

Background: Mitochondrial respiration plays an important role in alcohol metabolism by regenerating NAD+ needed for alcohol/acetaldehyde metabolism.
Results: Chronic alcohol feeding caused many mitochondrial alterations, such as increased mitochondrial respiration, that enhanced acetaldehyde metabolism.
Conclusion: Mitochondria in the liver adapt to the metabolic stress of alcohol.
Significance: Mitochondrial alterations may play a role in many vital functions of the liver.
Keywords: Alcohol, Liver Injury, Liver Metabolism, Mitochondria, Mitochondrial Metabolism
Abstract
Liver mitochondria undergo dynamic alterations following chronic alcohol feeding to mice. Intragastric alcohol feeding to mice resulted in 1) increased state III respiration (109% compared with control) in isolated liver mitochondria, probably due to increased levels of complexes I, IV, and V being incorporated into the respiratory chain; 2) increased mitochondrial NAD+ and NADH levels (∼2-fold), with no change in the redox status; 3) alteration in mitochondrial morphology, with increased numbers of elongated mitochondria; and 4) enhanced mitochondrial biogenesis in the liver, which corresponded with an up-regulation of PGC-1α (peroxisome proliferator-activated receptor γ coactivator-1α). Oral alcohol feeding to mice, which is associated with less liver injury and steatosis, slightly enhanced respiration in isolated liver mitochondria (30.8% compared with control), lower than the striking increase caused by intragastric alcohol feeding. Mitochondrial respiration increased with both oral and intragastric alcohol feeding despite extensive N-acetylation of mitochondrial proteins. The alcohol-induced mitochondrial alterations are probably an adaptive response to enhance alcohol metabolism in the liver. Isolated liver mitochondria from alcohol-treated mice had a greater rate of acetaldehyde metabolism and respiration when treated with acetaldehyde than control. Aldehyde dehydrogenase-2 levels were unaltered in response to alcohol, suggesting that the greater acetaldehyde metabolism by isolated mitochondria from alcohol-treated mice was due to increased mitochondrial respiration that regenerated NAD+, the rate-limiting substrate in alcohol/acetaldehyde metabolism. Overall, our work suggests that mitochondrial plasticity in the liver may be an important adaptive response to the metabolic stress caused by alcohol intake and could potentially play a role in many other vital functions performed by the liver.
Introduction
Mitochondria are very dynamic and adaptive organelles that alter in response to stress and metabolic changes. In skeletal muscle, it has been shown that the energy demands of acute or chronic exercise causes mitochondrial biogenesis and remodeling (i.e. increased components of the mitochondrial respiratory chain and proteins involved in β-oxidation) (1–3). Alteration in metabolic fuels, such as increased fatty acid intake, also increases mitochondrial biogenesis and β-oxidation capacity in muscle cells (4). Mitochondrial biogenesis and remodeling in most cells is mediated through PGC-1α (peroxisome proliferator-activated receptor γ coactivator-1α), the master regulator of mitochondria. PGC-1α knock-out mice have decreased levels of many key mitochondrial proteins (i.e. cytochrome c, ATP synthase, and cytochrome oxidase) (5, 6), whereas overexpression of PGC-1α promotes mitochondrial biogenesis (7). “Mitochondrial plasticity” has been used to describe the dynamic and adaptive nature of mitochondria in muscle cells (8, 9). A different type of dynamic adaptation in mitochondria involves morphological changes due to alteration in mitochondrial fusion-fission rates. Mitochondria constantly undergo fusion and fission to exchange mitochondrial DNA, proteins, and other constituents (10, 11). Stress can dramatically alter fusion-fission rates and consequently mitochondrial morphology. In embryonic fibroblasts, starvation causes a decrease in mitochondrial fission to produce elongated mitochondria that are more resistant to mitophagy (mitochondrial autophagy) and have a greater cristae surface area and thereby greater mitochondrial respiration (12). Mitochondrial plasticity has been characterized in muscle and cell lines, but has not received much attention in other organs, such as the liver. As the major organ that regulates metabolism in the body, mitochondrial plasticity may be an important adaptive phenomenon in the liver in response to stress and metabolic changes.
The liver is the major organ responsible for metabolism of alcohol, with mitochondria playing a central role in the process. Alcohol is metabolized primarily by two enzymes in the liver, alcohol dehydrogenase (ADH)2 in the cytoplasm and aldehyde dehydrogenase 2 (ALDH2) in mitochondria, respectively (Reactions 1 and 2).
 |
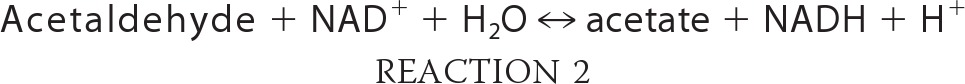 |
Both ADH and ALDH2 are kinetically limited by NAD+, with the levels of NAD+ being dependent on mitochondrial respiration, which oxidizes NADH to NAD+ (13–15). The liver adapts to alcohol and develops an enhanced capacity to metabolize alcohol following feeding (13, 16, 17). This enhanced capacity to metabolize alcohol is associated with increased oxygen uptake by the liver (13, 18, 19), suggesting that there is greater mitochondrial respiration in these hepatocytes. ADH and ALDH2 levels do not change with alcohol feeding, suggesting that adaptation to alcohol occurs through mitochondrial alterations or other pathways (15–17). Could mitochondria in the liver undergo plasticity changes to help enhance alcohol metabolism? Some studies of alcohol feeding in mice have hinted that mitochondrial alterations may occur as the liver adapts to alcohol. One study showed some enhancement of mitochondrial respiration in the liver of mice fed alcohol orally (20), whereas another study found that alcohol feeding to mice increases complex I levels in liver mitochondria (21). These studies suggest that chronic alcohol feeding to mice may trigger mitochondrial alterations as an adaptive response, which needs to be further explored.
Most of the effects of alcohol on mitochondrial respiration were described in rats fed the Lieber-DeCarli Diet. Oral alcohol feeding to rats results in a decline in mitochondrial respiration (state III) and the respiratory control ratio (RCR; defined as state III/state IV) in isolated liver mitochondria (22–24). The decline in mitochondrial respiration is accompanied by a decline in respiratory complex proteins in the liver of rats fed alcohol orally (25, 26). This suggests that alcohol feeding to mice and rats may induce different types of mitochondrial alterations in the liver. The species difference in response to alcohol is not completely surprising, because rats and mice differ in their sensitivity to alcohol, with mice being more susceptible to alcohol-induced liver injury than rats. In addition, our laboratory has shown differences in alcohol-induced ER stress response between rats and mice, due to the differential expression of betaine homocysteine methyltransferase (27). In addition to species differences, we must also consider that mitochondrial changes in the liver may also be affected by the doses of alcohol administered. Mice and rats do not consume high amounts of alcohol. Oral alcohol administration (Lieber-DeCarli oral diet) to mice and rats therefore only produces mild liver injury and fatty liver (steatosis), pathology associated with mild or moderate alcohol intake in humans (28). The intragastric alcohol infusion model, on the other hand, infuses high doses of alcohol into the stomach of rats and mice and therefore produces more severe liver injury with pathophysiologic features, such as fibrosis and inflammation, that are observed in human alcoholics (29, 30). To our knowledge, there have been no studies performed investigating changes to liver mitochondria following intragastric alcohol feeding in any animal models.
In this study, we investigated the effects of alcohol, administered both orally and intragastrically, on mitochondrial respiration in mice. We focused on the mouse model, where mitochondrial alterations following oral or intragastric alcohol feedings have not been extensively explored. The hypothesis that liver mitochondria respond to the increased demands of alcohol metabolism by undergoing adaptive plastic changes was examined in this work.
EXPERIMENTAL PROCEDURES
Animals
Male C57BL/6J mice (8 weeks of age) were obtained from Jackson Laboratory (Bar Harbor, ME), whereas Wistar rats (150 g) were obtained from Charles River (Wilmington, MA). The animals were housed in a temperature-controlled room and were acclimatized for a minimum of 3 days prior to use in experiments. Blood was obtained after mice were anesthetized at the indicated time periods, and serum alanine transaminase (ALT) was measured at the University of Southern California Pathology Reference Laboratory. All animals received care according to the methods approved under institutional guidelines for the care and use of laboratory animals in research.
Oral Alcohol Feeding
Mice and rats were fed a commercially available liquid diet (Bioserve), to which ethanol was added to yield a 5.4% (w/v) dose. Pair feeding was done to a control animal by feeding an amount equal to that consumed by ethanol-fed mice and rats, except that ethanol was isocalorically replaced with dextrin.
Intragastric Alcohol Feeding
Intragastric alcohol-fed mice and control mice were provided by the Southern California Research Center for Alcoholic Liver and Pancreatic Diseases and Cirrhosis. Male C57 BL/6 mice (8 weeks) were implanted with a long term gastrostomy catheter for alcohol infusion as described previously (29, 30). Briefly, after 1 week of acclimatization, mice were infused with a controlled high fat diet with or without alcohol. Alcohol infusion was initiated at a dose of 22.7 g/kg/day and gradually increased. Alcohol accounted for ∼32.9% of the total caloric intake after 1 week of intragastric alcohol feeding. By 4 weeks of intragastric alcohol feeding, alcohol accounted for ∼38.4% of the total caloric intake.
