As expected, all four antibodies recognized CDKAL1-HA3 (Fig. 2B, arrowhead). Ab1, Ab2, and Ab3 recognized the 61-kDa CDKAL1-immunoreactive species in all samples (Fig. 2B, lanes 1–12, asterisks), whereas Ab4 detected a 61-kDa protein in human HeLa cells (lanes 13 and 16) but not in rat INS-1 cells and only weakly in mouse insulinoma MIN6 cells. Although Ab2 reacted with a 55-kDa protein, Ab1 only weakly and inconsistently recognized a protein of approximately this size (Fig. 2B, dot), and Ab3 did not react at all (see also Fig. 2C). Ab1, Ab3, and Ab4, but not Ab2, recognized an ∼40-kDa protein in all samples, either as a single species or a doublet (Fig. 2, B and C, arrows). Thus, only the 61-kDa species is consistently recognized by all four antibodies.
FIGURE 2.

Specificity of anti-CDKAL1 antibodies. A, schematic representation of CDKAL1 recombinant fragments used to generate the four antibodies tested. The numbers on line edges correspond to amino acid positions in isoform 1. Notably, Ab3 (ab#3) can only recognize isoform 1 due to the lack of exon13 (coding residues 413–433) in isoform 2. The position of Cys (C) residues forming the two 4Fe-4S clusters, which are essentials for CDKAL1 enzymatic activity, is indicated for both isoforms. Protein domains are named at the bottom of the panel. B, immunoblot analysis of protein extracts from different cell lines and from CDKAL1-HA3-transfected INS-1 cells. Four different anti-CDKAL1 antibodies are compared. Endogenous CDKAL1 band is marked with *, CDKAL1-HA3 with >, the 55-kDa, and the 40-kDa proteins with ● and →, respectively. γ-Tubulin was used as a loading control. C, INS-1 cells transfected with control or CDKAL1-siRNA oligos (oligo1 + oligo2), combined with CDKAL1-HA3 or empty plasmid transfection, were analyzed by immunoblotting with anti-CDKAL1 Ab1 and Ab2 (Ab#1 and Ab#2) and shown in the upper or middle panel, respectively. Endogenous CDKAL1 is marked with * and CDKAL1-HA3 with >. Note that both 40-kDa (←) and 55-kDa (●) proteins, recognized by Ab1 and Ab2 (Ab#1 and Ab#2), respectively, are unaffected by CDKAL1 siRNA. γ-Tubulin was used as a loading control. D, cell extracts of INS-1 cells transfected with CDKAL1-HA3, or with empty plasmid, were subjected to IAA treatment, analyzed by SDS-PAGE. and immunoblotted with anti-CDKAL1#2. Endogenous CDKAL1 is marked with *, CDKAL1-HA3 with <, and the 55-kDa protein with ●. E, autoradiographic detection of samples ± IAA treatment after in vitro synthesis with [35S]Met. Control reactions without cDNA were analyzed in parallel. As a size indicator, luciferase cDNA was translated and analyzed in parallel by SDS-PAGE.
To further analyze the relationship of the 61-, 55-, and 40-kDa species with CDKAL1, we co-transfected INS-1 (Fig. 2C) and MIN6 (supplemental Fig. S2) cells with two siRNA oligos complementary to sequences in the 5′-UTR and coding region of rat Cdkal1 or with a siRNA oligo targeting firefly luciferase as a control. Cdkal1-directed siRNAs reduced expression of the 61-kDa protein (Fig. 2C, asterisks), but not of the 40-kDa (Fig. 2C, arrow) or 55-kDa species (Fig. 2C, dot). These results corroborate the conclusion that the 61-kDa species corresponds to CDKAL1.
To evaluate if the faster than expected electrophoretic mobility of the 61-kDa CDKAL1-immunoreactive species was due to aberrant migration upon SDS-PAGE, extracts of INS-1 cells or in vitro [35S]Met-translated rat CDKAL1 were treated with IAA, which prevents disulfide bond formation. Upon this treatment, the endogenous 61-kDa species (Fig. 2D, lanes 3 and 4, asterisk) and transfected CDKAL1-HA3 (Fig. 2D, lanes 1 and 2, arrowhead) shifted upward. Likewise, IAA treatment retarded the electrophoretic mobility of 35S-CDKAL1 (Fig. 2E, lanes 2 and 3), although its migration in comparison with that of the 61-kDa control firefly luciferase was still faster than expected (Fig. 2E, lane 1). Notably, the reactivity of the anti-CDKAL1 antibody to the 55-kDa species, unlike the 61-kDa species or CDKAL1-HA3, was sensitive to IAA treatment (Fig. 2D, lanes 1 and 3).
Taken together, the results shown in Figs. 1 and 2 indicate that rodent insulinoma cells express CDKAL1 isoform 1 as a protein with an apparent molecular mass of 61 kDa. The possibility that the 55- and 40-kDa species are additional CDKAL1 isoforms seems unlikely, although it is not formally excluded. The 61-kDa CDKAL1 species was also detected in human islets as well as in mouse pancreas, liver, and skeletal muscle, especially in white muscle fibers (supplemental Fig. S3A). In pancreatic islets, CDKAL1 expression is conceivably not limited to β-cells, as we also detected it in the glucagonoma α-TC cells originating from mouse pancreatic α-cells (supplemental Fig. S3B).
CDKAL1 Is a New Tail-anchored ER Protein
The primary sequence of CDKAL1 includes a 16-amino acid hydrophobic domain at its very C terminus, followed by a stretch of 4–6 residues in the human or rat protein (Fig. 2 and supplemental Fig. S1). This hydrophobic domain is required for the protein to associate with membrane, as its deletion led to the redistribution of EGFP-CDKAL1 from the ER to the cytosol (11). In view of these findings, CDKAL1 could be a new tail-anchored (TA) protein. TA proteins are characterized by a single transmembrane domain (TMD) anchoring them to intracellular membranes, mainly on the cytosolic side, leaving only a few residues in the lumen of the organelle of residence (Fig. 3A).
FIGURE 3.
