

Background: Nontruncating SCN1A mutations can cause severe myoclonic epilepsy of infancy (SMEI).
Results: Some SMEI-associated mutations exhibit reduced cell surface expression, which is reversible for certain alleles by pharmacological treatment.
Conclusion: Nontruncating SCN1A mutations with reduced cell surface expression may be amenable to pharmacological rescue.
Significance: Although rescue of trafficking-impaired SCN1A alleles may be beneficial, increased surface expression of dysfunctional channels could exacerbate SMEI.
Keywords: Epilepsy, Ion Channels, Molecular Pharmacology, Sodium Channels, Trafficking, Anti-epileptic Drugs, Dravet Syndrome
Abstract
Mutations in SCN1A, encoding the voltage-gated sodium channel NaV1.1, are the most common cause of severe myoclonic epilepsy of infancy (SMEI) or Dravet syndrome. SMEI is most often associated with premature truncations of NaV1.1 that cause loss of function, but nontruncating mutations also occur. We hypothesized that some nontruncating mutations might impair trafficking of NaV1.1 to the plasma membrane. Here we demonstrated that seven nontruncating missense or in-frame deletion mutations (L986F, delF1289, R1648C, F1661S, G1674R, and G1979E) exhibited reduced cell surface expression relative to wild type (WT) NaV1.1 consistent with impaired trafficking. We tested whether two commonly prescribed antiepileptic drugs (phenytoin, lamotrigine), as well as the cystic fibrosis transmembrane conductance regulator (CFTR) trafficking corrector VRT-325, could rescue cell surface and functional expression of two representative NaV1.1 mutants (R1648C, G1674R). Treatment of cells with phenytoin increased cell surface expression of WT-NaV1.1 and both mutant channels, whereas lamotrigine only increased surface expression of R1648C. VRT-325 did not alter surface expression of WT-NaV1.1 or mutant channels. Although phenytoin increased surface expression of G1674R, channel function was not restored, suggesting that this mutation also causes an intrinsic loss of function. Both phenytoin and lamotrigine increased functional expression of R1648C, but lamotrigine also increased persistent sodium current evoked by this mutation. Our findings indicate that certain nontruncating SCN1A mutations associated with SMEI have impaired cell surface expression and that some alleles may be amenable to pharmacological rescue of this defect. However, rescue of dysfunctional NaV1.1 channels to the plasma membrane could contribute to exacerbating rather than ameliorating the disease.
Introduction
Voltage-gated sodium (NaV)2 channels mediate cellular excitability by providing the molecular basis for generation and propagation of action potentials (1). Sodium channels exist as heteromultimeric complexes formed by a pore-forming α-subunit and one or more auxiliary β-subunits (2). Nine genes (e.g. SCN1A, SCN2A) encode distinct pore-forming subunits (e.g. NaV1.1, NaV1.2), whereas four genes encode β-subunits (2, 3). Mutations in NaV channels give rise to an expanding list of channelopathies including epilepsy (2).
More than 700 SCN1A mutations have been identified in association with various genetic epilepsy syndromes representing a wide range of clinical severity from the relatively benign genetic epilepsy with febrile seizures plus (GEFS+) to the more malignant severe myoclonic epilepsy of infancy (SMEI or Dravet syndrome) (2, 4–7). The majority of mutations associated with SMEI are predicted to cause protein truncation and loss of function, which has led to the hypothesis that SMEI stems from SCN1A haploinsufficiency (2, 4). Mice with heterozygous deletion of Scn1a have a phenotype resembling SMEI and have been used to investigate the neurophysiological mechanisms for epilepsy in this disorder. These studies have suggested that the mechanism responsible for this disorder is reduced sodium current density in GABAergic interneurons, resulting in reduced activity in inhibitory networks and causing neuronal hyperexcitability (8, 9). However, several missense mutations associated with SMEI and borderline SMEI generate functional NaV channels in heterologous cells (10–12), suggesting that other disease-producing channel mechanisms may exist in addition to overt loss of function.
Impaired trafficking of mutant ion channels to the plasma membrane is a well established pathophysiological mechanism for other channelopathies, including cystic fibrosis, resulting from mutations in the cystic fibrosis transmembrane conductance regulator (CFTR), and congenital long QT syndrome, resulting from mutations in the hERG potassium channel (13, 14). Pharmacological rescue of trafficking-impaired mutant ion channels has been demonstrated in these disorders using either known high affinity channel blockers or other small molecules. For example, the high affinity hERG blockers cisapride and E-4031 can rescue trafficking-defective hERG mutants possibly because channel blockers stabilize protein structure and allow proper trafficking (15, 16). Small molecule trafficking correctors of mutant CFTR have been identified that increase CFTR cell surface expression and restore membrane chloride conductance (14, 17). Interestingly, VRT-325 (4-cyclohexyloxy-2-{1-[4-(4-methoxy-benzenesulfonyl)-piperazin-1-yl]-ethyl}-quinazoline) also rescues the trafficking-defective hERG mutation G601S (14).