Mitochondrial Isolation and Respiration Measurements
Isolation of Liver Mitochondria
Liver mitochondria from oral fed mice and rats were isolated using differential centrifugation as described previously (31). Liver mitochondria from intragastric mice were isolated by differential centrifugation or Percoll discontinuous gradient as described previously (32). No differences in mitochondrial respiration were observed between the two methods.
Differential Centrifugation
Livers were excised, washed with 0.25 m sucrose, and homogenized in H-medium (210 mm mannitol, 70 mm sucrose, 2 mm HEPES, 0.05% bovine serum albumin (w/v), plus protease and phosphatase inhibitors). The homogenate was centrifuged at 850 × g for 10 min, the pellet was removed, and the centrifugation process was repeated. The resulting supernatant was centrifuged at 8,500 × g for 15 min. The supernatant (“cytoplasmic fraction”; post-mitochondrial S9 fraction) was collected and saved at −80° C for future analysis. The pellet, which represents the mitochondrial fraction, was washed with H-medium, and the centrifugation was repeated. The mitochondria were resuspended in H-medium without BSA before oxygen electrode and Western blot analyses.
Discontinuous Percoll Gradient
Livers were excised, washed, and homogenized in isolation buffer (H-medium) using a loose Teflon pestle. The homogenate was centrifuged at 1000 × g for 10 min at 4 °C, the pellet was removed, and the centrifugation process was repeated. The resulting supernatant was centrifuged at 9,000 × g for 15 min to generate the mitochondrial pellet. The mitochondrial pellet was dissolved in isolation buffer containing 18% Percoll and centrifuged at 10,000 × g for 10 min. The mitochondrial pellet was gently removed from the Percoll solution and layered on top of three discontinuous Percoll gradient tubes (18, 30, and 60%). The Percoll gradient was spun at 30,700 × g for 5 min at 4 °C. The mitochondrial layer, which resides in the interface between 60 and 30% Percoll was carefully removed using a pipette and suspended in isolation buffer. To remove the Percoll, mitochondria were spun at 10,000 × g for 10 min and washed, and the process was repeated twice. Mitochondria were suspended in isolation buffer (without BSA) before respiration measurements. Immunoblotting of isolated liver mitochondria showed minimal cytoplasmic contamination (actin), enrichment of complex IV, and some ER contamination (calnexin) (data not shown). Some of the ER is attached to mitochondria, so ER contamination always occurs to some degree with isolation of mitochondria.
Measurements of Respiration in Isolated Mitochondria
Respiration was measured in freshly isolated mitochondria by monitoring oxygen consumption with a Clark-type electrode (Hanstech) in respiration buffer containing 230 mm mannitol, 70 mm sucrose, 30 mm Tris-HCl, 5 mm KH2PO4, 1 mm EDTA, pH 7.4 (33). Isolated mitochondria (0.50–0.70 mg) were added to 1 ml of respiration buffer, and oxygen consumption was monitored in the presence of complex I substrates (glutamate/malate 7.5 mm) or complex II substrate (succinate 7.5 mm) with or without ADP (250 μm). In some experiments, acetaldehyde (125–375 μm) was used as a substrate (complex I) for mitochondrial respiration measurements (15).
Electron Microscopy and Histology
Electron Microscopy
Small pieces of freshly isolated liver (< 2 mm3) from control and alcohol-treated mice were immersed in 2.5% glutaraldehyde in 0.1 m sodium cacodylate, pH 7.4, and stored at 4 °C for 1–3 weeks. Tissues were then washed in 0.1 m sodium cacodylate, post-fixed with 1% OsO4 in 0.1 m sodium cacodylate for 1 h, stained en bloc in 3% uranyl acetate in 0.1 m sodium acetate buffer, dehydrated through a series of ethanol washes, and infiltrated and embedded in Spurr's plastic. Thick sections were stained with methylene blue, examined with a Zeiss Labrolux brightfield microscope, and photographed using a Spot Insight digital camera. Hepatocyte areas were measured using Vistametrix software. For transmission electron microscopy, thin sections (70 nm) of the same blocks were examined by LM stained with lead citrate and examined with a Zeiss EM109 transmission electron microscope.
Measurement of Mitochondrial Major and Minor Axes and Mitochondrial Density in Electron Microscopy Scans
Mitochondrial major and minor axes and density in vivo were measured using the ImageJ program from the National Institutes of Health. In hepatocytes, the major and minor axes in each mitochondrion (50–100 mitochondria in 5–7 different fields) were measured using the measurement function of ImageJ. For mitochondrial density measurements, each mitochondrion and the total cytoplasmic area (nucleus area was subtracted) were traced by hand to estimate the percentage of the cytoplasm that mitochondria occupied (10 hepatocytes/alcohol and control mice).
Estimation of Hepatocyte Size
Hepatocyte size was estimated in H&E-stained liver sections using ImageJ. The area for each hepatocyte was traced by hand and calculated in millimeters using a ruler as reference. Measurements were made in five random slide sections (∼40–50 hepatocytes counted in each slide) in alcohol and control mice (n = 4–5 mice).
Immunoblotting and Biochemical Assays
Immunoblotting
Aliquots of cytoplasmic or mitochondrial extracts were fractionated by electrophoresis on 8–12% SDS-polyacrylamide gels (Bio-Rad). Subsequently, proteins were transferred to nitrocellulose or PVDF membranes, and blots were blocked with 5% (w/v) nonfat milk dissolved in Tris-buffered saline (TBS) with Tween 20. Complex I (NDUFS3 subunit), II (SDHA subunit), complex III (subunit 1), V (a subunit), cytochrome c, and medium chain acyl-CoA dehydrogenase (MCAD) antibodies were obtained from Mitosciences (Eugene, OR). Complex IV, N-acetylation, actin, and prohibitin-1 antibodies were obtained from Cell Signaling Technology (Danvers, MA). Antibodies to aldehyde dehydrogenase 2 and PGC-1α were obtained from Abcam (Cambridge, MA). The antibody to glutamate dehydrogenase was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), and TFAM antibody was obtained from Avia Systems Biology (San Diego, CA). All blots shown are representative samples from 3–7 experiments. Densitometry was performed using ImageJ and normalized for complex III for mitochondrial proteins and actin for cytoplasmic proteins.
Acetaldehyde Metabolism
Acetaldehyde metabolism was measured by using an NADH-coupled assay. Isolated liver mitochondria (500 mg) from intragastric alcohol-fed and control mice (2 weeks of treatment) were treated with acetaldehyde (188 or 250 mm) plus ADP (250 mm). After 5 min of incubation, mitochondria were centrifuged (10,000 × g for 5 min at 4 °C), and the supernatant containing acetaldehyde was immediately frozen. Acetaldehyde measurements were made in buffer (50 mm sodium pyrophosphate, pH 8.5) plus NAD+ (500 mm) and ALDH 2 (5 units) (Sigma). Acetaldehyde levels in the supernatant were measured by following NADH production at 340 nm.
NAD+-NADH Measurements
NAD+ and NADH levels were measured by HPLC as described previously (34) in intragastric (2 weeks) and oral alcohol-fed (5 weeks) mice. Briefly, liver homogenate and isolated mitochondria were homogenized in buffer (0.06 m KOH, 0.2 m KCN, and 1 mm bathophenanthroline disulfonic acid), followed by chloroform extraction. Chloroform extraction was carried out by centrifugation at 14,000 rpm in a microcentrifuge at 4 °C; the resulting aqueous supernatant with soluble pyridine nucleotides was collected and extracted three times to remove lipids and proteins. Finally, it was filtered with a 0.45-μm positively charged filter (Pall Life Sciences) to remove RNA and DNA in a microcentrifuge at 4 °C. The mobile phase consisted of 0.2 m ammonium acetate (buffer A) at pH 5.5 and HPLC-grade methanol (buffer B). A gradient program with initial conditions (100% buffer A, 0% buffer B) was adjusted as follows: 1) buffer B was increased from 0 to 3% during 0–4 minutes of the run; 2) buffer B was further increased from 3 to 6.8% during 4–23 minutes of the run; and 3) at the end of the run the column was re-equilibrated. Quantitation of pyridine nucleotides was performed by integrating the peaks and adding the cyanide adducts, as detected by the fluorescence spectrophotometer (λex = 330 nm; λem = 460 nm).
Statistical Analyses
Statistical analyses were performed using Student's t test for unpaired data or analysis of variance for comparison of multiple groups. p < 0.05 was defined as statistically significant.
RESULTS
Effects of Chronic Alcohol Feeding on Mitochondrial Respiration in Mice and Rats
Mitochondrial respiration was measured in isolated liver mitochondria following alcohol feeding to mice and rats. The feeding of alcohol to mice, both orally and intragastrically, caused increased state III respiration (respiration in the presence of substrates and ADP) in isolated liver mitochondria (Fig. 1). Mice fed alcohol orally (Lieber-DeCarli diet) had a statistically significant increase in state III respiration with complex I substrates (glutamate/malate (29.8%)) or complex II substrate (succinate (30.8%)) treatment of isolated liver mitochondria (Fig. 1, A–C). State IV respiration (substrate without ADP) and the RCR (state III/state IV) were not significantly altered. The increase in state III respiration in isolated liver mitochondria was more pronounced when alcohol was given intragastrically to mice for 4 weeks (Fig. 1, D–F). Mice fed alcohol intragastrically had a statistically significant increase in state III respiration with glutamate/malate (85.3%) and succinate (86.9%) compared with control mitochondria. State IV respiration also increased, whereas the RCR did not significantly change. The intragastric feeding of alcohol caused greater hepatomegaly (increased size), as shown by increased liver weight, and liver/body ratio (8.75% for intragastric alcohol fed mice, 5.78% for oral alcohol fed mice, 3.78% for oral rats), accompanied by greater liver injury, manifested as increased serum ALT (Table 1). In contrast to mice, oral alcohol feeding of rats (Lieber-DeCarli diet; 6 weeks) caused a decline in state III respiration; a 26.7% decline in respiration using glutamate/malate and a 42.1% decline in respiration using succinate occurred compared with control rats (data not shown), in close agreement with previously published results (22–24). These data suggest that the mitochondrial response to alcohol in rats and mice is dramatically different. Interestingly, mitochondrial state III respiration was the highest in animals with the greatest amount of liver injury (ALT levels).