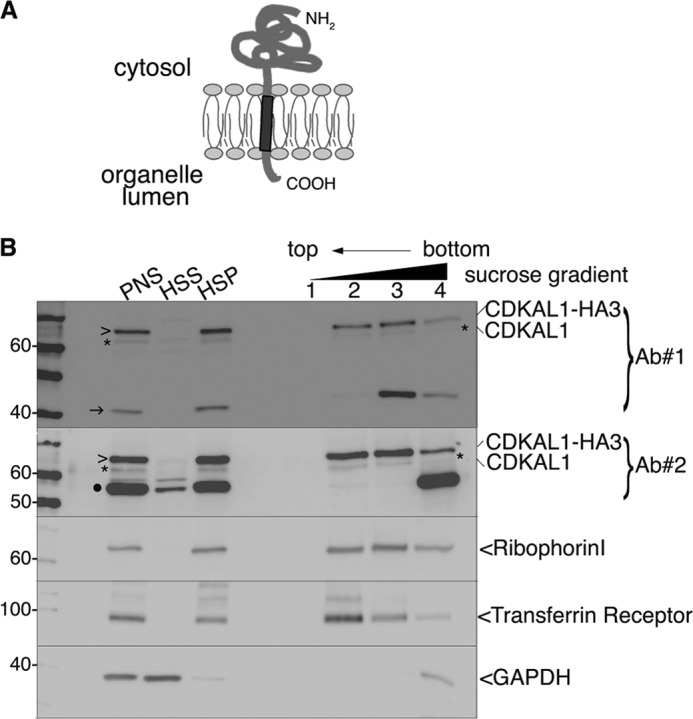
Tight association of CDKAL1 with intracellular membranes. A, schematic representation of the membrane topology of a TA protein. The single TMD (dark gray) at the C terminus is followed by only a few residues in the lumen of the organelle. B, immunoblot analysis of fraction separated by flotation through an alkaline sucrose density gradient. Homogenates from CDKAL1-HA3-transfected INS-1 cells were separated into high speed supernatant (HSS) and HSP fractions. The HSP was treated with Na2CO3 and loaded at the bottom of a discontinuous alkaline sucrose gradient. Membrane vesicles float to the top fractions of the gradient. Both anti-CDKAL1 Ab1 and Ab2 (Ab#1 and Ab#2) were used (top two panels). Ribophorin I and transferrin receptor are control integral membrane proteins; GAPDH is a control soluble cytosolic protein. Endogenous CDKAL1 (*) and transfected CDKAL1-HA3 (>) display an overlapping distribution profile, similar to the ones of control membrane proteins. Note that both the 40-kDa (→) and 55-kDa (●) proteins detected with anti-CDKAL1 antibodies are restricted to the bottom of the gradient, implying only a peripheral or indirect association with membranes. PNS, post-nuclear supernatant.
To test whether CDKAL1 is a TA protein, we first analyzed whether its membrane association is resistant to alkaline treatment with Na2CO3, which dissociates peripherally associated proteins from membranes. First, endogenous CDKAL1 and overexpressed CDKAL1-HA3, similar to bona fide transmembrane proteins ribophorin I and the transferrin receptor, were quantitatively recovered in the high speed pellet (Fig. 3B, HSP), whereas the control cytosolic marker GAPDH was predominantly found in the high speed supernatant (Fig. 3B, HSS). Second, both CDKAL1 proteins, again similar to ribophorin I and the transferrin receptor, floated with light membrane fractions upon fractionation on a sucrose density gradient of Na2CO3-treated HSP (Fig. 3B, lanes 2 and 3). Thus, the 61-kDa CDKAL1 species displayed tight, alkaline-resistant binding to membranes, consistent with its hydrophobic stretch being buried in the lipid bilayer. Notably, the 40- and 55-kDa CDKAL1-immunoreactive species were also fully recovered in the HSP, but after Na2CO3 treatment they remained at the bottom of the gradient (Fig. 3B, lanes 3 and 4), suggesting they are only peripherally or indirectly associated with membranes.
Except for mitochondrial and, possibly, peroxisomal membrane proteins, which are directly inserted into the membrane of residence, all membrane proteins are inserted first into the ER bilayer then sorted to their final destination (reviewed in Ref. 28). According to the length and hydrophobicity of the TMD, the final compartment of residence for TA proteins can be predicted with good degree of confidence (29). Because of the physical-chemical characteristics of its hydrophobic domain, we expected CDKAL1 to be retained in the ER membrane after insertion. In agreement with previous data (10, 11), immunostaining for CDKAL1-HA3 in INS-1 and HeLa cells indeed produced a reticular pattern, with a clear labeling of the nuclear envelope. This pattern coincided with that of ER markers (Fig. 4A). To corroborate the association of CDKAL1 with the ER, we transfected INS-1 cells with a CDKAL1 construct containing an N-glycosylation site at the very C terminus (CDKAL1-Nglyc). This construct was N-glycosylated, as indicated by the electrophoretic shift compared with the same samples treated with N-endoglycosidases PNGaseF and Endo-H (Fig. 4B, lanes 4–6, arrowhead). As expected, endogenous 61-kDa CDKAL1 was not affected by these treatments (Fig. 4B, lanes 1–3), as an N-glycosylation consensus sequence is not present in the 6-residue stretch C-terminal to the hydrophobic domain. This analysis also demonstrated that the C-terminal hydrophobic domain of CDKAL1 spans the lipid bilayer, i.e. is a bona fide TMD, because the engineered N-glycosylation site reached the ER lumen where the oligosaccharyltransferase complex is located. Moreover, CDKAL1-Nglyc was fully Endo-H-sensitive. These findings, together with the ER-restricted immunostaining pattern, indicate that CDKAL1 is a TA protein that does not reach compartments of the secretory pathway downstream of the ER.
FIGURE 4.