Here, we examined trafficking of seven SMEI-associated mutations, which represent both functional and nonfunctional proteins, and found that many exhibited reduced plasma membrane expression. We further studied two representative mutations (R1648C, G1674R) to test whether small molecules could rescue NaV1.1 cell surface and functional expression. We found that two widely used antiepileptic drugs, phenytoin and lamotrigine, had different effects on wild type (WT) and mutant channels. Although phenytoin increased surface and functional expression of WT and both mutant channels, lamotrigine only increased surface and functional expression of the R1648C mutant channel. VRT-325, a CFTR trafficking corrector, had no effect on NaV1.1 cell surface expression or function. Our findings indicate that nontruncating SCN1A missense mutations or an in-frame deletion associated with SMEI have impaired cell surface trafficking and that certain alleles may be amenable to pharmacological rescue.
MATERIALS AND METHODS
Mutagenesis and Heterologous Expression of NaV1.1
Mutagenesis of recombinant NaV1.1 to create L986F, ΔF1289, R1648C, F1661S, G1674R, G1979E, and T1901I channels was described previously (10, 11, 18). To minimize spontaneous mutagenesis of NaV1.1 cDNA in bacterial culture, recombinants were always propagated in Stbl2 cells (Invitrogen) at 30 °C.
Heterologous expression of NaV1.1 in tsA201 cells was performed as described previously (19, 20). Transient expression of WT or mutant NaV1.1 with the accessory β1 and β2 subunits was achieved using Qiagen SuperFect reagent (2 μg of total plasmid DNA was transfected with a cDNA ratio of 10:1:1 for α:β1:β2 subunits). Human β1 and β2 cDNAs were cloned into plasmids also encoding the CD8 receptor (CD8-IRES-hβ1) or enhanced green fluorescent protein (EFGP-IRES-hβ2), respectively, as co-transfection markers (18).
Stable expression of WT and mutant NaV1.1 in HEK-293 cells was achieved using the piggyBac transposon-mediated integration system as described previously (21). Cells were co-transfected with plasmids encoding WT or mutant NaV1.1 (pT-SCN1A:3X-FLAG), human sodium channel β1 and β2 subunits (pT-SCN1B:cMyc-IRES-SCN2B:HA), and the piggyBac transposase (pCMV-piggyBac). Epitope tags are encoded on the NaV1.1 C terminus. Clonal populations of cells were selected based on dual puromycin (NaV1.1) and G418 (β1, β2) resistance. For each cell line, 8–10 individual clonal lines were tested by Western blot analysis to evaluate NaV1.1 and β-subunit protein expression levels, and a single clonal cell line was selected for experiments. The specific cell lines used for this study were chosen based on protein expression level of NaV1.1 within the upper 50th percentile among clones and approximately equal expression of the two β-subunits. All cells expressing either WT or mutant NaV1.1 exhibited measurable sodium current (>50 pA) when tested electrophysiologically.
Cell Surface Biotinylation
Cells were washed twice with 4 °C PBS and then treated with 3.3 mm sulfo-NHS-biotin (Thermo Scientific) in PBS for 1 h at 4 °C. Excess biotinylating reagent was quenched by washing cells twice with 100 mm glycine in PBS followed by a 10-min incubation in 100 mm glycine at 4 °C. Cells were washed twice with PBS to remove excess glycine and then lysed with radioimmune precipitation assay (RIPA) (150 mm NaCl, 50 mm Tris base, 1% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, pH 7.5) supplemented with Complete Mini protease inhibitor mixture (Roche Applied Science). Cell lysate containing 15 μg of protein was saved for total protein immunoblots, whereas 600 μg of cell lysate protein was bound to 50 μl of streptavidin beads (Thermo Scientific) overnight at 4 °C. After incubation, beads were washed three times with RIPA, and then proteins were eluted in 2× Laemmli buffer containing 5% β-mercaptoethanol. The entirety of eluted protein per sample was subjected to SDS-PAGE and immunoblotting.