FIGURE 1.

Isolated liver mitochondria from oral alcohol and intragastric fed mice have a greater respiration than control. A, state III respiration in oral alcohol fed mice. B, state IV respiration in oral alcohol-fed mice. C, RCR in oral alcohol-fed mice. D, state III respiration in intragastric alcohol-fed mice. E, state IV respiration in intragastric alcohol-fed mice. F, RCR in intragastric alcohol-fed mice. Solid bars, control; empty bars, alcohol treatment. State III respiration was measured using either complex I substrates (glutamate/malate, 7.5 mm) or complex II substrate (succinate, 7.5 mm) plus ADP (250 μm) with an oxygen electrode. State IV respiration was measured in the presence of substrates but no ADP. RCR is defined as the state III respiration/state IV respiration ratio. Following 5 weeks of oral alcohol feeding or 4 weeks of intragastric alcohol feeding, liver mitochondria were isolated using differential centrifugation or a Percoll gradient as described under “Experimental Procedures.” n = 5–8 mice/group. Results are mean ± S.D. (error bars); *, p < 0.05 versus control.
TABLE 1.
Effects of alcohol feeding on rats and mice
n = 4–8 mice/group. Results are mean ± S.D.
| Oral alcohol rats | Oral alcohol mice | Intragastric mice | |
|---|---|---|---|
| Final body weight (g) | |||
| Control | 426 ± 4 | 32.0 ± 3.7 | 22.5 ± 0.55 |
| Alcohol | 375 ± 40 | 30.2 ± 1.3 | 21.5 ± 0.35 |
| Liver weight (g) | |||
| Control | 13.0 ± 1.6 | 1.61 ± 0.29 | 1.20 ± 0.15 |
| Alcohol | 14.1 ± 1.1 | 1.74 ± 0.08 | 1.88 ± 0.12a |
| Liver/body weight (%) | |||
| Control | 3.06 ± 0.36 | 5.01 ± 0.38 | 5.33 ± 0.58 |
| Alcohol | 3.78 ± 0.32a | 5.78 ± 0.20a | 8.75 ± 0.52a |
| ALT (units/liter) | |||
| Control | 30.0 ± 11.5 | 56.7 ± 10.0 | 17.9 ± 4.2 |
| Alcohol | 47.5 ± 6.19a | 70.5 ± 21.6 | 223 ± 108a |
a p < 0.05 versus control.
Time Course of Mitochondrial and Liver Changes following Intragastric Alcohol Feeding to Mice
A time course of liver injury and mitochondrial respiratory changes were examined in mice following intragastric alcohol feeding. Fig. 2A shows that an increase in state III respiration in isolated liver mitochondria, using either glutamate/malate or succinate as substrate, occurred even after only 1 week of intragastric alcohol feeding. State III respiration in isolated liver mitochondria following intragastric alcohol feeding appeared to peak at 2 weeks, with state III respiration from alcohol-treated samples being 109% (glutamate/malate) and 97.4% (succinate) increased versus control mice. State IV respiration and RCR also peaked at 2 weeks following intragastric alcohol feeding (data not shown). Increases in liver size also began early, with liver/body ratios peaking after 2 weeks of intragastric alcohol treatment (Fig. 2B). Serum ALT levels began to increase after 1 week of intragastric alcohol feeding and increased with time (Fig. 2C). These data suggest that an increase in mitochondrial respiration is an early event in the liver following intragastric alcohol feeding.
FIGURE 2.
Time course of liver injury and mitochondrial changes following intragastric alcohol feeding to mice. A, state III respiration with complex I or II substrates. Solid lines, glutamate/malate treatment; dashed lines, succinate treatment. B, liver/body ratio; C, serum ALT levels (units/liter (international units/liter)). ●, control; □, intragastric alcohol feeding. Mice were fed alcohol intragastrically for 1, 2, or 4 weeks, and liver mitochondria was isolated using a Percoll gradient as described under “Experimental Procedures.” State III respiration was measured using either complex I substrates (glutamate/malate, 7.5 mm) or complex II substrate (succinate, 7.5 mm) plus ADP (250 μm) with an oxygen electrode. n = 4–8 mice/group. Results are mean ± S.D. (error bars); *, p < 0.05 versus control.
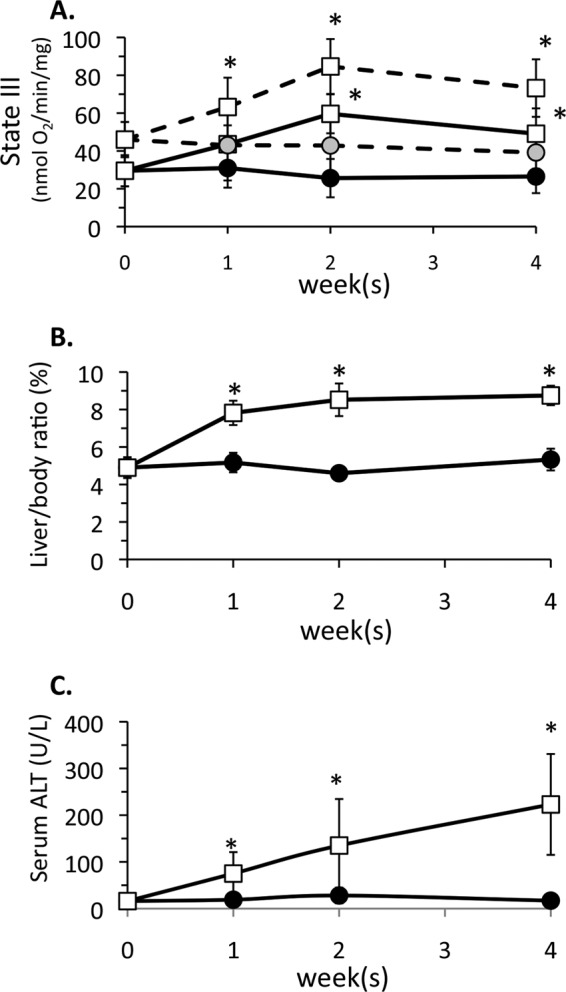
Alcohol Feeding Alters Respiratory Complex Protein Levels in Liver Mitochondria
We next investigated the mechanism(s) underlying the increase in mitochondrial respiration in mouse liver induced by intragastric and oral alcohol feeding. The protein levels of respiratory complexes in the electron transport chain play an important role in determining the respiration rate of mitochondria. The levels of complex I (NDUFS3 subunit), complex II (SHDA subunit), complex III (subunit 1), complex IV (subunit IV), and complex V (a subunit), all essential subunits necessary for function of the respiratory complexes, were examined. Levels of complex I, IV, and V increased after 2 weeks of intragastric alcohol feeding, when state III respiration is maximal (Fig. 3). Blue native-stained gels served as loading controls (data not shown). Levels of complex III showed no change in response to alcohol feeding; therefore, it was used as the reference in all densitometry measurements. The levels of complexes I and V significantly increased after 2 weeks of intragastric alcohol feeding, whereas complex IV showed a notable increase even after only 1 week of alcohol feeding. Levels of complex II also trended toward an increase at all time points, but the increase was not statistically significant. The levels of glutamate dehydrogenase, a mitochondrial matrix protein important in forming NADH using glutamate as a substrate, showed no change following intragastric alcohol feeding. Overall, the selective up-regulation of complexes I, IV, and V in the respiratory chain may contribute to the observed increase in mitochondrial respiration in the liver of intragastric alcohol-fed mice. In oral alcohol-fed mice, where mitochondrial respiratory increases were modest, levels of only complex I were enhanced (Fig. 3).
FIGURE 3.
Alcohol feeding increases protein levels of respiratory complexes in the electron transport chain of liver mitochondria. Solid bars, control; empty bars, alcohol treatment. Mice were fed alcohol intragastrically (1–4 weeks) or orally (5 weeks), and liver mitochondria were isolated using a Percoll gradient or differential centrifugation. Mitochondrial protein levels were assessed by immunoblotting. Densitometry was performed using ImageJ. Complex III was used as the loading control after observing that it was not changed with alcohol feeding. n = 4–7 mice/group. Results are mean ± S.D. (error bars); *, p < 0.05 versus control.