Subcellular localization of CDKAL1 in the ER. A, immunocytochemical detection of CDKAL1-HA3. INS-1 and HeLa cells were transfected with CDKAL1-HA3 and immunostained with the anti-HA antibody (green). In HeLa cells, the ER (red) is visualized by ConA-AlexaFluor594 staining and in INS-1 cells by co-transfection of the fluorescent ER reporter protein mVenus-17 (19). Nuclei are visualized with DAPI (blue). Single confocal sections are shown. Scale bar, 10 μm. B, cell extracts of INS-1 cells transfected with CDKAL1-Nglyc, or the empty vector, were treated with N-endoglycosidases Endo-H and PNGaseF to verify the N-glycosylation of the recombinant protein. Samples were analyzed by SDS-PAGE and immunoblot with anti-CDKAL1 Ab1. Glycosylated CDKAL1-Nglyc is marked with >.
CDKAL1 Is Inserted into ER Membranes via the TRC40/GET3 Pathway
Because CDKAL1 is a TA protein, it can only be inserted into the ER membrane via a post-translational pathway (28). To define which pathway CDKAL1 uses for its membrane insertion, we again used the CDKAL1-Nglyc construct in a well established protease protection assay (23). Briefly, the 19-residue sequence from bovine opsin that was placed at the very C terminus of CDKAL1-Nglyc, in addition to the N-glycosylation site, includes an epitope recognized by a monoclonal antibody. Translocation of this C-terminal 19-residue sequence across a vesicle lipid bilayer generates a fragment protected from protease K (PK) digestion; the protected fragment (PF) can be recovered by immunoprecipitation with the opsin antibody. If the C terminus is translocated across pure lipid vesicles, a nonglycosylated PF migrates as an ∼5-kDa peptide by SDS-PAGE (23). When the vesicles are ER-derived and contain the oligosaccharyltransferase complex, the PF migrates instead as an ∼9-kDa peptide (glyc-PF) due to its N-glycosylation (Fig. 5A) (23). When human CDKAL1-Nglyc was exposed to RM membranes, we observed the appearance of a slower migrating form (Fig. 5B, upper panel, −PK, asterisk), and in parallel, PK digestion produced PFs that mostly migrated as ∼9-kDa peptides, consistent with N-glycosylation (Fig. 5B, bottom panel, +PK, lane 3). A minor amount of nonglycosylated PF was also recovered. This corresponded to the fraction of CDKAL1 that was translocated but not glycosylated. As controls, PF was neither recovered from samples incubated in the absence of membranes (Fig. 5B, lane 1) nor from samples incubated with membranes and then digested with PK in the presence of detergent (Fig. 5B, lane 4). In contrast to cytochrome b5, a TA protein utilizing the unassisted pathway (Ref. 23 and supplemental Fig. S4A, lane 2), we found poor insertion of hCDKAL1-Nglyc into protein-free liposomes, as a low amount of PF was recovered after incubation with pure phosphatidylcholine vesicles (Fig. 5B, bottom panel, lane 2). These findings point to the requirement of chaperones for translocation of CDKAL1 into ER membranes.
FIGURE 5.

CDKAL1 is inserted into the ER via the TRC40/Get3 pathway. A, schematic representation of the constructs b5-Nglyc and hCDKAL1-Nglyc and assay design for transmembrane insertion of TA proteins. Constructs were engineered at their C terminus with a 19-residue sequence derived from bovine opsin and containing an N-glycosylation site. Upon incubation with vesicles, reactions were digested with PK prior to immunoprecipitation with an anti-opsin antibody to recover any PF generated. B, in vitro translated hCDKAL-Nglyc was post-translationally incubated with liposomes (PC) or rough microsomes (RM) for 1 h. After translocation, a small amount of the reactions was directly analyzed by SDS-PAGE and phosphorimaging (upper panel, −PK); the remaining part was PK-digested in the presence or absence of detergent (0.5% sodium deoxycholate, lane 4) and immunoprecipitated with anti-opsin Ab to recover any protected fragments (lower panel, +PK, PF, or glyc-PF). A control translocation reaction in the absence of vesicles was also performed (lane 1). In both panels, glycosylated forms of the protein or protected fragment are marked with *. C, immunoblot detection of TRC40 in cell extracts from insulinoma INS-1 and MIN6 cells. γ-tubulin was used as a loading control. D, hCDKAL1-Nglyc and b5-Nglyc constructs were in vitro translated in complete or TRC40-immunodepleted reticulocyte lysates (lanes 5 and 10). Standard translocation reactions were then performed with untreated RM (lanes 2 and 3, 7, and 8) or high salt-treated RM (strip) to remove peripherally associated proteins (lanes 4 and 5, 9, and 10). Control reactions with no vesicles (lanes 1 and 6) or with PK digestion in the presence of detergent (det, lanes 2 and 7) were also included. The translocation efficiency into RM in different conditions, calculated as described previously (23), is reported below each corresponding lane. E, immunoblot detection of Sec61α in membrane-detergent extract prior to (tot extract) or after (ΔSec61α extract) centrifugation. Equal volumes of extracts were loaded on SDS-PAGE, as indicated, before reconstitution into proteoliposomes. Note that Sec61α is retained in the insoluble pelleted fraction. F, in vitro translated Sec61β-Nglyc was post-translationally incubated with wild-type RM (wt, lanes 3 and 4) or with proteoliposomes reconstituted from Sec61α-depleted membrane-detergent extract (lanes 5 and 6). PK digestion was then performed as described in B, and the reaction was directly analyzed by SDS-PAGE and phosphorimaging. A control reaction without vesicles was also included (lanes 1 and 2). G, in vitro translated hCDKAL1-Nglyc was post-translationally incubated with RM (lane 2) or Sec61α-depleted proteoliposomes (lane 3) as described in F. Upon PK digestion, PF and glyc-PF were recovered by immunoprecipitation (lanes 2 and 3, lower panel, +PK). Note that the glycosylation activity was inefficiently reconstituted into proteoliposomes, and thus they only generated a PF (lane 3), in contrast to wild-type microsomes (lane 2). A control reaction with PK digestion in the presence of detergent was also included (lane 1).
