

Background: CYP26A1 is a critical enzyme in the metabolism of all-trans-retinoic acid (atRA).
Results: CYP26A1 metabolizes atRA stereoselectively to (4S)-OH-RA, which corresponds to atRA orientation within the CYP26A1 active site.
Conclusion: Preference for (4S)-OH-RA is conserved in atRA metabolism and in elimination of 4-OH-RA.
Significance: Determination of stereoselectivity in atRA metabolism is important for better understanding of retinoid biology and CYP26A1 biochemistry.
Keywords: Cytochrome P450, Enzyme Kinetics, Metabolism, Retinoid, Retinoid-binding Protein
Abstract
All-trans-retinoic acid (atRA), the major active metabolite of vitamin A, plays a role in many biological processes, including maintenance of epithelia, immunity, and fertility and regulation of apoptosis and cell differentiation. atRA is metabolized mainly by CYP26A1, but other P450 enzymes such as CYP2C8 and CYP3As also contribute to atRA 4-hydroxylation. Although the primary metabolite of atRA, 4-OH-RA, possesses a chiral center, the stereochemical course of atRA 4-hydroxylation has not been studied previously. (4S)- and (4R)-OH-RA enantiomers were synthesized and separated by chiral column HPLC. CYP26A1 was found to form predominantly (4S)-OH-RA. This stereoselectivity was rationalized via docking of atRA in the active site of a CYP26A1 homology model. The docked structure showed a well defined niche for atRA within the active site and a specific orientation of the β-ionone ring above the plane of the heme consistent with stereoselective abstraction of the hydrogen atom from the pro-(S)-position. In contrast to CYP26A1, CYP3A4 formed the 4-OH-RA enantiomers in a 1:1 ratio and CYP3A5 preferentially formed (4R)-OH-RA. Interestingly, CYP3A7 and CYP2C8 preferentially formed (4S)-OH-RA from atRA. Both (4S)- and (4R)-OH-RA were substrates of CYP26A1 but (4S)-OH-RA was cleared 3-fold faster than (4R)-OH-RA. In addition, 4-oxo-RA was formed from (4R)-OH-RA but not from (4S)-OH-RA by CYP26A1. Overall, these findings show that (4S)-OH-RA is preferred over (4R)-OH-RA by the enzymes regulating atRA homeostasis. The stereoselectivity observed in CYP26A1 function will aid in better understanding of the active site features of the enzyme and the disposition of biologically active retinoids.
Introduction
All-trans-retinoic acid (atRA)2 is the major biologically active metabolite of vitamin A (retinol). It is an essential component of signaling networks in chordates. During embryonic and fetal development atRA regulates cell differentiation, and during adult life it is important for maintaining healthy epithelia, immunity, fertility, and in regulating cell proliferation (1). atRA also plays a role in neurogenesis in adults and in cell survival (2), and it contributes to regulation of glucose homeostasis (3). These effects of atRA result mainly from its binding to the three nuclear retinoic acid receptors (RARα, RARβ, and RARγ) that are activated by atRA resulting in induction of gene transcription (4).
Therapeutically atRA (Tretinoin®), 13-cis-retinoic acid (Isotretinoin®), and 9-cis-retinoic acid (Alitretinoin®) are used in the treatment of various cancers and skin diseases (4). All three RA isomers are cleared mainly by metabolic oxidation and glucuronidation (5–8). The main circulating metabolite of 13-cis-RA and atRA detected in human plasma is 4-oxo-RA (9, 10), the subsequent metabolite of 4-OH-RA. 4-OH-RA is the primary oxidized metabolite of atRA. It is formed by several cytochrome P450 enzymes including CYP26s, CYP3As, and CYP2C8 (6–8, 11–13). Of these P450 enzymes, CYP26A1 appears to be the main isoform responsible for both atRA clearance and formation of 4-OH-RA in human liver (13). CYP26A1 and CYP26B1 have also been identified as the key enzymes that regulate atRA metabolism in other tissues during development and in adulthood (11, 12, 14, 15).