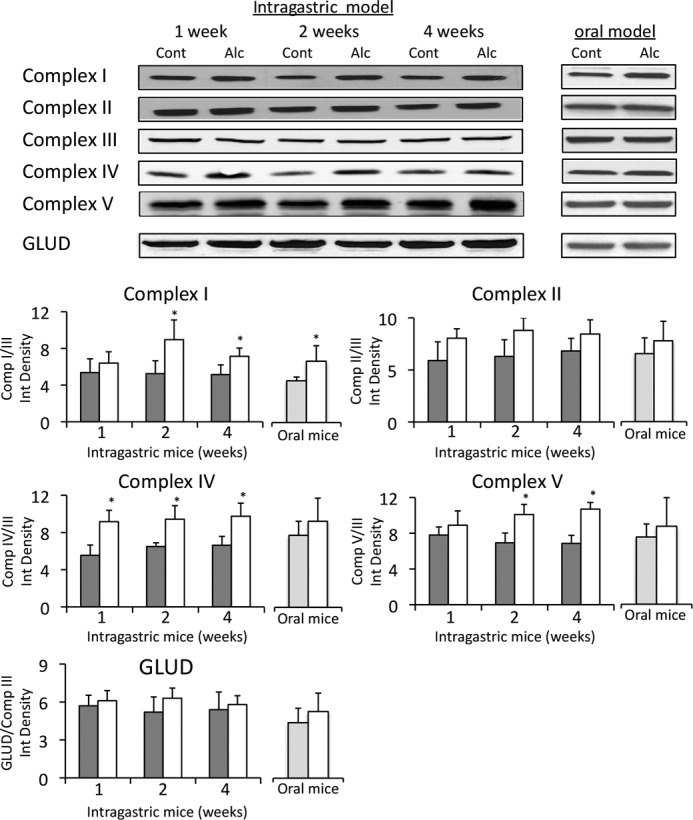
Effect of Alcohol Feeding on NAD+-NADH Levels and Redox Status in Liver Homogenate and Mitochondria
Because NAD+ is the rate-limiting substrate in alcohol metabolism, NAD+-NADH levels and redox status were examined in liver homogenate and mitochondria following alcohol feeding, both oral and intragastric. Intragastric alcohol feeding did not alter NAD+, NADH, total NAD+-NADH levels, or the NAD+/NADH ratio in the liver homogenate (Fig. 4A). However, in isolated mitochondria from intragastric alcohol-fed mice, NAD+ and total NAD+-NADH levels (nmol/mg mitochondrial protein) were increased more than 2-fold. The NAD+/NADH ratio was not significantly altered in liver mitochondria of mice fed alcohol intragastrically. Similarly, mice fed alcohol orally had a significant increase in NAD+ and total NAD+-NADH levels in isolated mitochondria, but not in the liver homogenate (Fig. 4B). There was a 73.4% increase in NAD+-NADH levels in liver mitochondria following oral alcohol feeding compared with control, which was lower than the 132% increase in mitochondrial NAD+-NADH levels in intragastric alcohol-fed mice. Oral alcohol feeding did not significantly alter the NAD+/NADH ratio in isolated liver mitochondria. Overall, an increase in mitochondrial NAD+-NADH levels is the major effect observed with alcohol feeding, both oral and intragastric.
FIGURE 4.
Effects of alcohol feeding on NAD+-NADH levels and redox status in liver homogenate and mitochondria. A, intragastric alcohol model. B, oral alcohol model. Solid bars, control; empty bars, alcohol treatment. Mice were fed alcohol intragastrically or orally, and liver mitochondria were isolated using a Percoll gradient or differential centrifugation. NAD+ and NADH levels were measured by HPLC with the fluorescence detector. NAD+-NADH levels in liver homogenate are expressed as nmol/mg liver wet weight, whereas mitochondrial NAD+-NADH levels are expressed as nmol/mg of protein. n = 4–5 mice/group. Results are mean ± S.D. (error bars); *, p < 0.05 versus control.
N-Acetylation of Mitochondrial Proteins in the Liver following Alcohol Feeding
Feeding alcohol orally to mice and rats has been shown to increase N-acetylation of mitochondrial proteins (35, 36). N-Acetylation of mitochondrial proteins, including the respiratory complexes, is generally considered inhibitory (37). We examined if intragastric alcohol feeding to mice similarly increased N-acetylation of mitochondrial proteins. Fig. 5 demonstrates that there was a time-dependent increase in N-acetylation of mitochondrial proteins that peaked at 4 weeks. Oral alcohol feeding caused extensive N-acetylation of mitochondrial proteins but at lower levels than observed with intragastric alcohol feeding. Alcohol feeding, therefore, enhances mitochondrial respiration in the liver despite extensive N-acetylation of mitochondrial proteins.
FIGURE 5.
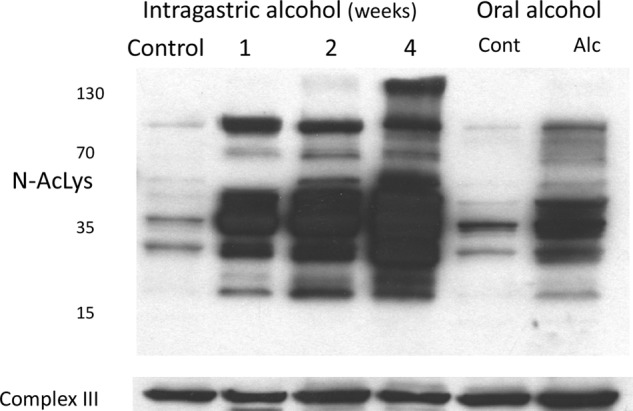
Alcohol feeding causes N-acetylation of mitochondrial proteins in the liver. Mice were fed alcohol intragastrically (1–4 weeks) or orally (5 weeks). N-Acetylation of mitochondrial proteins were assessed by immunoblotting using antisera against N-acetylated lysine proteins. Complex III was used as loading control. Liver mitochondria were isolated using differential centrifugation.
Effect of Intragastric Alcohol Feeding on Mitochondrial Morphology and Density in Hepatocytes
Intragastric alcohol feeding also altered mitochondrial morphology in hepatocytes in vivo. Fig. 6A shows that the shape of mitochondria in the liver of control mice was mainly round or oval. With intragastric alcohol feeding, a greater diversity of mitochondrial shapes was observed, including increases in elongated tubular mitochondria. The larger tubular mitochondria were mainly observed in hepatocytes where lipid droplets were prominent. The increased diversity of mitochondrial shape with intragastric alcohol feeding was confirmed by plotting the major axis (length) and minor axis (width) of mitochondria (Fig. 6B). The major and minor reference lines on the graph represents the averages for control mitochondria. Intragastric alcohol feeding increased the distribution of mitochondrial shape with the number of elongated mitochondria increasing (lower right quadrant; control = 23.5% versus alcohol = 35.5%). Very elongated mitochondria (major axis >2.5 μm) were rare in control mice, but became more frequent with intragastric alcohol feeding. The number of smaller round mitochondria also increased with alcohol feeding (lower left quadrant; control 26.9% versus alcohol 47.1%). Mitochondria from intragastric alcohol-fed mice were thinner overall (minor axis diameter, control = 0.86 ± 0.10 μm versus alcohol = 0.66 ± 0.11 μm). Alcohol feeding altered mitochondrial length distribution, but the average length did not change (major axis diameter, control = 1.14 ± 0.06 μm versus alcohol = 1.19 ± 0.09 μm).
FIGURE 6.

Effects of intragastric alcohol feeding on mitochondrial morphology in hepatocytes in vivo. A, electron microscopy image of hepatocytes from intragastric or oral alcohol-fed mice and their respective controls. B, assessment of mitochondrial major and minor axes following intragastric alcohol feeding (2 weeks). Major and minor mitochondrial axes were measured using the ImageJ program as described under “Experimental Procedures.” The reference lines on the graph represent the averages of major and minor axes for control mitochondria. n = 4 mice/group. Results are mean ± S.D.; *, p < 0.05 versus control.
Intragastric alcohol feeding altered mitochondrial morphology in the liver, but mitochondrial density in hepatocytes in vivo remained unaltered. The total percentage of cytoplasm (cell area minus nuclear area) that mitochondria occupied was observed to be 18.4 ± 2.9% for intragastric control and 20.8 ± 5.4% for intragastric alcohol-fed mice. Oral alcohol feeding did not significantly alter mitochondrial morphology (Fig. 6A, bottom panels) or density (data not shown).
Intragastric Alcohol Feeding Increases the Number of Mitochondria per Hepatocyte in Vivo
Although the density of mitochondria in hepatocytes did not change following intragastric alcohol feeding, hepatocyte size increased with intragastric alcohol feeding (Fig. 7, A and B). Following 2 weeks of intragastric alcohol feeding, hepatocyte size increased ∼75% compared with controls. This finding is in agreement with previous findings, which showed that hepatocyte size increases with alcohol treatment, due to increased accumulation of proteins such as albumin (38, 39). If alcohol treatment is causing hepatocyte enlargement without changing mitochondrial density, then the number of mitochondria per hepatocyte and per liver must increase. Although not a perfect sphere, the volume equation of a sphere ( × π × r3) is the best method to approximate the volume of hepatocytes. Based on measurements of mitochondrial density and hepatocyte area and using the equation of the sphere, the total volume of mitochondria in hepatocytes was estimated to be ∼2.78 times greater in alcohol-treated mice than in control mice. The actual mitochondrial yield (mg of mitochondrial protein/liver) was 6.32 ± 0.64 for control mice and 14.9 ± 2.97 for intragastric alcohol-fed mice. Thus, the mitochondrial protein yield was 2.35-fold greater with alcohol feeding, which was close to the 2.78-fold increase predicted by the volume calculations. The dramatic increase in mitochondria yield per liver with alcohol feeding can be primarily attributed to the fact that livers from alcohol-fed mice were significantly larger. However, even calculating for a greater liver size, intragastric alcohol feeding significantly increases mitochondrial protein levels/g of liver (Fig. 7C; 9.5 ± 1.7 mg/g for alcohol versus 6.94 ± 0.61 mg/g for control). No differences in hepatocyte size and mitochondria yield were observed between oral alcohol-fed and pair-fed control mice (data not shown).