We next investigated whether CDAKL1 exploits the newly identified TRC40/Get3-assisted pathway for translocation into the ER (18, 30). First we ascertained that TRC40 is expressed in INS-1 and MIN6 cells (Fig. 5C) and then tested the role of TRC40 in the cell-free system. To this aim, TRC40 bound to membranes was partially stripped off by treatment of RM with high salts and immunodepleted from reticulocyte lysates before in vitro translation of hCDKAL1-Nglyc. The efficiency of TRC40 removal from microsomes and reticulocyte lysates was monitored by immunoblotting (supplemental Fig. S4B). The translocation of hCDKAL1-Nglyc in stripped RM was increased compared with untreated RM, as assessed by quantitation of glyc-PF recovery (Fig. 5D, lanes 8 and 9). However, combined stripping and immunodepletion of TRC40 reduced the translocation efficiency of hCDKAL1-Nglyc by 45% compared with translocation in stripped RM alone (Fig. 5D, lanes 8 and 9). Similar results were obtained with rat CDKAL1-Nglyc (supplemental Fig. S3, D and E). Conversely, TRC40 depletion or microsomal stripping did not affect cytochrome b5 translocation (Fig. 5D, lanes 3–5). The increased translocation of CDKAL1 upon stripping of RM most likely reflects the increased fraction of TRC40 receptors available on ER membranes to form new translocation complexes.
An alternative insertion pathway for TA proteins has been described to be mediated by the signal recognition particle and the Sec61 complex in an unusual post-translational mode (31). To rule out the possibility that CDKAL1-Nglyc could exploit this less prominent insertion mechanism, we obtained Sec61α-depleted proteoliposomes (Fig. 5E, ΔSec61α), in which the lack of Sec61α abolishes the function of the Sec61 translocation channel. These proteoliposomes were indeed unable to sustain the translocation of a classical Sec61-dependent substrate, preprolactin, as assessed by the lack of processing of the prolactin signal sequence and the susceptibility to protease digestion of the mature form (supplemental Fig. S4C, lanes 7 and 8 compared with lanes 5 and 6). However, the same proteoliposomes efficiently supported translocation of a well defined substrate of the TRC40/Get3 pathway, Sec61β (Fig. 5F) (18). Although we observed inefficient reconstitution of the glycosylating complex in the ΔSec61α proteoliposomes, as shown by the lack of glycosylated Sec61β and the respective glyc-PF (Fig. 5F, lanes 5 and 6), the Sec61-independent translocation activity was retained, as indicated by the efficient recovery of a PF originating from Sec61β-Nglyc translocation (Fig. 5F, lane 6). Similarly, hCDKAL1-Nglyc translocated in ΔSec61α proteoliposomes, generating a PF (Fig. 5G, lane 3).
Taken together, these data show that the behavior of CDKAL1 resembles that of other TRC40-assisted TA proteins. Translocation is never completely blocked due to the redundancy of the system and the availability of salvage pathways to compensate for TRC40 impairment (20, 32).
Role of CDKAL1 in the Expression of Insulin Secretory Granule Components
CDKAL1 has recently been shown to be a modifying enzyme specific for tRNALys, and its loss affects the conversion of proinsulin into insulin (11). Consistently with these findings, we observed a drastic reduction of total insulin levels after CDKAL1 silencing in INS-1 cells incubated in resting (0 mm glucose, p = 0.0003) or stimulating (25 mm glucose, p = 0.001) conditions (Fig. 6A, left panel). This deficit was mirrored by an equivalent loss of total proinsulin in resting (p = 0.002) and stimulating (p = 0.0008) conditions (Fig. 6B). We also observed a reduction of glucose-induced insulin secretion (Fig. 6A, right panel, p = 0.01). An ∼50% loss of proinsulin/insulin in lysates of CDKAL1-silenced cells was also verified by immunoblotting (Fig. 6C). Likewise, knockdown of CDKAL1 reduced the expression of granule proteins prochromogranin A (pro-CGA), proICA512/IA-2, and its mature form ICA512-TMF, but not that of other granule components such as carboxypeptidase H, prohormone convertase 1/3 (PC1/3) (Fig. 6D), prohormone convertase 2 (PC2), and Phogrin (supplemental Fig. S5A). In a complementary manner, overexpression of CDKAL1-HA3 or a CDKAL1 construct with a triple HA tag at its N terminus (3HA-CDKAL1) increased the levels of proinsulin, insulin, pro-CGA, and both ICA512 species (Fig. 7A and supplemental Fig. S5C). Moreover, the phenotype observed upon CDKAL1 silencing was almost completely rescued by overexpression of a siRNA-resistant CDKAL1-HA3 construct (Fig. 7B, lanes 3 and 4). This analysis suggests that CDKAL1 promotes the translation of several other secretory granule proteins in addition to proinsulin (11).
FIGURE 6.

Effect of CDKAL1 silencing on insulin granule protein content. A, glucose-stimulated insulin secretion upon CDKAL1 knockdown. INS-1 cells were transfected with control (ctr) or CDKAL1-specific (kd) siRNA oligos. 48 h after transfection, cells were either kept at rest with 0 mm glucose (white bar, Rest) or stimulated with 25 mm glucose (black bar, Stim.) for 2 h. The amount of insulin released in the medium and remaining in the cells was determined by RIA. Total insulin (ins) (left panel) was calculated as insulin(medium) + insulin(cell). Secreted insulin (right panel) was calculated as insulin(medium)/total insulin. The values in control-transfected cells at 0 mm glucose equal 1. n = 6, each independent experiment performed in triplicate. Error bars in A–C show mean ± S.D., and asterisks indicate statistical significance as determined by Student's t test. **, p ≤ 0.01; *, 0.01< p < 0.05. B, INS-1 cells were transfected and treated as in A. Proinsulin content in the medium and cells was determined by ELISA. The amount of total proinsulin in control-transfected cells at 0 mm glucose equals 1. n = 3, each independent experiment was performed in triplicate. C, cell extracts from INS-1 cells 48 h after CDKAL1-siRNA or control-siRNA oligos transfection were analyzed by immunoblot with anti-insulin Ab; two independent experiments (Exp) are shown (upper panel). In the lower panel, quantification of signals for proinsulin/insulin after CDKAL1 knockdown and normalization for the corresponding signal of γ-tubulin. Values in control-transfected cells equal 1. n = 5. D, immunoblot analysis of cell extracts as in C, for the indicated granule proteins. CDKAL1 was detected with Ab1. ICA512-TMF is the transmembrane fragment generated by convertase-mediated cleavage of the luminal domain of ICA512. γ-Tubulin was used as loading control. Three independent experiments are shown.