Immunoblot Analysis
Immunoblot analysis was performed as described previously (21). Cells were washed twice with 4 °C PBS and lysed with RIPA (150 mm NaCl, 50 mm Tris base, 1% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, pH 7.5) supplemented with Complete Mini protease inhibitor mixture (Roche Applied Science). Solubilized lysate containing 30 μg of protein was diluted in 2× Laemmli buffer containing 5% β-mercaptoethanol, subjected to 4–20% gradient SDS-PAGE, and transferred to a PVDF membrane. Proteins were detected with primary antibodies directed against the FLAG epitope (mouse, anti-FLAG M2, 1:15,000, Sigma-Aldrich). Immunoreactive bands were detected using horseradish peroxidase-conjugated secondary antibody directed against the primary antibody (goat, anti-mouse, 1:10,000, Santa Cruz Biotechnology, Santa Cruz, CA) coupled with ECL Plus reagent and then imaged using hypersensitive ECL film (GE Healthcare) exposed for five separate time intervals ranging from 10 s to 45 min. Densitometric analysis was performed using an exposure time in which there was no signal saturation. Densitometric quantification of Western blots was performed using ImageJ software (National Institutes of Health). Band intensity of NaV1.1 was normalized to that of transferrin (loading control). All data were background-subtracted using a region with no immunodetected protein. Cell surface expression was determined by subsequent normalization of biotinylated protein to total protein.
Cell Surface Sandwich ELISA
Untreated Costar 96-well assay plates were coated with 50 μl of 1.0 ng/μl mouse anti-FLAG M2 IgG (Sigma-Aldrich). Plates were blocked for 1 h at room temperature with TBS+ (20 mm Tris base, 137 mm NaCl, pH 7.6, 0.15% Tween 20, 0.35% BSA) and 0.25 ng/μl streptavidin. Biotinylated protein lysate (50 μl), prepared as described above, was plated in triplicate at a concentration of 8 ng/μl in TBS+ and incubated for 2 h at room temperature. The plate was incubated with TBS+ for 30 min followed by a 2-h incubation with 50 μl of high sensitivity streptavidin-HRP (0.25 ng/μl in TBS+). The plate was then incubated with TBS+ for 30 min and incubated with 100 μl of SuperSignal Femto substrate for 1 min. Chemiluminescence was measured using a Packard LumiCount detector. For each sample, chemiluminescence was measured in triplicate, averaged, and counted as a single biological replicate. Background luminescence was subtracted from each sample using luminescence measured from empty wells. Chemiluminescence was measured for cells expressing WT and mutant sodium channels grown in the presence of 0.1% DMSO, 50 μm phenytoin, or 50 μm lamotrigine.
Electrophysiology and Data Analysis
Whole-cell voltage clamp recordings of HEK-293 cells stably transfected with WT or mutant NaV1.1 channels were performed as described previously (10, 18, 20). Patch pipettes were fabricated from thin wall borosilicate glass (Warner Instruments) with a P-97 multistage Flaming-Brown micropipette puller (Sutter Instruments) and fire-polished using a microforge (MF 830, Narishige, Tokyo, Japan). Final pipette resistance was between 1.0 and 2.0 megaohms. The pipette solution consisted of (in mm) 110 CsF, 10 NaF, 20 CsCl, 2 EGTA, 10 HEPES, with pH 7.35 and osmolality 300 mosmol/kg. Bath solution contained (in mm) 145 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES with pH 7.35 and osmolality 310 mosmol/kg. Cells were allowed to stabilize for 10 min after establishment of the whole-cell configuration before currents were measured. Persistent sodium current was isolated by digital subtraction following the addition of 0.5 μm tetrodotoxin. Whole-cell capacitance and access resistance were determined by integrating capacitive transients in response to a +10-mV voltage step from −120 mV filtered at 50 kHz. Series resistance was compensated 90%, with 70% prediction, to ensure that the command potential was reached within microseconds with a voltage error <2 mV. Cells that did not meet these criteria because of excessively large current amplitudes (>12 nA) were excluded from final analysis. Leak currents were subtracted by using an online P/4 procedure. All whole-cell currents were low-pass Bessel-filtered at 5 kHz and digitized at 50 kHz. Specific voltage clamp protocols assessing channel activation and inactivation are depicted in the figures.
Pharmacology
Phenytoin and lamotrigine were purchased from Sigma-Aldrich. VRT-325 was synthesized by the Vanderbilt Institute for Chemical Biology chemical synthesis core facility according to the published synthetic scheme (14). The final product was verified by NMR and mass spectroscopy.
All drug stock solutions were prepared in DMSO. Stock concentrations of phenytoin and lamotrigine were 50 mm, whereas VRT-325 was 10 mm. All drugs were diluted in cell culture media on the day of the experiment. Final concentrations for phenytoin and lamotrigine were 50 μm, and VRT-325 was 10 μm. The concentration of DMSO did not exceed 0.1%.