The oxidative metabolism of atRA by CYP26A1 is believed to be the first step in its degradation and inactivation (14). However, the main atRA metabolites, 4-OH-RA and 4-oxo-RA, also bind to the three RAR isoforms with affinities similar to atRA (16), and they induce gene transcription, inhibit proliferation, and induce differentiation in various atRA susceptible cell lines and models (17, 18). Formation of metabolites from atRA by CYP26A1 also appears to be required for differentiation of P19 EC cells into neurons, and for atRA-induced differentiation of embryonic stem cells (19, 20). These data suggest that metabolites of atRA may be important in retinoid function when sufficient concentrations are reached in vivo. However, transgenic animal studies have shown that CYP26 enzymes are required for removal of atRA rather than for formation of active metabolites during mouse development (21).
The hydroxylation of atRA to 4-OH-RA results in the formation of a chiral center (Fig. 1) but all studies characterizing formation of 4-OH-RA and its activity have been conducted using racemic 4-OH-RA. Because formation of 4-OH-RA could be stereospecific and the biological activity may reside in a single 4-OH-RA enantiomer, understanding the functional role(s) of atRA metabolites requires characterization of the stereochemical course of metabolism. In addition, stereospecificity of the enzymes involved in RA biotransformation may help in better understanding of the structure-function of the proteins involved in regulating RA homeostasis.
FIGURE 1.

Synthesis of (+)-(4S)- and (−)-(4R)-all-trans-RA and the 4-dimethylaminocinnamoate esters of methyl 4-OH-retinoate. Reaction conditions were: a, (R)-2-methyl-CBS-oxazaborolidine ((R)-CBS), BH3·S(CH3)2, THF, 16 h, yield 85%; b, (S)-2-methyl-CBS-oxazaborolidine ((S)-CBS), BH3·S(CH3)2, THF, 16 h, yield 79%; c, 1 m KOH in water:MeOH (8:1) room temperature, 4 h; citrate buffer (pH 6); and d, 4-(dimethylamino)cinnamoyl-1H-1,2,4-tetrazole amide, 1,8-diazabicyclo[5.4.0]undec-7-ene.
Stereoselectivity appears to be a key characteristic of retinoid biotransformation. Hydroxylation of retinol in F9 cells results predominantly in the formation of (4S)-OH-retinol (22), and in the firefly squid, the visual pigment is (4R)-OH-retinal (23). The mouse and zebrafish retinol saturase selectively forms (R)-all-trans-13,14-dihydroretinol, and the biological activity (adipocyte differentiation and RAR activation) of this product resides in the (R)-enantiomer (24).
Based on the observed stereoselectivity associated with other retinoids and the tight binding of atRA to CYP26A1, it was hypothesized that formation of 4-OH-RA by CYP26A1 would be stereoselective. To test this hypothesis, a chiral column HPLC method was developed to separate the 4-OH-RA enantiomers, standards of the (4S)- and (4R)-OH-RA enantiomers were synthesized, and the formation of 4-OH-RA enantiomers by CYP26A1, CYP2C8, and CYP3A4 enzymes was characterized. The formation of 4-OH-RA by CYP26A1 was stereoselective resulting in formation of (4S)-OH-RA, whereas the other P450s formed both 4-OH-RA enantiomers with varying stereoselectivities. (4S)-OH-RA was also a better substrate of CYP26A1 than (4R)-OH-RA resulting in formation of different metabolites and suggesting that the observed stereoselectivity may have broader biological importance.
EXPERIMENTAL PROCEDURES
Separation of (4S)- and (4R)-OH-RA Enantiomers
The 4-OH-RA enantiomers were separated using an Agilent Technologies (Palo Alto, CA) 1200 series HPLC system equipped with a Chiralcel OD-RH column (5 μm, 2.1 × 150 mm), an Agilent temperature controlled auto-sampler, a multiple wavelength detector, and HP Chemstation software. The mobile phase flow was 0.17 ml/min. A linear gradient from initial 50:50 water:acetonitrile for 14 min to 20:80 over 10 min was used, followed by 20:80 water:acetonitrile for 10 min. Analytes were quantified by determining the UV absorbance at 360 nm.