FIGURE 7.

Effect of CDKAL1 overexpression on insulin granule protein content. A, INS-1 cells were transfected by electroporation with different amounts of CDKAL1-HA3. 72 h after transfection, cell extracts were analyzed by immunoblot for the indicated granule proteins. As a short exposure with anti-CDKAL1 Ab1 is shown, endogenous CDKAL1 is not visible. γ-Tubulin was used as loading control. B, rescue of CDKAL1 silencing by overexpression of CDKAL1-HA3. INS-1 cells were transfected with CDKAL1-HA3 or the empty plasmid and 1 day later with CDKAL1-siRNA or control-siRNA. 48 h after transfection of siRNA oligo, cell extracts were analyzed by immunoblot for the indicated granule proteins. Note that CDKAL1-HA3 is not targeted by the specific siRNA oligos. Overexpression of CDKAL1-HA3 in the context of CDKAL1 silencing (lane 4) counteracts the decrease of the three proteins examined. γ-Tubulin was used as loading control.
CDKAL1 and the Unfolded Protein Response
Because of its proofreading role during translation, down-regulation of CDKAL1 could lead to increased production of aberrant/misfolded proteins in the ER, and hence trigger the unfolded protein response (UPR) to temporarily reduce protein synthesis and alleviate ER stress (33). In agreement with a previous study (11), we indeed found that silencing CDKAL1 in INS-1 cells induced expression of the ER stress marker CHOP10 (Fig. 8A). Because the DECipherment Of DNA Elements (DECODE) database points to the inclusion of a putative binding site for the transcription factors C/EBPα and its homologue CHOP10 in the promoter region of human CDKAL1, we wondered whether the latter is itself a target of the UPR. Rat CDKAL1 transcript and protein levels were measured at different time points after CHOP10 induction by treatment of INS-1 cells with thapsigargin (Fig. 8, B–D). Although the levels of CHOP10 increased ∼15-fold after a 6-h treatment, CDKAL1 did not significantly vary relative to untreated cells (Fig. 8, B and C). However, Cdkal1 mRNA levels increased up to 3-fold after 6 h compared with time 0, in parallel to robust production of CHOP10 mRNA (Fig. 8D). We then tested whether CHOP10 regulates expression of Cdkal1 mRNA by inducing the UPR in CHOP10-silenced cells (Fig. 8, E–G). Transfection of specific siRNA oligos efficiently blocked CHOP10 induction by thapsigargin, as assessed by protein (Fig. 8, E, lanes 2 and 4, and F) and mRNA (Fig. 8G) quantitation. However, CDKAL1 protein levels were again unaffected (Fig. 8, E and F) despite an ∼10-fold increase in mRNA levels compared with control cells upon UPR induction (Fig. 8G). Moreover, overexpression of CHOP10 did not alter CDKAL1 protein or mRNA levels (data not shown). These results suggest that rat CDKAL1 is up-regulated during UPR, but not in a CHOP10-dependent fashion.
FIGURE 8.

CDKAL1 and the unfolded protein response. A, immunoblot analysis of INS-1 cell extracts, after CDKAL1-siRNA or control-siRNA transfection, with anti-CHOP antibody. γ-Tubulin was used as a loading control. Four independent experiments are shown. B, INS-1 cells treated with 1 μm thapsigargin were harvested at different time points and analyzed by SDS-PAGE and immunoblot with anti-CHOP10 antibody and anti-CDKAL1 Ab1. γ-Tubulin was used as a loading control. C, quantification of Western blots as in A for CHOP10 and CDKAL1 from three independent experiments. Pixel intensity values for the two proteins at time point 0 equal 1. D, quantitative RT-PCR for mRNA levels of CHOP10 and CDKAL1 in cells treated as in A. Values for mRNA levels at time point 0 equal 1. n = 3. E, CHOP10 knockdown. INS-1 cells were transfected with CHOP10-siRNA or control-siRNA. 48 h after transfection, cells were treated for 4.5 h with 1 μm thapsigargin, or DMSO, to induce CHOP10 expression. Cells extracts were analyzed by SDS-PAGE and immunoblot with anti-CHOP10 antibody and CDKAL1 Ab1. γ-Tubulin was used as loading control. F, quantification of Western blots as in E for CHOP10 and CDKAL1 from three independent experiments. The white bar corresponds to lane 1 in E, the dotted bar to lane 2, the gray bar to lane 3, and the black bar to lane 4. Pixel intensity values for the two proteins as in lane 2 equal 1. G, quantitative RT-PCR for mRNA levels of CHOP10 and CDKAL1 in cells treated as in E. Values for mRNA level with control-siRNA and DMSO treatment equal 1. n = 3.
DISCUSSION
Here, we have characterized several novel aspects of CDKAL1 biology in insulin-producing cells. We show that human islets and rat insulinoma INS-1 cells express only the transcript for isoform 1, which migrates as a 61-kDa protein by SDS-PAGE. In all tissues investigated, isoform 1 has been the only form detected (34). It would be interesting to determine whether this is also the case in subjects with single nucleotide polymorphisms in CDKAL1, which increase the risk for T2D and could affect its splicing, possibly in a tissue-specific manner. Notably, all these single nucleotide polymorphisms map to intron 5 along with other single nucleotide polymorphisms found to confer susceptibility to psoriasis and Crohn disease (34–36).