RESULTS
SMEI-associated Mutations R1648C and G1674R Impair NaV1.1 Trafficking
Although most SCN1A mutations associated with SMEI are predicted to cause truncation of the coding region leading to loss of function, many missense alleles have also been identified that have less certain functional effects. We have previously determined that the SMEI-associated missense mutations illustrated in Fig. 1A exhibited a range of functional properties in vitro (10–12, 22), but none of these alleles has been studied for cell surface expression. To evaluate expression at the plasma membrane, we performed cell surface biotinylation and Western blot analysis on cells transiently expressing either WT or mutant NaV1.1 channels. Examination of total protein immunoblots indicated that many mutations had reduced expression when compared with WT, whereas biotinylation experiments demonstrated reduced cell surface expression for some mutants (Fig. 1B). To evaluate trafficking efficiency semiquantitatively, we normalized cell surface expression to total protein and found that all mutations have variable degrees of impaired trafficking to the membrane relative to WT (Fig. 1, B and C).
FIGURE 1.

SMEI-associated NaV1.1 mutants exhibit reduced cell surface expression. A, topology diagram of NaV1.1 illustrating the location of SMEI-associated mutations. B, cell surface biotinylation was used to examine plasma membrane expression of WT-NaV1.1, L986F, ΔF1289, R1648C, F1661S, G1674R, G1749E, and T1909I in transiently transfected cells. Representative immunoblots of total protein (left) and biotinylated cell surface protein (right) are illustrated. The lower panel in each blot is the transferrin loading control. C, semiquantitative analysis of cell surface protein expressed as a ratio of recovered biotinylated protein to total protein. Data are expressed as mean ± S.E. for 6 replicates.
Quantitative Analysis of Cell Surface Expression of WT and Mutant NaV1.1
To further investigate impaired cell surface expression of SMEI-associated NaV1.1 alleles, we selected two representative mutants (R1648C, G1674R), as well as WT, and created stable cell lines expressing these channels for additional studies. We compared the electrophysiological properties of WT-NaV1.1 with mutations R1648C and G1764R expressed stably in HEK-293 cells. When compared with WT NaV1.1, both R1648C and G1674R exhibited significantly reduced current density (Fig. 2) consistent with reduced cell surface expression observed in the transient expression experiments.
FIGURE 2.

SMEI-associated mutations exhibit reduced whole-cell current. A, representative whole-cell current from cells stably expressing WT-NaV1.1 (top), R1648C (middle), or G1674R (bottom). B, current-voltage relationship of WT (filled circles), R1648C (open circles), and G1674R (filled triangles). All data are expressed as mean ± S.E. for 6–10 replicates. pF, picofarads.
To better quantify plasma membrane NaV1.1 expression and to improve experimental throughput, we developed an enzyme-linked immunosorbent assay (ELISA) for detection of biotinylated cell surface proteins (see “Materials and Methods”) and used this approach to compare cell surface expression of WT, R1648C, and G1674R (Fig. 3A). As we observed with Western blot analysis, R1648C and G1674R have reduced surface expression when compared with WT (Fig. 3B). This method also enabled experiments designed to screen selected compounds for their effects on cell surface expression as a strategy to identify pharmacological approaches to rescue trafficking-impaired NaV1.1 mutants.
FIGURE 3.
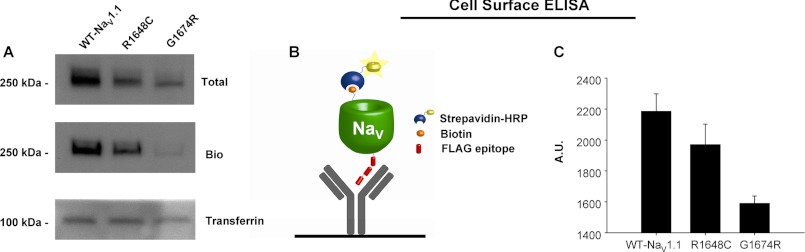
Quantitative analysis of cell surface expression. A, total and cell surface protein expression of WT-NaV1.1, R1648C, and G1674R detected by Western blot analysis. Proteins were detected with antibodies directed against the FLAG epitope. Cell surface protein was probed following cell surface biotinylation (Bio) and recovery of biotinylated proteins by streptavidin affinity chromatography. Detection of the endogenous protein transferrin was done to demonstrate equal protein loading in each lane. B, graphic representing cell surface ELISA showing immobilized anti-FLAG antibody and antibody bound to FLAG-tagged NaV channel that has been biotinylated and conjugated to streptavidin-HRP. C, average cell surface ELISA data showing relative expression of WT-NaV1.1, R1648C, and G1674R. Data are expressed as mean ± S.E. for 6 replicates. A. U., absorbance units.