FIGURE 7.

Intragastric alcohol feeding increases hepatocyte size and the number of mitochondria in the liver. A, H&E staining of liver from intragastric alcohol (2 weeks) and control mice. B, changes in hepatocyte area with alcohol feeding. Solid bars, control; empty bars, alcohol treatment. Hepatocyte area was measured using ImageJ as described under “Experimental Procedures.” C, mitochondria yield, protein levels/g of liver (wet weight). n = 4–8 mice/group. Results are mean ± S.D. (error bars); *, p < 0.05 versus control.
Intragastric Alcohol Feeding Increases PGC-1α Levels in the Liver of Mice
We next examined whether PGC-1α, the master regulator of mitochondrial biogenesis, was altered by intragastric alcohol feeding in mice. PGC-1α levels in the liver exhibited a time-dependent increase beginning at 1 week with intragastric alcohol feeding (Fig. 8A), in concert with a previous finding that suggests intragastric alcohol feeding increases PGC-1α mRNA levels (40). The expression of PGC-1α-regulated proteins, TFAM, MCAD, and cytochrome c were all significantly increased with intragastric alcohol feeding (Fig. 8B). TFAM, a key mitochondrial transcription factor that is encoded in the nucleus through action of PGC-1α, increased significantly (densitometry of TFAM/complex III ratio: week 1, control = 12.1 ± 3.2 versus alcohol = 17.5 ± 2.2*; week 2, control = 10.7 ± 4.0 versus alcohol = 20.6 ± 3.8*; week 4, control = 13.1 ± 4.2 versus alcohol = 24.1 ± 3.1* (n = 4, mean ± S.D.; *, p < 0.05 versus control)) in isolated liver mitochondria, suggesting that intragastric alcohol feeding enhances mitochondrial transcriptional activity in the liver. Similar significant increases in densitometry measurements of MCAD and cytochrome c were observed in intragastric alcohol-fed mice at all time points (calculations not shown). Taken together, our data suggest that increases in PGC-1α protein levels may underlie increased expression of mitochondrial proteins and mitochondrial biogenesis with intragastric alcohol feeding.
FIGURE 8.
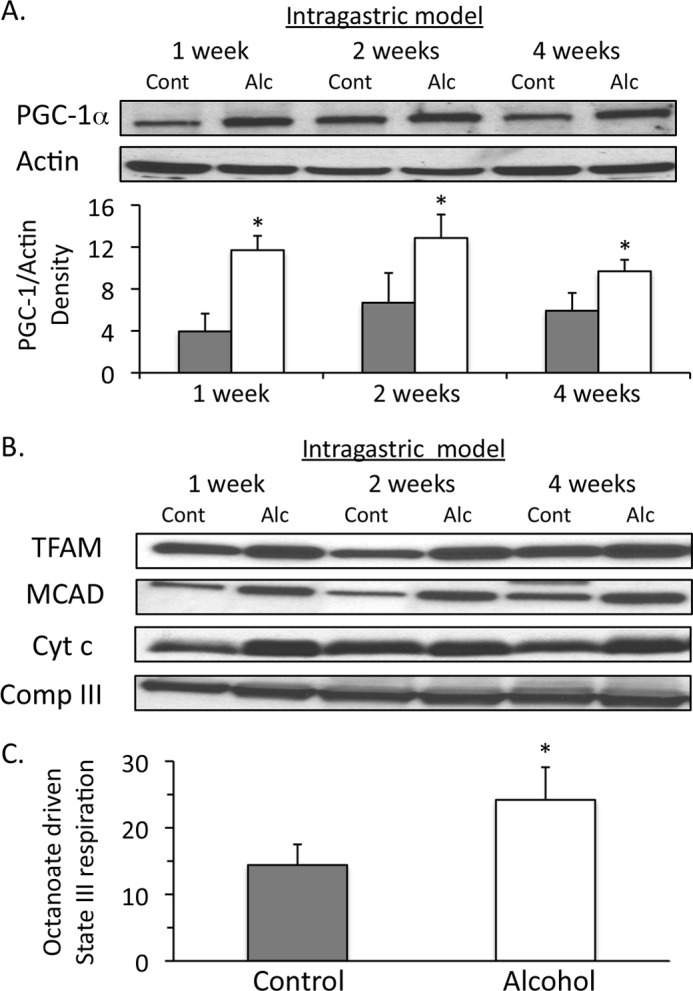
Effect of intragastric alcohol feeding on expression of PGC-1α and PGC-1α-regulated genes in the liver. A, intragastric alcohol feeding enhances PGC-1α expression in the liver. Solid bars, control (Cont); empty bars, alcohol treatment (Alc). B, intragastric alcohol feeding increases expression of PGC-1α-regulated genes TFAM, MCAD, and cytochrome c. PGC-1α levels in liver homogenate and TFAM, MCAD, and cytochrome c levels in isolated liver mitochondria were assessed by immunoblotting. Densitometry was performed using ImageJ. Actin was used as loading control in liver homogenate, and complex III was used as the loading control in isolated liver mitochondria. C, Mitochondrial respiration utilizing octanoate, a medium chain fatty acid, is enhanced in isolated liver mitochondria intragastric alcohol-fed mice. State III respiration was measured using octanoate (200 μm) plus ADP (250 μm) with an oxygen electrode. n = 4–6 mice/group. Results are mean ± S.D. (error bars); *, p < 0.05 versus control.
MCAD is a key mitochondrial protein involved in β-oxidation that produces FADH2 consumed by the respiratory chain. Intragastric alcohol feeding increased MCAD levels, suggesting that β-oxidation may also be increased with alcohol feeding. To test the effect of alcohol feeding on β-oxidation in mitochondria, isolated liver mitochondria from alcohol-fed and control mice were incubated with octanoate, a medium chain fatty acid. Mitochondrial respiration on octanoate was greater in intragastric alcohol fed mice compared with control (Fig. 8C), suggesting an enhanced β-oxidation capacity with alcohol feeding.
Intragastric Alcohol Feeding Increases Acetaldehyde Metabolism in Isolated Liver Mitochondria
Finally, we examined the possibility that increased mitochondrial respiration could enhance alcohol metabolism in the liver. Acetaldehyde can act as a mitochondrial substrate because ALDH2 generates NADH that feeds into complex I of the respiratory chain (15), similar to glutamate/malate and pyruvate. Previous studies have shown that acetaldehyde metabolism is limited by the rate of NAD+ regeneration in the electron transport chain and thus is coupled to mitochondrial respiration. Therefore, we measured state III and IV respirations using acetaldehyde as a substrate with isolated liver mitochondria. In intragastric alcohol-fed mice, we observed an enhanced state III respiration (∼71–141% depending on dose) using acetaldehyde (Fig. 9A). Isolated liver mitochondria from intragastric alcohol fed mice also had a higher RCR than control mitochondria (Fig. 9B). Both state III respiration and RCR declined at high concentrations of acetaldehyde, an effect that was more prominent in mitochondria from control mice. At high concentrations, acetaldehyde may be damaging components of the respiratory chain, as well as uncoupling respiration (15). Mitochondria from mice fed alcohol intragastrically seem more resistant to acetaldehyde-induced damage than control mitochondria because their RCR remained elevated even with 375 mm acetaldehyde. Mice fed alcohol orally also had an increase in state III respiration and RCR using acetaldehyde (Fig. 9, C and D), but these rates were lower than those observed with isolated mitochondria from intragastric fed mice. Alcohol feeding, both intragastric and oral, increased the capacity of isolated liver mitochondria to metabolize acetaldehyde (Fig. 9E), corroborating respiration measurements. These data demonstrate that mitochondria from alcohol-fed mice can metabolize acetaldehyde at a greater rate than control mitochondria because of an enhanced respiratory capacity.
FIGURE 9.

Isolated liver mitochondria from alcohol-fed mice (intragastric and oral fed) have a greater capacity to metabolize acetaldehyde without changes in aldehyde dehydrogenase 2 levels. Control (●) and alcohol feeding (□). Effect of intragastric alcohol feeding (2 weeks) on state III respiration (A) and RCR (B) using acetaldehyde as substrate. Effect of oral alcohol feeding (5 weeks) on state III respiration (C) and RCR (D) using acetaldehyde as substrate. E, acetaldehyde metabolism by isolated liver mitochondria from intragastric and oral alcohol-fed mice. F, ALDH2 protein levels following intragastric alcohol feeding. Solid bars, control; empty bars, alcohol treatment. Mitochondrial respiration was performed using various concentrations of acetaldehyde as substrate in the presence or absence of ADP (250 μm) using an oxygen electrode. Acetaldehyde metabolism by liver mitochondria was measured by monitoring NADH formation as described under “Experimental Procedures.” ALDH2 levels were measured using immunoblotting. n = 5–6 mice/group. Results are mean ± S.D. (error bars); *, p < 0.05 versus control. #, p < 0.05 for intragastric versus oral alcohol-fed mice.