Our results corroborate previous findings localizing CDKAL1 to the ER (10, 11). Although these studies indicated that CDKAL1 is associated with intracellular membranes, it was unclear if it is an integral membrane protein or is peripherally associated with the lipid bilayer (10, 11). We now show that CDKAL1 is a true transmembrane protein belonging to the TA protein family. This family includes several key enzymes and regulatory proteins, whose activities are inextricably linked to their localization. Among them are SNARE proteins, members of the Bcl-2 family and components of the translocation machinery in the ER, mitochondria, and peroxisomes (28). The majority of these transmembrane proteins rely on the TRC40/Get3 pathway for their ER membrane insertion. Notably, the Caenorhabditis elegans TRC40 homologue, Asna1, was reported to positively modulate insulin secretion (37). Our data may explain this observation given the broad role of TRC40 in the correct targeting of CDKAL1 and other SNARE proteins (27), which are essentials for vesicle fusion.
The ER localization of CDKAL1 implies the coupling of this methylthiotransferase with polysomes engaged in the translation of proteins destined only for the secretory pathway. Consistent with this conclusion, CDKAL1 knockdown decreases the levels of at least two other secretory granule components, pro-CGA and proICA512/ICA512-TMF, in addition to proinsulin/insulin (11). However, not all transcripts translated at the ER seem susceptible to regulation by CDKAL1, as its silencing did not alter the expression of other secretory proteins, e.g. the SNAREs syntaxin1a and VAMP2/synaptobrevin, as well as Kir6.2, SUR1 (10), PC1/3, PC2, and Phogrin/IA2-β (this study). Notably, these last six proteins are all found on insulin secretory granules (38–40). Thus efficient translation of granule proteins does not always depend on CDKAL1 activity. Indeed, the number of insulin granules in CDKAL1−/− mice was not reduced (10).
Bioinformatics analysis revealed no significant differences in the frequency of Lys residues among proteins translocated through the ER versus those that are cytosolic (data not shown). Lys is the target of many post-translational modifications and is also crucial for the correct sorting and maturation of proteins along the secretory pathway (41, 42). In professional secretory cells, such as insulin-producing β-cells, proteins destined for secretion can account for up to 50% of the total protein content (43). Lower Lys incorporation into proinsulin and ER stress of β-cells upon deletion of CDKAL1 could result in the misfolding of proinsulin (11). Proteins misfolded in the ER are retrotranslocated to the cytosol for proteasomal degradation (44). By increasing the translation fidelity of mRNAs coding for proteins in transit through the ER, and thus minimizing misfolding and energy consumption involved in ER-associated protein degradation, CDKAL1 may be critical in professional secretory cells. Accordingly, lymphocytes also display high CDKAL1 expression levels (34).
The need for CDKAL1 may be more overt in conditions that place a high metabolic load on the ER and trigger a stress response. Indeed β-cell-restricted CDKAL1−/− mice showed UPR activation and profound glucose intolerance when fed a high fat diet (11). Here, we showed silencing CDKAL1 in INS-1 cells was sufficient to up-regulate the ER stress marker CHOP10. Intriguingly, we found that the CDKAL1 mRNA content was also enhanced by thapsigargin-induced ER stress, although the protein levels were unchanged. It is plausible that increased CDKAL1 transcription prompts the cell for more efficient translation, once the constraints imposed by the UPR are alleviated.
Reduction of CDKAL1 expression in INS-1 cells further correlated with reduced expression of the granule precursor proteins proinsulin, pro-CGA, and pro-ICA512. As proposed for proinsulin (11), the lower levels of pro-CGA and pro-ICA512 could result from aberrant folding and processing consequent to incorrect Lys incorporation. Reduced expression of these granule proteins could account, at least in part, for the decreased insulin secretion of CDKAL1-depleted INS-1 (this study) and mouse β-cells (11). Alternatively, CDKAL1 may affect secretion by regulating β-cell processes other than insulin granule biogenesis. Islet cells of systemic (10) and β-cell-restricted (11) CDKAL1−/− mice displayed lower ATP levels due to mitochondrial dysfunction. Reduced ATP generation could impair the function of KATP channels, which are key for depolarization of β-cells and insulin secretion. Thus, both β-cell ER stress and deficits in later stages of granule exocytosis could contribute to deficient insulin release in carriers of CDKAL1 alleles associated with an increased risk for T2D.
In conclusion, elucidation of CDKAL1 regulation in normal and pathological conditions and identification of the protein subset that depends on CDKAL1 for efficient translation will help reveal how its genetic variants confer susceptibility to diabetes and other disorders, such as Crohn disease and psoriasis.
Acknowledgments
We are grateful to Barbara Ludwig, Anja Steffen, and Stefan Bornstein for providing human islets and Klaus Knoch for sharing RNA extracts. We thank Ramanujan S. Hegde (Medical Research Council, Cambridge, UK) and Stephen High (University of Manchester, UK) for the preprolactin and Sec61b-Nglyc constructs, respectively; Gert Kreibich (New York University School of Medicine), and Paul Hargrave (University of Florida, Gainsville), and Richard Zimmermann (Saarland University, Homburg, Germany) for the anti-ribophorin I, anti-opsin, and anti-Sec61a antibodies, respectively. We thank all members of the Solimena laboratory for advice and support.
This work was supported in part by funds from the German Ministry for Education and Research to the German Centre for Diabetes Research, a research grant from Boehringer Ingelheim (to M. S.), and by Italian Association for Cancer Research Grant IG9040 (to the N. B. laboratory).

This article contains supplemental Experimental Procedures, Figs. S1–S5, Tables S1 and S2, and additional references.
The nucleotide sequence(s) reported in this paper has been submitted to the GenBankTM/EBI Data Bank with accession number(s) JQ935002
- T2D
- type 2 diabetes
- Cdkal1
- Cdk5 regulatory subunit-associated protein 1-like 1
- TA
- tail-anchored
- UPR
- unfolded protein response
- RM
- rough microsomes
- IAA
- iodoacetamide
- Ab
- antibody
- TMD
- transmembrane domain
- ER
- endoplasmic reticulum
- PF
- protected fragment
- CGA
- chromogranin A
- ICA512
- islet cells autoantigen 512
- Endo-H
- endoglycosidase H
- PNGaseF
- peptide:N-glycosidase F
- oligo
- oligonucleotide
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine
- Nglyc
- N-glycosylated
- HSP
- high speed pellet.