Antiepileptic Drugs but Not VRT-325 Increase Cell Surface Expression of NaV1.1 Mutants
We tested whether the antiepileptic drugs phenytoin and lamotrigine or the investigational compound VRT-325 can promote increased cell surface expression of WT-NaV1.1 or mutants R1684C and G1674R. Cells were grown 24 h in the presence of vehicle (0.1% DMSO), 10 μm VRT-325, 50 μm phenytoin, or 50 μm lamotrigine. ELISA measurements of cell surface expression were then normalized to untreated HEK cells. We observed no effect of VRT-325 on cell surface expression of WT, R1648C, or G1674R channels (data not shown). However, both antiepileptic drugs promoted increased surface expression for one or more alleles.
For WT channels, phenytoin significantly increased surface expression by 2.6 ± 0.3-fold (p < 0.01) when compared with 1.1 ± 0.1-fold induced by vehicle, whereas lamotrigine had no significant effect (1.1 ± 0.2-fold increase, not significant; Fig. 4A). Both phenytoin and lamotrigine significantly increased surface expression of R1648C (2.1 ± 0.1 and 2.9 ± 0.2-fold respectively, p < 0.01) when compared with 1.0 ± 0.03-fold for the vehicle control (Fig. 4B). By contrast, G1674R exhibited increased surface expression only following phenytoin treatment (2.9 ± 0.3-fold, p = 0.01) when compared with 1.1 ± 0.03-fold for the vehicle control with a nonsignificant trend following lamotrigine (1.2 ± 0.4; Fig. 4C).
FIGURE 4.

Antiepileptic drugs can rescue cell surface expression of NaV1.1 channels. A–C, cell surface ELISA data from cells stably expressing WT (A), R1648C (B), and G1674R (C) grown in the presence of 0.1% DMSO, 50 μm phenytoin (Phe), or 50 μm lamotrigine (Lam). Data are expressed as mean ± S.E. for 6 replicates. An asterisk indicates p < 0.05 when compared with DMSO-treated cells.
We next determined whether increased cell surface protein expression strictly correlates with an increase in functional expression. To examine functional expression of WT and mutant channels, we performed whole-cell patch clamp experiments on cells stably expressing WT-NaV1.1, R1648C, or G1674R channels treated 24 h with vehicle, 50 μm phenytoin, or 50 μm lamotrigine (cells were equilibrated in drug-free solution 30 min before recording experiments). Although we observed a 2.6-fold increase in surface expression of WT by cell surface ELISA, we detected a 1.4-fold increase in peak current density when cells were grown in the presence of phenytoin when compared with vehicle-treated cells (Fig. 5A). The fold increase in peak current density was proportionally lower than the change in surface expression in part because we had to exclude some cells in the phenytoin-treated group that exhibited extremely large current amplitudes and compromised voltage control. No cells in the vehicle-treated group were excluded for this reason. By contrast, phenytoin-treated R1648C cells exhibited a significant 2.6-fold increase in functional expression (p < 0.01) when compared with vehicle treatment (Fig. 5B), and this was comparable with the 2.1-fold increase in surface expression (Fig. 4B). Finally, phenytoin had no effect on G1674R functional expression (1.3-fold increase in current density, p = 0.1) when compared with vehicle-treated controls (Fig. 5C) despite the robust increase in cell surface expression determined with ELISA (Fig. 4C). The disparity between cell surface and functional expression rescue observed for phenytoin-treated G1674R cells suggested that this mutation causes both functional and trafficking defects.
FIGURE 5.

Phenytoin rescues NaV1.1 channel function. A–C, representative whole-cell currents recorded from cells stably expressing WT-NaV1.1 (A), R1648C (B), or G1674R (C) grown in the presence of 0.1% DMSO (left) or 50 μm phenytoin (middle). The right panel represents the average current voltage relationship for cells grown in the presence of either 0.1% DMSO (filled circles) or 50 μm phenytoin (open circles). Data are expressed as mean ± S.E. for 8–18 replicates. An asterisk indicates p < 0.05 when compared with DMSO-treated cells. pF, picofarads.
Lamotrigine treatment of R1648C-expressing cells evoked a 2.9-fold (p < 0.01) increase in peak current density when compared with vehicle-treated cells (Fig. 6B), and this was concordant with the observed increase in cell surface expression (Fig. 4B). By contrast, lamotrigine did not significantly increase functional expression of either WT (1.33-fold, p = 0.33) or G1674R (1.1-fold, p = 0.86) when compared with vehicle-treated controls (Fig. 6, A and C), and this was in line with the cell surface expression data (Fig. 4, B and C).