The increased acetaldehyde metabolism in isolated mitochondria from alcohol-treated mice is probably due to increased mitochondrial respiration that regenerates NAD+ faster due to increased levels of complexes I, IV, and V and increased total NAD+-NADH levels in isolated liver mitochondria. However, it is also possible that alcohol treatment induced ALDH2, which could theoretically increase acetaldehyde metabolism. We measured ALDH2 protein levels by immunoblotting and observed that there was no difference between mitochondria from intragastric alcohol-fed and control mice (Fig. 9F), confirming other studies suggesting that no changes in ALDH2 activity occur with alcohol treatment (15). Similarly, no differences in ALDH2 levels were observed in isolated mitochondria of mice fed alcohol orally (data not shown). Consequently, increased acetaldehyde metabolism in isolated liver mitochondria from alcohol-fed mice is probably due to an enhanced respiration that regenerates NAD+ faster rather than changes in ALDH2 levels.
DISCUSSION
Mitochondrial Plasticity in the Liver following Chronic Alcohol Feeding to Mice
This study demonstrates that mitochondria in the liver are dynamic and adapt to the metabolic stress caused by alcohol feeding. Mitochondrial changes and adaptation were greater and more extensive with higher alcohol intake (intragastric feeding) than with lower alcohol intake (Lieber-DeCarli oral diet). Intragastric alcohol feeding was observed to cause four major changes to liver mitochondria, including increased respiration, increased mitochondrial NAD+-NADH levels, altered mitochondrial morphology, and increased mitochondrial biogenesis. Oral alcohol feeding, on the other hand, primarily increased mitochondrial respiration and increased mitochondrial NAD+-NADH levels, both at levels much lower than observed with intragastric alcohol feeding.
Increased Mitochondrial Respiration and Increased Components of the Respiratory Complexes
Intragastric alcohol feeding dramatically increased mitochondrial respiration (up to 141% after 2 weeks with intragastric feeding), creating “super-respiring mitochondria.” The increase in mitochondrial respiration caused by intragastric alcohol feeding is probably due to a remodeling of mitochondria with increased levels of respiratory complexes I, IV, and V being incorporated into the electron transport chain and possibly due to increased levels of the mitochondrial NAD+-NADH pool. Levels of TFAM were increased, confirming that transcriptional activity in mitochondria was dramatically increased with intragastric alcohol feeding. Oral alcohol feeding to mice caused a ∼30% increase in respiration, which was primarily associated with increased levels of complex I and increases in mitochondrial NAD+-NADH levels. However, given the semiquantitative nature of immunoblotting, small increases to other complexes, such as complex IV, may be occurring without being detected. Complex I is stoichiometrically at low levels compared with the other complexes, so it is likely that increased expression of complex I contributes to increased mitochondria respiration (41, 42). Similarly, increased expression of subunits of complex IV has also been shown to increase mitochondrial respiration in cells (43). The increased expression of respiratory complex proteins was selective, with matrix proteins, such as ALDH2 and glutamate dehydrogenase, not exhibiting any increase in expression.
The increase in mitochondrial respiration by alcohol treatment occurred although alcohol feeding induced extensive N-acetylation of mitochondrial proteins. N-Acetylation of mitochondrial proteins is generally believed to inhibit protein activity, including the activity of respiratory proteins, such as complex I (37). Because N-acetylation of mitochondrial proteins has been observed previously in mice and rats fed alcohol orally, it was believed to play a role in inhibiting mitochondrial respiration. However, our findings show that despite extensive N-acetylation, mitochondrial respiration is enhanced with alcohol feeding.
Increased NAD+ and NADH Levels in Mitochondria
The levels of the NAD+ and the NAD+-NADH pool were dramatically increased in liver mitochondria with both oral (∼73.4%) and intragastric alcohol feeding (∼132%). Because NAD+ is the limiting substrate for alcohol/acetaldehyde metabolism, the increase in mitochondrial NAD+ and total NAD+-NADH levels with alcohol feeding may be an important adaptation to help enhance alcohol/acetaldehyde metabolism. Greater NAD+ availability and cycling would enhance ADH and ALDH2 activity in the liver. It is also possible that greater NAD+-NADH levels may also be contributing to the enhanced mitochondrial respiration observed in isolated mitochondria treated with glutamate/malate by increasing the capacity of NAD+-NADH cycling. Greater NAD+-NADH levels allow greater NAD+ availability for glutamate/malate dehydrogenases to generate NADH for mitochondrial respiration. Complex I respiration is mainly dependent on NADH levels (44), not NADH redox status. Thus, having greater mitochondrial NAD+ levels will not inhibit respiration, but could possibly increase availability of NAD+ to various mitochondrial dehydrogenases to form NADH. Because mitochondrial levels of the NAD+-NADH pool were increased with both oral and intragastric alcohol feeding, this enhancement of mitochondrial NAD+-NADH levels appears to be an important adaptation to alcohol feeding.
Mitochondrial Morphological Alterations
Intragastric alcohol feeding caused mitochondria to be more variable, particularly with the formation of longer tubular mitochondria in hepatocytes that contained lipid droplets. This suggests that intragastric alcohol feeding alters fusion-fission rates in mitochondria. Mitochondrial morphological changes were observed primarily in intragastric alcohol-fed mice, with oral alcohol causing minimal change. It is therefore possible that mitochondrial morphological changes may be contributing to enhanced respiration observed with intragastric alcohol feeding, possibly through alterations in the cristae surface area (12). Determination of how intragastric alcohol feeding alters mitochondrial fusion-fission rates and how much morphological alterations affect mitochondrial respiration in the liver requires further investigation.
Mitochondrial Biogenesis in the Liver
Intragastric alcohol feeding to mice also caused significant mitochondrial biogenesis to occur in the liver. As hepatocytes increased in size, the density of mitochondria in hepatocytes remained constant. Thus, the number of mitochondria per hepatocyte increased as hepatocytes enlarged with alcohol feeding. It has previously been shown that hepatocytes increase in size following alcohol intake due to increased levels of proteins, in part due to retention of serum proteins, such as albumin in the liver (38, 39). Our work suggests that increases in mitochondrial proteins also contribute to the enlargement of hepatocytes with alcohol feeding. The early increase in PGC-1α, the master regulator of mitochondrial biogenesis, further supports the notion that mitochondrial biogenesis was occurring with intragastric alcohol feeding. It is also possible that a decline in autophagy of mitochondria (mitophagy) could be contributing to the increased mitochondria number caused by intragastric alcohol feeding. With acute alcohol feeding, mitophagy seems to increase in the liver (45). However, the impact of chronic alcohol feeding on the clearance of mitochondria remains to be determined. Recently, it has been suggested that elongated mitochondria, due to their shape, are less susceptible to mitophagy (12). Consequently, the increased number of mitochondria in hepatocytes with intragastric alcohol feeding could also be due to increased numbers of mitophagy-resistant, elongated mitochondria.
It is likely that the increased mitochondrial biogenesis and increased levels of respiratory complexes in the liver caused by intragastric alcohol feeding are being mediated by PGC-1α. We observed increased levels of PGC-1α following intragastric alcohol feeding, which complements a previous study demonstrating that intragastric alcohol feeding increased PGC-1α mRNA levels (40). Proteins regulated by PGC-1α, complex I, complex IV, TFAM, MCAD, and cytochrome c were all observed to increase with alcohol. Although PGC-1α is the master regulator of mitochondrial biogenesis, many other factors have also been shown to contribute to mitochondrial biogenesis and remodeling, including nitric oxide, AMPK, HIF, and NRF-1 (46–48). Whether these factors work with PGC-1α to regulate mitochondrial composition and biogenesis following alcohol treatment needs to be further investigated.
Mitochondrial Plasticity Enhances Alcohol Metabolism by the Liver
The dynamic changes induced in liver mitochondria by alcohol feeding are probably an adaptive response to deal with the metabolic stress caused by chronic alcohol feeding (Fig. 10). NAD+ is the limiting substrate for alcohol metabolism by ADH in the cytoplasm and acetaldehyde metabolism by ALDH2 in mitochondria (13–15). Consequently, an increase in mitochondrial respiration following alcohol treatment can increase NADH oxidation by complex I to enhance alcohol/acetaldehyde metabolism. In support of this notion, we observed that isolated liver mitochondria from alcohol-treated mice had a greater capacity to metabolize acetaldehyde. Furthermore, the metabolism of acetaldehyde was still coupled to ADP and therefore completely dependent on mitochondrial respiration. ALDH2 levels were unaltered in alcohol-treated mice, suggesting that the greater acetaldehyde respiration and metabolism in isolated liver mitochondria from alcohol-treated mice was mainly due to the increased respiration. These findings are in agreement with previous findings demonstrating that acetaldehyde metabolism in mitochondria is rate-limited by mitochondrial respiration and not rate-limited by ALDH2 (13–15). Although we were unable to directly show that increased mitochondrial respiration and NAD+ formation increases alcohol metabolism by ADH, other studies of ADH kinetics suggest that enhancing NAD+ cycling by mitochondrial respiration enhances alcohol metabolism by ADH (13, 14).
FIGURE 10.

Role of mitochondria in alcohol metabolism in the liver. The metabolism of alcohol in the liver is dependent on three major pathways: 1) the conversion of ethanol to acetaldehyde by ADH, utilizing NAD+ that occurs in the cytoplasm; 2) the conversion of acetaldehyde to acetate by ALDH2, utilizing NAD+ that occurs in the mitochondrial matrix; and 3) mitochondrial respiration, which oxidizes NADH to regenerate NAD+ for alcohol metabolism that occurs in the mitochondrial inner membrane. Both alcohol and acetaldehyde metabolism are rate-limited by the availability of NAD+. Thus, the rate-limiting step in alcohol metabolism is mitochondrial respiration to regenerate NAD+. Chronic alcohol feeding to mice increases mitochondrial respiration, due to increased levels of complexes I, IV, and V, thus enhancing the capacity of liver to metabolize alcohol. Increased mitochondrial respiration caused by alcohol may, however, lead to hypoxia in the liver.