REFERENCES
- 1. Bonnefond A., Froguel P., Vaxillaire M. (2010) The emerging genetics of type 2 diabetes. Trends Mol. Med. 16, 407–416 [DOI] [PubMed] [Google Scholar]
- 2. Kirchhoff K., Machicao F., Haupt A., Schäfer S. A., Tschritter O., Staiger H., Stefan N., Häring H. U., Fritsche A. (2008) Polymorphisms in the TCF7L2, CDKAL1, and SLC30A8 genes are associated with impaired proinsulin conversion. Diabetologia 51, 597–601 [DOI] [PubMed] [Google Scholar]
- 3. Groenewoud M. J., Dekker J. M., Fritsche A., Reiling E., Nijpels G., Heine R. J., Maassen J. A., Machicao F., Schäfer S. A., Häring H. U., 't Hart L. M., van Haeften T. W. (2008) Variants of CDKAL1 and IGF2BP2 affect first-phase insulin secretion during hyperglycaemic clamps. Diabetologia 51, 1659–1663 [DOI] [PubMed] [Google Scholar]
- 4. Ching Y. P., Pang A. S., Lam W. H., Qi R. Z., Wang J. H. (2002) Identification of a neuronal Cdk5 activator-binding protein as Cdk5 inhibitor. J. Biol. Chem. 277, 15237–15240 [DOI] [PubMed] [Google Scholar]
- 5. Dhavan R., Tsai L. H. (2001) A decade of CDK5. Nat. Rev. Mol. Cell Biol. 2, 749–759 [DOI] [PubMed] [Google Scholar]
- 6. Lilja L., Yang S. N., Webb D. L., Juntti-Berggren L., Berggren P. O., Bark C. (2001) Cyclin-dependent kinase 5 promotes insulin exocytosis. J. Biol. Chem. 276, 34199–34205 [DOI] [PubMed] [Google Scholar]
- 7. Lilja L., Johansson J. U., Gromada J., Mandic S. A., Fried G., Berggren P. O., Bark C. (2004) Cyclin-dependent kinase 5 associated with p39 promotes munc18-1 phosphorylation and Ca2+-dependent exocytosis. J. Biol. Chem. 279, 29534–29541 [DOI] [PubMed] [Google Scholar]
- 8. Wei F. Y., Nagashima K., Ohshima T., Saheki Y., Lu Y. F., Matsushita M., Yamada Y., Mikoshiba K., Seino Y., Matsui H., Tomizawa K. (2005) Cdk5-dependent regulation of glucose-stimulated insulin secretion. Nat. Med. 11, 1104–1108 [DOI] [PubMed] [Google Scholar]
- 9. Schubert S., Knoch K. P., Ouwendijk J., Mohammed S., Bodrov Y., Jäger M., Altkrüger A., Wegbrod C., Adams M. E., Kim Y., Froehner S. C., Jensen O. N., Kalaidzidis Y., Solimena M. (2010) β2-Syntrophin is a Cdk5 substrate that restrains the motility of insulin secretory granules. PLoS ONE 5, e12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ohara-Imaizumi M., Yoshida M., Aoyagi K., Saito T., Okamura T., Takenaka H., Akimoto Y., Nakamichi Y., Takanashi-Yanobu R., Nishiwaki C., Kawakami H., Kato N., Hisanaga S., Kakei M., Nagamatsu S. (2010) Deletion of CDKAL1 affects mitochondrial ATP generation and first-phase insulin exocytosis. PLoS ONE 5, e15553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wei F. Y., Suzuki T., Watanabe S., Kimura S., Kaitsuka T., Fujimura A., Matsui H., Atta M., Michiue H., Fontecave M., Yamagata K., Suzuki T., Tomizawa K. (2011) Deficit of tRNALys modification by Cdkal1 causes the development of type 2 diabetes in mice. J. Clin. Invest. 121, 3598–3608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arragain S., Handelman S. K., Forouhar F., Wei F. Y., Tomizawa K., Hunt J. F., Douki T., Fontecave M., Mulliez E., Atta M. (2010) Identification of eukaryotic and prokaryotic methylthiotransferase for biosynthesis of 2-methylthio-N6-threonylcarbamoyladenosine in tRNA. J. Biol. Chem. 285, 28425–28433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gustilo E. M., Vendeix F. A., Agris P. F. (2008) tRNA's modifications bring order to gene expression. Curr. Opin. Microbiol. 11, 134–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Trajkovski M., Mziaut H., Altkrüger A., Ouwendijk J., Knoch K. P., Müller S., Solimena M. (2004) Nuclear translocation of an ICA512 cytosolic fragment couples granule exocytosis and insulin expression in β-cells. J. Cell Biol. 167, 1063–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Trajkovski M., Mziaut H., Schubert S., Kalaidzidis Y., Altkrüger A., Solimena M. (2008) Regulation of insulin granule turnover in pancreatic β-cells by cleaved ICA512. J. Biol. Chem. 283, 33719–33729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pedrazzini E., Villa A., Longhi R., Bulbarelli A., Borgese N. (2000) Mechanism of residence of cytochrome b5, a tail-anchored protein, in the endoplasmic reticulum. J. Cell Biol. 148, 899–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ronchi P., Colombo S., Francolini M., Borgese N. (2008) Transmembrane domain-dependent partitioning of membrane proteins within the endoplasmic reticulum. J. Cell Biol. 181, 105–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stefanovic S., Hegde R. S. (2007) Identification of a targeting factor for post-translational membrane protein insertion into the ER. Cell 128, 1147–1159 [DOI] [PubMed] [Google Scholar]
- 19. Hermel J. M., Dirkx R., Jr., Solimena M. (1999) Post-translational modifications of ICA512, a receptor tyrosine phosphatase-like protein of secretory granules. Eur. J. Neurosci. 11, 2609–2620 [DOI] [PubMed] [Google Scholar]
- 20. Colombo S. F., Longhi R., Borgese N. (2009) The role of cytosolic proteins in the insertion of tail-anchored proteins into phospholipid bilayers. J. Cell Sci. 122, 2383–2392 [DOI] [PubMed] [Google Scholar]