FIGURE 6.

Lamotrigine rescues NaV1.1 channel function. A–C, representative whole-cell currents recorded from cells stably expressing WT-NaV1.1 (A), R1648C (B), or G1674R (C) grown in the presence of 0.1% DMSO (left) or 50 μm lamotrigine (middle). The right panel represents the average current voltage relationship of cells grown in the presence of either 0.1% DMSO (filled circles) or 50 μm lamotrigine (open circles). Data are expressed as mean ± S.E. for 10–12 replicates. An asterisk indicates p < 0.05 when compared with DMSO-treated cells. pF, picofarads.
Antiepileptic Drug Treatment Increases Persistent Current Mediated by R1648C
Exacerbation of epilepsy by commonly used antiepileptic drugs, such as phenytoin and lamotrigine, has been described in SMEI (23, 24). It is conceivable that in some SMEI cases, epilepsy severity could be aggravated by increasing cell surface expression of a dysfunctional mutant channel. We previously demonstrated that heterologously expressed R1648C channels exhibit significantly increased persistent sodium current when compared with WT channels (10). Therefore, we examined whether persistent current density increased when cells expressing R1648C were treated with either phenytoin or lamotrigine. Fig. 7 illustrates that cells exposed to either of these antiepileptic drugs exhibited significantly larger persistent current density than vehicle-treated controls. Further, lamotrigine treatment also increased the proportional level of persistent current when normalized to peak current (1.5 ± 0.3% versus 0.9 ± 0.1% for DMSO and 0.9 ± 0.1 for phenytoin). These findings suggest that pharmacological rescue of trafficking-impaired NaV1.1 mutants may carry the liability of increasing functional expression of dysfunctional channel behaviors.
FIGURE 7.

Lamotrigine treatment increases persistent current density of R1648C. A, average current traces of cells stably expressing R1648C grown in the presence of 0.1% DMSO (black trace), 50 μm phenytoin (blue trace), or 50 μm lamotrigine (red trace). Whole-cell currents were normalized to cell size and peak current density of the DMSO control. B, persistent current density of R1648C stably expressing cells grown in the presence of 0.1% DMSO, 50 μm phenytoin (Phe), or 50 μm lamotrigine (Lam). Data are expressed as mean ± S.E. for 9–17 replicates. An asterisk indicates p < 0.05 when compared with DMSO-treated cells. pF, picofarads.
DISCUSSION
Mutations in genes encoding brain NaV channels are associated with a spectrum of epilepsy syndromes, and investigations into the molecular pathogenesis of these disorders may help inspire new therapeutic strategies. The profound allelic heterogeneity reported for SCN1A mutations associated with SMEI and related conditions emphasizes the complexity of genotype-phenotype correlations among these disorders that may reflect diverse underlying disease mechanisms. In this study, we sought to explore the contribution of impaired cell surface expression to the pathophysiology of SMEI and to determine whether pharmacological strategies could reverse this type of molecular defect in the context of NaV1.1 channels.
We demonstrated that seven nontruncating SCN1A mutations exhibit variable degrees of reduced cell surface expression (Fig. 1), and this finding is consistent with a loss of NaV channel function, the most likely molecular mechanism to explain SMEI (1). Missense mutations in integral membrane proteins can disrupt intrinsic protein folding and assembly that lead to abnormal protein trafficking or degradation during biogenesis (25). In our experiments, we observed reduced total protein expression for more than half of the tested mutants, suggesting that protein degradation may be evoked by certain mutations. Other alleles that exhibited more robust total protein expression have reduced cell surface expression presumably because the mutant protein gets trapped between the endoplasmic reticulum and plasma membrane or is prematurely removed from the plasma membrane following normal insertion. However, for most of the mutations we studied, this impairment in reaching the cell surface is not complete, raising the possibility that the defect is partial and possibly remediable.
We used a combination of biochemical and electrophysiological approaches to test the ability of three compounds to rescue cell surface expression of two representative SMEI mutations. For these experiments, we implemented stable cell lines expressing WT-NaV1.1 or mutant channels and thus avoided the highly variable cell-to-cell and transfection-to-transfection nature of transient expression. Our results demonstrated that R1648C, a mutant with partial impairment of cell surface expression, could be rescued biochemically and electrophysiologically using the antiepileptic drugs phenytoin and lamotrigine. However, the mutant G1674R exhibits minimal cell surface expression and could only be biochemically rescued by phenytoin but without a positive electrophysiological response. We interpret these data to indicate that multiple mechanisms lead to trafficking-impaired SMEI mutations and that functional rescue must be investigated in parallel. Indeed, a SEMI-associated SCN1A mutation (M1841T) has been previously shown amenable to pharmacological rescue of impaired functional expression, but this previous study did not directly investigate cell surface expression, creating uncertainty about the mechanism underlying this phenomenon (26).