Our findings also help to explain why hypoxia may occur in the liver with alcohol feeding (49, 50). Increased mitochondrial respiration and increases in the number of mitochondria consuming oxygen for alcohol metabolism may underlie hypoxia in the liver with alcohol feeding. Alcohol intake has also been shown to cause a hypermetabolic state (sometimes referred to as a swift increase in alcohol metabolism (SIAM effect)), a condition where oxygen consumption increases within 2–6 h after a bolus dose of alcohol in mice and rats (13, 51, 52). The increased oxygen consumption in liver during the hypermetabolic state may be an activation of the spare respiratory capacity of hepatocytes rather than mitochondrial biogenesis and up-regulation of respiratory substrates, which would take a greater amount of time. The super-respiring mitochondria and greater number of mitochondria induced by alcohol are probably long term changes in the liver as it adapts to chronic alcohol intake. These mitochondrial plasticity changes could eventually lead to centrilobular hypoxia, which may contribute to liver injury. Although little data are available in humans, some evidence suggests that hypoxia occurs in the liver of alcoholic patients, suggesting that mitochondrial plasticity changes may also occur in humans in response to chronic alcohol intake (53).
Alcohol feeding to mice, in addition to enhancing acetaldehyde metabolism, also increased the β-oxidation capacity of liver mitochondria. We observed that intragastric alcohol feeding to mice increases octanoate-driven respiration and expression of MCAD, an important dehydrogenase involved in β-oxidation, in isolated liver mitochondria. Whether mitochondrial changes, such as increased mitochondrial pyridine nucleotide levels, are related to the changes in β-oxidation capacity of liver mitochondria remains to be determined. Although the β-oxidation capacity of liver mitochondria increased with alcohol feeding, it remains uncertain whether the β-oxidation rate in intact liver, in vivo, is actually enhanced with alcohol. Previous studies have shown that alcohol feeding induces fatty acid synthesis, through a sterol response element-binding protein-1c (SREBP-1c)-regulated pathway (54). Because fatty acid synthesis generally inhibits β-oxidation (55), β-oxidation may be inhibited in the liver although the β-oxidation capacity of liver mitochondria increases with alcohol feeding. We speculate that as fatty acids accumulate in the liver, mitochondria develop an enhanced β-oxidation capacity to be able to rapidly utilize fatty acids for energy utilization. In muscle cells, it has been similarly shown that increased fatty acid intake increases mitochondrial β-oxidation capacity (4).
Species Differences in Mitochondria in Response to Alcohol Feeding
In contrast to the findings in mice, we observed that oral alcohol feeding to rats caused a decline in mitochondrial respiration, in agreement with previous results (22–24). Why mice and rats respond differently in terms of mitochondrial changes to alcohol is not clear. The mechanism for species difference in response to alcohol will require future investigation. Based on alcohol feeding studies with rats, it has been suggested that mitochondrial dysfunction may be important in mediating alcoholic liver disease. However, our study suggests the opposite; mice with the greatest increase in mitochondrial respiration (intragastric alcohol-fed) had the worst liver injury, whereas oral alcohol feeding had a modest increase in respiration associated with modest injury, and rats, which had a decline in mitochondrial respiration, had the least amount of liver injury. This suggests a correlation between increased mitochondrial respiration and liver injury. We therefore speculate that super-respiring mitochondria may predispose mice to hypoxic injury and/or possibly are a source of increased reactive oxygen species production (56, 57).
In conclusion, in recent years, there has been a greater recognition of the dynamic nature of mitochondria in adapting to stress and metabolic changes. As shown in this work, alcohol feeding induces mitochondrial plasticity in mouse liver, characterized by dynamic changes in mitochondrial number, morphology, and respiratory capacity in the liver. Although mitochondrial plasticity changes may lead to pathological consequences, such as hypoxia, our work suggests the relevance of mitochondrial plasticity as an important physiological phenomenon and adaptive mechanism in response to the metabolic demands of alcohol in hepatocytes.
Acknowledgments
We acknowledge the Analytical/Instrumentation Core of the University of Southern California Research Center for Liver Diseases (P30DK48522) for the use of various instruments. We also acknowledge the Animal and Morphology Core facilities of the NIAAA-supported Southern California Research Center for Alcoholic Liver and Pancreatic Diseases and Cirrhosis (P50 AA011999) for providing intragastric and oral alcohol-fed mice and oral alcohol-fed rats.
This work was supported, in whole or in part, by National Institutes of Health Grant AA016911 (to D. H.), AA14428 (to N. K.), and P50AA11999 (to H. T.).
- ADH
- alcohol dehydrogenase
- ALDH2
- aldehyde dehydrogenase 2
- MCAD
- medium chain acyl-CoA dehydrogenase
- TFAM
- transcriptional factor A
- ER
- endoplasmic reticulum
- RCR
- respiratory control ratio
- ALT
- alanine transaminase.
REFERENCES
- 1. Baar K., Wende A. R., Jones T. E., Marison M., Nolte L. A., Chen M., Kelly D. P., Holloszy J. O. (2002) Adaptations of skeletal muscle to exercise. Rapid increase in the transcriptional coactivator PGC-1. FASEB J. 16, 1879–1886 [DOI] [PubMed] [Google Scholar]
- 2. Holloszy J. O., Booth F. W. (1976) Biochemical adaptations to endurance exercise in muscle. Annu. Rev. Physiol. 38, 273–291 [DOI] [PubMed] [Google Scholar]
- 3. Molé P. A., Oscai L. B., Holloszy J. O. (1971) Adaptation of muscle to exercise. Increase in levels of palmityl Coa synthetase, carnitine palmityltransferase, and palmityl CoA dehydrogenase, and in the capacity to oxidize fatty acids. J. Clin. Invest. 50, 2323–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Garcia-Roves P., Huss J. M., Han D. H., Hancock C. R., Iglesias-Gutierrez E., Chen M., Holloszy J. O. (2007) Raising plasma fatty acid concentration induces increased biogenesis of mitochondria in skeletal muscle. Proc. Natl. Acad. Sci. U.S.A. 104, 10709–10713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Leone T. C., Lehman J. J., Finck B. N., Schaeffer P. J., Wende A. R., Boudina S., Courtois M., Wozniak D. F., Sambandam N., Bernal-Mizrachi C., Chen Z., Holloszy J. O., Medeiros D. M., Schmidt R. E., Saffitz J. E., Abel E. D., Semenkovich C. F., Kelly D. P. (2005) PGC-1α deficiency causes multi-system energy metabolic derangements. Muscle dysfunction, abnormal weight control and hepatic steatosis. PLoS Biol. 3, e101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lin J., Wu P. H., Tarr P. T., Lindenberg K. S., St-Pierre J., Zhang C. Y., Mootha V. K., Jäger S., Vianna C. R., Reznick R. M., Cui L., Manieri M., Donovan M. X., Wu Z., Cooper M. P., Fan M. C., Rohas L. M., Zavacki A. M., Cinti S., Shulman G. I., Lowell B. B., Krainc D., Spiegelman B. M. (2004) Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1α null mice. Cell 119, 121–135 [DOI] [PubMed] [Google Scholar]
- 7. Scarpulla R. C. (2008) Transcriptional paradigms in mammalian mitochondrial biogenesis and function. Physiol. Rev. 88, 611–638 [DOI] [PubMed] [Google Scholar]
- 8. Hoppeler H., Fluck M. (2003) Plasticity of skeletal muscle mitochondria. Structure and function. Med. Sci. Sports Exerc. 35, 95–104 [DOI] [PubMed] [Google Scholar]
- 9. Adhihetty P. J., Irrcher I., Joseph A. M., Ljubicic V., Hood D. A. (2003) Plasticity of skeletal muscle mitochondria in response to contractile activity. Exp. Physiol. 88, 99–107 [DOI] [PubMed] [Google Scholar]
- 10. Otera H., Mihara K. (2011) Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 149, 241–251 [DOI] [PubMed] [Google Scholar]
- 11. Reddy P. H., Reddy T. P., Manczak M., Calkins M. J., Shirendeb U., Mao P. (2011) Dynamin-related protein 1 and mitochondrial fragmentation in neurodegenerative diseases. Brain Res. Rev. 67, 103–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gomes L. C., Di Benedetto G., Scorrano L. (2011) During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 13, 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Israel Y., Orrego H. (1984) Hypermetabolic state and hypoxic liver damage. Recent Dev. Alcohol 2, 119–133 [DOI] [PubMed] [Google Scholar]
- 14. Videla L., Israel Y. (1970) Factors that modify the metabolism of ethanol in rat liver and adaptive changes produced by its chronic administration. Biochem. J. 118, 275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hasumura Y., Teschke R., Lieber C. S. (1976) Characteristics of acetaldehyde oxidation in rat liver mitochondria. J. Biol. Chem. 251, 4908–4913 [PubMed] [Google Scholar]