Protein Expression and Enzymatic Formation of 4-OH-RA Enantiomers
CYP26A1 microsomes were prepared and rat reductase was expressed and purified as previously described (13, 25). CYP3A4, CYP3A5, CYP3A7, and CYP2C8 were purchased from BD Gentest and co-expressed with P450 reductase and cytochrome b5. All incubations were conducted in 100 mm potassium phosphate buffer (pH 7.4) at 37 °C and initiated by addition of NADPH (1 mm final concentration). The reactions were quenched and metabolites were extracted using ethyl acetate as described previously (25, 26). The stereoselective formation of 4-OH-RA by P450 enzymes (5 nm) was characterized with 10 nm to 40 μm atRA as a substrate. P450 reductase (10 nm) was added to incubations with CYP26A1. The product formation data were analyzed and the Michaelis-Menten equation was fitted to the data using GraphPad Prism version 5 (La Jolla, CA).
Metabolism of (4R)- and (4S)-OH-RA by CYP26A1
The metabolites formed from (4R)-OH-RA and (4S)-OH-RA by CYP26A1 were identified using LC-MS/MS conditions identical to those previously reported for racemic 4-OH-RA (15). Five pmol of CYP26A1 and 10 pmol of P450 reductase were incubated with 1 μm of each enantiomer in 1 ml of potassium phosphate (100 mm, pH 7.4) buffer, and samples were extracted using ethyl acetate as described (15).
The time course of (4R)-OH-RA and (4S)-OH-RA depletion by CYP26A1 was determined at six substrate concentrations between 2 and 250 nm using a P450 reductase to CYP26A1 ratio of 2:1 (2 nm of P450 in each incubation), in 100 mm potassium phosphate buffer (pH 7.4) at 37 °C. All incubations were conducted under red light in triplicate and the experiments were repeated three times on separate days. The reactions were initiated by addition of NADPH (1 mm final concentration). At 0.5, 1, 2, and 5 min, 0.5-ml aliquots were collected and quenched with 3 ml of cold ethyl acetate. For a separate experiment at 2.5 nm substrate concentration, 1-ml aliquots were collected at 0.5, 1, 1.5, and 2 min. As an internal standard, 20 μl of a 2.5 μm solution of 4-oxo-RA-D3 (Toronto Research Chemicals, North York, Ontario) in ethanol was added, and samples were extracted as previously described (15). 4-OH-RA enantiomers were analyzed using an AB Sciex API 5500 Q/LIT mass spectrometer equipped with an Agilent 1290 Infinity UHPLC and an Agilent Zorbax C18 column (3.5 μm, 2.1 × 100 mm). The analytes were separated using a 10-min linear gradient from initial 5:95, acetonitrile, 0.1% aqueous formic acid to a final 95:5, acetonitrile, 0.1% aqueous formic acid. Analytes were detected using positive ion electrospray mass spectrometry with the declustering potential set to 80 V, collision energy to 33 eV (4-OH-RA) and 35 eV (4-oxo-RA-D3), and collision cell exit potential to 13 V (4-OH-RA) and 2 V (4-oxo-RA-D3). The parent fragment MS/MS transitions of m/z 299.2 → 121.2 Da for (4R)-OH-RA and (4S)-OH-RA and m/z 300.2 → 226.2 Da for 4-oxo-RA-D3 were used. The data were analyzed using Analyst software, and the 4-OH-RA concentration was plotted as a function of time. The concentration versus time data were log-linear for 2 min and only data up to 2 min were used. Points for which the concentration was below the limit of quantification of the assay (0.1 nm) were not included in the analysis. The depletion rate constant was obtained from the log-linear fit to the equation: C = C0e−kdept, in which C equals 4-OH-RA enantiomer concentration at a given time t, C0 the initial concentration of 4-OH-RA enantiomers, and kdep the depletion rate constant as described previously (25). The Km was obtained using the depletion data according to Equation 1 (27),
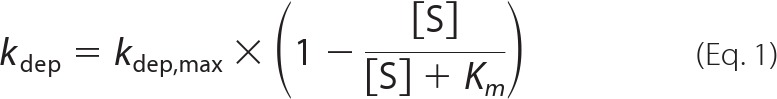 |
in which the substrate concentration [S] was corrected for depletion during the incubation (25). The maximum efficiency of depletion, the intrinsic clearance (Vmax/Km), was calculated as kdep, max divided by the protein concentration as described (25). All nonlinear fits were done using GraphPad Prism version 5 and values are reported as mean ± S.E.