The clinical utility of rescuing trafficking-impaired mutant ion channels is under evaluation for CFTR in cystic fibrosis (27), but we are uncertain as to the prospects for this therapeutic strategy in SMEI. Conceivably, treatment with agents that restore normal or near normal cell surface expression of mutant NaV1.1 channels or boost expression of WT-NaV1.1 would offer therapeutic benefit. However, rescuing impaired cell surface expression with antiepileptic agents such as phenytoin and lamotrigine might be limited by their sodium channel-blocking activity based on anecdotal reports that such agents may exacerbate epilepsy in SMEI. For these reasons, we attempted to rescue cell surface expression of R1648C and G1674R using VRT-325, a somewhat promiscuous corrector of trafficking-deficient mutant CFTR and hERG channels (14, 17). Unfortunately, VRT-325 showed no activity against the mutant NaV1.1 channels we tested.
Even if normal cell surface expression could be restored for mutant NaV1.1 channels, there is uncertainty as to the pathophysiological impact of intrinsic channel defects such as incomplete inactivation leading to increased persistent sodium current. As we demonstrated in Fig. 7, rescuing cell surface expression by phenytoin and lamotrigine also promoted greater persistent current density in cells expressing R1648C channels. Most likely, sodium channel-blocking drugs capable of rescuing surface expression would also be capable of suppressing this abnormal sodium current. However, this raises concern that restoring dysfunctional channels to the plasma membrane by an agent without sodium channel-blocking activity could exacerbate the disease.
Acknowledgment
We are grateful to Dr. Carlos Vanoye for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant NS032387 (to A. L. G.).
- NaV
- voltage-gated sodium
- SMEI
- severe myoclonic epilepsy of infancy
- CFTR
- cystic fibrosis transmembrane conductance regulator
- hERG
- human ether-a-gogo-related gene
- VRT-325
- 4-cyclohexyloxy-2-{1-[4-(4-methoxy-benzenesulfonyl)-piperazin-1-yl]-ethyl}-quinazoline
- DMSO
- dimethyl sulfoxide
- RIPA
- radioimmune precipitation assay.
REFERENCES
- 1. Catterall W. A., Kalume F., Oakley J. C. (2010) NaV1.1 channels and epilepsy. J. Physiol. 588, 1849–1859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. George A. L., Jr. (2005) Inherited disorders of voltage-gated sodium channels. J. Clin. Invest. 115, 1990–1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Isom L. L. (2001) Sodium channel β subunits: anything but auxiliary. Neuroscientist 7, 42–54 [DOI] [PubMed] [Google Scholar]
- 4. Meisler M. H., O'Brien J. E., Sharkey L. M. (2010) Sodium channel gene family: epilepsy mutations, gene interactions, and modifier effects. J. Physiol. 588, 1841–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Meisler M. H., Kearney J. A. (2005) Sodium channel mutations in epilepsy and other neurological disorders. J. Clin. Invest. 115, 2010–2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mulley J. C., Scheffer I. E., Petrou S., Dibbens L. M., Berkovic S. F., Harkin L. A. (2005) SCN1A mutations and epilepsy. Hum. Mutat. 25, 535–542 [DOI] [PubMed] [Google Scholar]
- 7. Lossin C. (2009) A catalog of SCN1A variants. Brain Dev. 31, 114–130 [DOI] [PubMed] [Google Scholar]
- 8. Yu F. H., Mantegazza M., Westenbroek R. E., Robbins C. A., Kalume F., Burton K. A., Spain W. J., McKnight G. S., Scheuer T., Catterall W. A. (2006) Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 9, 1142–1149 [DOI] [PubMed] [Google Scholar]
- 9. Ogiwara I., Miyamoto H., Morita N., Atapour N., Mazaki E., Inoue I., Takeuchi T., Itohara S., Yanagawa Y., Obata K., Furuichi T., Hensch T. K., Yamakawa K. (2007) Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J. Neurosci. 27, 5903–5914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rhodes T. H., Lossin C., Vanoye C. G., Wang D. W., George A. L., Jr. (2004) Noninactivating voltage-gated sodium channels in severe myoclonic epilepsy of infancy. Proc. Natl. Acad. Sci. U.S.A. 101, 11147–11152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohmori I., Kahlig K. M., Rhodes T. H., Wang D. W., George A. L., Jr. (2006) Nonfunctional SCN1A is common in severe myoclonic epilepsy of infancy. Epilepsia 47, 1636–1642 [DOI] [PubMed] [Google Scholar]