- 16. Videla L., Bernstein J., Israel Y. (1973) Metabolic alterations produced in the liver by chronic ethanol administration. Increased oxidative capacity. Biochem. J. 134, 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tobon F., Mezey E. (1971) Effect of ethanol administration on hepatic ethanol and drug-metabolizing enzymes and on rates of ethanol degradation. J. Lab. Clin. Med. 77, 110–121 [PubMed] [Google Scholar]
- 18. Israel Y., Kalant H., Orrego H., Khanna J. M., Videla L., Phillips J. M. (1975) Experimental alcohol-induced hepatic necrosis. Suppression by propylthiouracil. Proc. Natl. Acad. Sci. U.S.A. 72, 1137–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ji S., Lemasters J. J., Christenson V., Thurman R. G. (1982) Periportal and pericentral pyridine nucleotide fluorescence from the surface of the perfused liver. Evaluation of the hypothesis that chronic treatment with ethanol produces pericentral hypoxia. Proc. Natl. Acad. Sci. U.S.A. 79, 5415–5419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Venkatraman A., Shiva S., Wigley A., Ulasova E., Chhieng D., Bailey S. M., Darley-Usmar V. M. (2004) The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology 40, 565–573 [DOI] [PubMed] [Google Scholar]
- 21. Zhang X., Tachibana S., Wang H., Hisada M., Williams G. M., Gao B., Sun Z. (2010) Interleukin-6 is an important mediator for mitochondrial DNA repair after alcoholic liver injury in mice. Hepatology 52, 2137–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Spach P. I., Cunningham C. C. (1987) Control of state 3 respiration in liver mitochondria from rats subjected to chronic ethanol consumption. Biochim. Biophys. Acta 894, 460–467 [DOI] [PubMed] [Google Scholar]
- 23. Cederbaum A. I., Lieber C. S., Rubin E. (1974) Effects of chronic ethanol treatment of mitochondrial functions damage to coupling site I. Arch. Biochem. Biophys. 165, 560–569 [DOI] [PubMed] [Google Scholar]
- 24. Bernstein J. D., Penniall R. (1978) Effects of chronic ethanol treatment upon rat liver mitochondria. Biochem. Pharmacol. 27, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 25. Cunningham C. C., Coleman W. B., Spach P. I. (1990) The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol 25, 127–136 [DOI] [PubMed] [Google Scholar]
- 26. Venkatraman A., Landar A., Davis A. J., Chamlee L., Sanderson T., Kim H., Page G., Pompilius M., Ballinger S., Darley-Usmar V., Bailey S. M. (2004) Modification of the mitochondrial proteome in response to the stress of ethanol-dependent hepatotoxicity. J. Biol. Chem. 279, 22092–22101 [DOI] [PubMed] [Google Scholar]
- 27. Shinohara M., Ji C., Kaplowitz N. (2010) Differences in betaine-homocysteine methyltransferase expression, endoplasmic reticulum stress response, and liver injury between alcohol-fed mice and rats. Hepatology 51, 796–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Arteel G. E. (2010) Animal models of alcoholic liver disease. Dig. Dis. 28, 729–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tsukamoto H., French S. W., Benson N., Delgado G., Rao G. A., Larkin E. C., Largman C. (1985) Severe and progressive steatosis and focal necrosis in rat liver induced by continuous intragastric infusion of ethanol and low fat diet. Hepatology 5, 224–232 [DOI] [PubMed] [Google Scholar]
- 30. Tsukamoto H., Towner S. J., Ciofalo L. M., French S. W. (1986) Ethanol-induced liver fibrosis in rats fed high fat diet. Hepatology 6, 814–822 [DOI] [PubMed] [Google Scholar]
- 31. Han D., Antunes F., Canali R., Rettori D., Cadenas E. (2003) Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol. J. Biol. Chem. 278, 5557–5563 [DOI] [PubMed] [Google Scholar]
- 32. Garcia J., Han D., Sancheti H., Yap L. P., Kaplowitz N., Cadenas E. (2010) Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J. Biol. Chem. 285, 39646–39654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Han D., Williams E., Cadenas E. (2001) Mitochondrial respiratory chain-dependent generation of superoxide anion and its release into the intermembrane space. Biochem. J. 353, 411–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Klaidman L. K., Leung A. C., Adams J. D., Jr. (1995) High-performance liquid chromatography analysis of oxidized and reduced pyridine dinucleotides in specific brain regions. Anal. Biochem. 228, 312–317 [DOI] [PubMed] [Google Scholar]
- 35. Picklo M. J., Sr. (2008) Ethanol intoxication increases hepatic N-lysyl protein acetylation. Biochem. Biophys. Res. Commun. 376, 615–619 [DOI] [PubMed] [Google Scholar]
- 36. Fritz K. S., Galligan J. J., Hirschey M. D., Verdin E., Petersen D. R. (2012) Mitochondrial acetylome analysis in a mouse model of alcohol-induced liver injury utilizing SIRT3 knockout mice. J. Proteome Res. 11, 1633–1643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Verdin E., Hirschey M. D., Finley L. W., Haigis M. C. (2010) Sirtuin regulation of mitochondria. Energy production, apoptosis, and signaling. Trends Biochem. Sci. 35, 669–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Baraona E., Leo M. A., Borowsky S. A., Lieber C. S. (1977) Pathogenesis of alcohol-induced accumulation of protein in the liver. J. Clin. Invest. 60, 546–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Baraona E., Lieber C. S. (1982) Effects of alcohol on hepatic transport of proteins. Annu. Rev. Med. 33, 281–292 [DOI] [PubMed] [Google Scholar]
- 40. Oliva J., French B. A., Li J., Bardag-Gorce F., Fu P., French S. W. (2008) Sirt1 is involved in energy metabolism. The role of chronic ethanol feeding and resveratrol. Exp. Mol. Pathol. 85, 155–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yap L. P., Garcia J. V., Han D., Cadenas E. (2009) The energy-redox axis in aging and age-related neurodegeneration. Adv. Drug Deliv. Rev. 61, 1283–1298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schägger H., Pfeiffer K. (2001) The ratio of oxidative phosphorylation complexes I-V in bovine heart mitochondria and the composition of respiratory chain supercomplexes. J. Biol. Chem. 276, 37861–37867 [DOI] [PubMed] [Google Scholar]
- 43. Campian J. L., Gao X., Qian M., Eaton J. W. (2007) Cytochrome c oxidase activity and oxygen tolerance. J. Biol. Chem. 282, 12430–12438 [DOI] [PubMed] [Google Scholar]
- 44. Vinogradov A. D. (2008) NADH/NAD+ interaction with NADH:ubiquinone oxidoreductase (complex I). Biochim. Biophys. Acta. 1777, 729–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ding W. X., Li M., Chen X., Ni H. M., Lin C. W., Gao W., Lu B., Stolz D. B., Clemens D. L., Yin X. M. (2010) Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology 139, 1740–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fukuda R., Zhang H., Kim J. W., Shimoda L., Dang C. V., Semenza G. L. (2007) HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 129, 111–122 [DOI] [PubMed] [Google Scholar]
- 47. Hock M. B., Kralli A. (2009) Transcriptional control of mitochondrial biogenesis and function. Annu. Rev. Physiol. 71, 177–203 [DOI] [PubMed] [Google Scholar]
- 48. Nisoli E., Tonello C., Cardile A., Cozzi V., Bracale R., Tedesco L., Falcone S., Valerio A., Cantoni O., Clementi E., Moncada S., Carruba M. O. (2005) Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310, 314–317 [DOI] [PubMed] [Google Scholar]
- 49. Arteel G. E., Iimuro Y., Yin M., Raleigh J. A., Thurman R. G. (1997) Chronic enteral ethanol treatment causes hypoxia in rat liver tissue in vivo. Hepatology 25, 920–926 [DOI] [PubMed] [Google Scholar]
- 50. French S. W., Benson N. C., Sun P. S. (1984) Centrilobular liver necrosis induced by hypoxia in chronic ethanol-fed rats. Hepatology 4, 912–917 [DOI] [PubMed] [Google Scholar]
- 51. Bradford B. U., Rusyn I. (2005) Swift increase in alcohol metabolism (SIAM). Understanding the phenomenon of hypermetabolism in liver. Alcohol 35, 13–17 [DOI] [PubMed] [Google Scholar]
- 52. Thurman R. G., Paschal D., Abu-Murad C., Pekkanen L., Bradford B. U., Bullock K., Glassman E. (1982) Swift increase in alcohol metabolism (SIAM) in the mouse. Comparison of the effect of short-term ethanol treatment on ethanol elimination in four inbred strains. J. Pharmacol. Exp. Ther. 223, 45–49 [PubMed] [Google Scholar]
- 53. Iturriaga H., Ugarte G., Israel Y. (1980) Hepatic vein oxygenation, liver blood flow, and the rate of ethanol metabolism in recently abstinent alcoholic patients. Eur. J. Clin. Invest. 10, 211–218 [DOI] [PubMed] [Google Scholar]
- 54. Ji C., Chan C., Kaplowitz N. (2006) Predominant role of sterol response element binding proteins (SREBP) lipogenic pathways in hepatic steatosis in the murine intragastric ethanol feeding model. J. Hepatol. 45, 717–724 [DOI] [PubMed] [Google Scholar]
- 55. Saggerson D. (2008) Malonyl-CoA, a key signaling molecule in mammalian cells. Annu. Rev. Nutr. 28, 253–272 [DOI] [PubMed] [Google Scholar]
- 56. Bailey S. M., Cunningham C. C. (2002) Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic. Biol. Med. 32, 11–16 [DOI] [PubMed] [Google Scholar]
- 57. Arteel G. E. (2003) Oxidants and antioxidants in alcohol-induced liver disease. Gastroenterology 124, 778–790 [DOI] [PubMed] [Google Scholar]