- 21. Yu Y. H., Sabatini D. D., Kreibich G. (1990) Antiribophorin antibodies inhibit the targeting to the ER membrane of ribosomes containing nascent secretory polypeptides. J. Cell Biol. 111, 1335–1342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lang S., Benedix J., Fedeles S. V., Schorr S., Schirra C., Schäuble N., Jalal C., Greiner M., Hassdenteufel S., Tatzelt J., Kreutzer B., Edelmann L., Krause E., Rettig J., Somlo S., Zimmermann R., Dudek J. (2012) Different effects of Sec61α, Sec62, and Sec63 depletion on transport of polypeptides into the endoplasmic reticulum of mammalian cells. J. Cell Sci. 125, 1958–1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brambillasca S., Yabal M., Soffientini P., Stefanovic S., Makarow M., Hegde R. S., Borgese N. (2005) Transmembrane topogenesis of a tail-anchored protein is modulated by membrane lipid composition. EMBO J. 24, 2533–2542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Adamus G., Arendt A., Hargrave P. (1991) Genetic control of antibody response to bovine rhodopsin in mice. Epitope mapping of rhodopsin structure. J. Neuroimmunol. 34, 89–97 [DOI] [PubMed] [Google Scholar]
- 25. Hegde R. S., Lingappa V. R. (1996) Sequence-specific alteration of the ribosome-membrane junction exposes nascent secretory proteins to the cytosol. Cell 85, 217–228 [DOI] [PubMed] [Google Scholar]
- 26. Ort T., Voronov S., Guo J., Zawalich K., Froehner S. C., Zawalich W., Solimena M. (2001) Dephosphorylation of β2-syntrophin and Ca2+/μ-calpain-mediated cleavage of ICA512 upon stimulation of insulin secretion. EMBO J. 20, 4013–4023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Livak K. J., Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 28. Borgese N., Colombo S., Pedrazzini E. (2003) The tale of tail-anchored proteins. Coming from the cytosol and looking for a membrane. J. Cell Biol. 161, 1013–1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Borgese N., Brambillasca S., Colombo S. (2007) How tails guide tail-anchored proteins to their destinations. Curr. Opin. Cell Biol. 19, 368–375 [DOI] [PubMed] [Google Scholar]
- 30. Hegde R. S., Keenan R. J. (2011) Tail-anchored membrane protein insertion into the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol. 12, 787–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abell B. M., Jung M., Oliver J. D., Knight B. C., Tyedmers J., Zimmermann R., High S. (2003) Tail-anchored and signal-anchored proteins utilize overlapping pathways during membrane insertion. J. Biol. Chem. 278, 5669–5678 [DOI] [PubMed] [Google Scholar]
- 32. Borgese N., Fasana E. (2011) Targeting pathways of C-tail-anchored proteins. Biochim. Biophys. Acta 1808, 937–946 [DOI] [PubMed] [Google Scholar]
- 33. Scheuner D., Kaufman R. J. (2008) The unfolded protein response. A pathway that links insulin demand with β-cell failure and diabetes. Endocr. Rev. 29, 317–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quaranta M., Burden A. D., Griffiths C. E., Worthington J., Barker J. N., Trembath R. C., Capon F. (2009) Differential contribution of CDKAL1 variants to psoriasis, Crohn's disease, and type II diabetes. Genes Immun. 10, 654–658 [DOI] [PubMed] [Google Scholar]
- 35. Wolf N., Quaranta M., Prescott N. J., Allen M., Smith R., Burden A. D., Worthington J., Griffiths C. E., Mathew C. G., Barker J. N., Capon F., Trembath R. C. (2008) Psoriasis is associated with pleiotropic susceptibility loci identified in type II diabetes and Crohn disease. J. Med. Genet. 45, 114–116 [DOI] [PubMed] [Google Scholar]
- 36. Li Y., Liao W., Chang M., Schrodi S. J., Bui N., Catanese J. J., Poon A., Matsunami N., Callis-Duffin K. P., Leppert M. F., Bowcock A. M., Kwok P. Y., Krueger G. G., Begovich A. B. (2009) Further genetic evidence for three psoriasis-risk genes. ADAM33, CDKAL1, and PTPN22. J. Invest. Dermatol. 129, 629–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kao G., Nordenson C., Still M., Rönnlund A., Tuck S., Naredi P. (2007) ASNA-1 positively regulates insulin secretion in C. elegans and mammalian cells. Cell 128, 577–587 [DOI] [PubMed] [Google Scholar]
- 38. Varadi A., Grant A., McCormack M., Nicolson T., Magistri M., Mitchell K. J., Halestrap A. P., Yuan H., Schwappach B., Rutter G. A. (2006) Intracellular ATP-sensitive K+ channels in mouse pancreatic beta cells. Against a role in organelle cation homeostasis. Diabetologia 49, 1567–1577 [DOI] [PubMed] [Google Scholar]
- 39. Steiner D. F., Rouillé Y., Gong Q., Martin S., Carroll R., Chan S. J. (1996) The role of prohormone convertases in insulin biosynthesis: evidence for inherited defects in their action in men and experimental animals. Diabetes Metab. 22, 94–104 [PubMed] [Google Scholar]
- 40. Bailyes E. M., Bennett D. L., Hutton J. C. (1991) Proprotein-processing endopeptidases of the insulin secretory granule. Enzyme 45, 301–313 [DOI] [PubMed] [Google Scholar]
- 41. Zhou A., Webb G., Zhu X., Steiner D. F. (1999) Proteolytic processing in the secretory pathway. J. Biol. Chem. 274, 20745–20748 [DOI] [PubMed] [Google Scholar]
- 42. Teasdale R. D., Jackson M. R. (1996) Signal-mediated sorting of membrane proteins between the endoplasmic reticulum and the Golgi apparatus. Annu. Rev. Cell Dev. Biol. 12, 27–54 [DOI] [PubMed] [Google Scholar]
- 43. Schuit F. C., In't Veld P. A., Pipeleers D. G. (1988) Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc. Natl. Acad. Sci. U.S.A. 85, 3865–3869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vembar S. S., Brodsky J. L. (2008) One step at a time. Endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]