- 12. Rhodes T. H., Vanoye C. G., Ohmori I., Ogiwara I., Yamakawa K., George A. L., Jr. (2005) Sodium channel dysfunction in intractable childhood epilepsy with generalized tonic-clonic seizures. J. Physiol. 569, 433–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. (1990) Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell 63, 827–834 [DOI] [PubMed] [Google Scholar]
- 14. Van Goor F., Straley K. S., Cao D., González J., Hadida S., Hazlewood A., Joubran J., Knapp T., Makings L. R., Miller M., Neuberger T., Olson E., Panchenko V., Rader J., Singh A., Stack J. H., Tung R., Grootenhuis P. D., Negulescu P. (2006) Rescue of ΔF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell. Mol. Physiol. 290, L1117–L1130 [DOI] [PubMed] [Google Scholar]
- 15. Zhou Z., Gong Q., January C. T. (1999) Correction of defective protein trafficking of a mutant HERG potassium channel in human long QT syndrome: pharmacological and temperature effects. J. Biol. Chem. 274, 31123–31126 [DOI] [PubMed] [Google Scholar]
- 16. Ficker E., Obejero-Paz C. A., Zhao S., Brown A. M. (2002) The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether-a-gogo-related gene (HERG) mutations. J. Biol. Chem. 277, 4989–4998 [DOI] [PubMed] [Google Scholar]
- 17. Van Goor F., Hadida S., Grootenhuis P. D., Burton B., Stack J. H., Straley K. S., Decker C. J., Miller M., McCartney J., Olson E. R., Wine J. J., Frizzell R. A., Ashlock M., Negulescu P. A. (2011) Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. U.S.A. 108, 18843–18848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lossin C., Wang D. W., Rhodes T. H., Vanoye C. G., George A. L., Jr. (2002) Molecular basis of an inherited epilepsy. Neuron 34, 877–884 [DOI] [PubMed] [Google Scholar]
- 19. Thompson C. H., Kahlig K. M., George A. L., Jr. (2011) SCN1A splice variants exhibit divergent sensitivity to commonly used antiepileptic drugs. Epilepsia 52, 1000–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kahlig K. M., Rhodes T. H., Pusch M., Freilinger T., Pereira-Monteiro J. M., Ferrari M. D., van den Maagdenberg A. M., Dichgans M., George A. L., Jr. (2008) Divergent sodium channel defects in familial hemiplegic migraine. Proc. Natl. Acad. Sci. U.S.A. 105, 9799–9804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kahlig K. M., Saridey S. K., Kaja A., Daniels M. A., George A. L., Jr., Wilson M. H. (2010) Multiplexed transposon-mediated stable gene transfer in human cells. Proc. Natl. Acad. Sci. U.S.A. 107, 1343–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Volkers L., Kahlig K. M., Verbeek N. E., Das J. H., van Kempen M. J., Stroink H., Augustijn P., van Nieuwenhuizen O., Lindhout D., George A. L., Jr., Koeleman B. P., Rook M. B. (2011) Nav1.1 dysfunction in genetic epilepsy with febrile seizures-plus or Dravet syndrome. Eur. J. Neurosci. 34, 1268–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Guerrini R., Dravet C., Genton P., Belmonte A., Kaminska A., Dulac O. (1998) Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia 39, 508–512 [DOI] [PubMed] [Google Scholar]
- 24. Guerrini R., Belmonte A., Genton P. (1998) Antiepileptic drug-induced worsening of seizures in children. Epilepsia 39, Suppl. 3, S2–10 [DOI] [PubMed] [Google Scholar]
- 25. Sanders C. R., Myers J. K. (2004) Disease-related misassembly of membrane proteins. Annu. Rev. Biophys. Biomol. Struct. 33, 25–51 [DOI] [PubMed] [Google Scholar]
- 26. Rusconi R., Scalmani P., Cassulini R. R., Giunti G., Gambardella A., Franceschetti S., Annesi G., Wanke E., Mantegazza M. (2007) Modulatory proteins can rescue a trafficking defective epileptogenic Nav1.1 Na+ channel mutant. J. Neurosci. 27, 11037–11046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Becq F., Mall M. A., Sheppard D. N., Conese M., Zegarra-Moran O. (2011) Pharmacological therapy for cystic fibrosis: from bench to bedside. J. Cyst. Fibros. 10, Suppl. 2, S129–S145 [DOI] [PubMed] [Google Scholar]