The kinetics of the formation of 4-oxo-RA and 4,16-OH2-RA were analyzed from (4R)-OH-RA and (4S)-OH-RA incubations, respectively, using identical LC-MS conditions as described (15). 2 pmol of CYP26A1 and 4 pmol of P450 reductase were incubated with 5–150 nm of each enantiomer in 1 ml of potassium phosphate (100 mm, pH 7.4) buffer, and samples were extracted using ethyl acetate. For 4-oxo-RA quantification a standard curve using a 4-oxo-RA standard was used and formation velocity was measured. The formation of 4,16-OH2-RA from (4S)-OH-RA was determined using relative quantification of the 4,16-OH2-RA peak height to internal standard ratio as a function of (4S)-OH-RA concentration.
The affinity (Kd) of the 4-OH-RA enantiomers to CYP26A1 was determined by fitting the Morrison equation (tight binding equation, Equation 2) to the data as previously described for atRA binding to CYP26A1 (25).
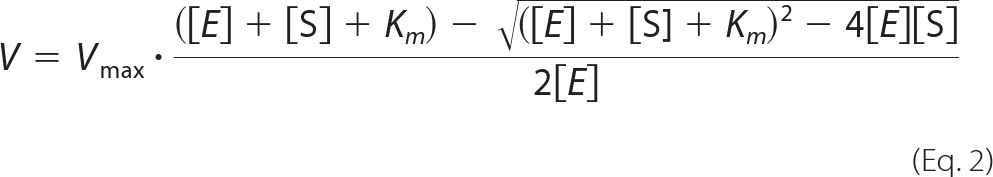 |
Significance of differences between determined kinetic parameters for the two enantiomers were evaluated using the Z-test and a p value <0.05 was considered significant.
Chemical Synthesis
The overall pathway for synthesis of 4-OH-RA enantiomers from methyl 4-oxo-retinoate is shown in Fig. 1. The synthesis of the 4-dimethylaminocinnamoate esters of methyl 4-OH-retinoate for CD spectroscopy is also shown. All syntheses are described in detail under the supplemental Data.
Assignment of Absolute Stereochemistry Using CD Spectroscopy
CD samples were prepared by dissolving weighed amounts of 4-dimethylaminocinnamoate esters (Fig. 1) of the enantiomeric methyl 4-hydroxyretinoates in acetonitrile to make 200 μm solutions (100 μg/ml). Spectra were recorded at 25 °C on a JASCO J715 spectropolarimeter (JASCO Corp., Tokyo, Japan) using a 0.10-cm path length cell with a nitrogen flow rate of 4 liters/min. The wavelength and degree ellipticity scales were calibrated using a reference (1S)-(+)-10-camphorsulfonic acid ammonium salt sample assuming that the (1S)-(+)-10-camphorsulfonic acid ammonium salt maximum corresponds to [ϴ]290.5 = +7910 (28). To minimize the effects of a possible photoisomerization, only a single scan for each spectrum was collected at a scan rate of 100 nm/min with a 2 nm bandwidth and a 0.2-nm step resolution over the wavelength range of 200–500 nm. The collected spectra were trimmed at a dynode voltage of 650 V (where required) prior to baseline subtraction (acetonitrile) and computationally smoothed using a two point fast Fourier transformation filter function (FFT filter) included within JASCO software (Spectra Manager 1.53.04). The raw ellipticity data (mdeg) were normalized by dividing them by the UV visible absorbance of the solutions at λmax (356 nm).
Generation and Validation of a CYP26A1 Homology Model
A CYP26A1 homology model was made using Modeler (29, 30). The homology model was built using the amino acid sequence of human CYP26A1 (NCBI sequence number NP_000774.2). Two cytochrome P450 crystal structures, cyanobacterial CYP120A1 (PDB code 2VE3 (31)) with atRA bound and mammalian CYP2C8 (PDB code 2NNH (32)) with two 9-cis-RAs bound, were used as structural templates. The CYP120A1 structure and the homology model were aligned and heme and atRA in the CYP120A1 structure were roughly docked to the homology model using PyMOL version 1.2 (DeLano Scientific LLC, Palo Alto, CA). To model the predominant structure in solution, molecular dynamics simulations were performed of the complex using GROMACS version 4.07 (33). The topology files were modified to reflect the cysteinyl ligation as described previously (34, 35). atRA was used to form the active site of the homology model of CYP26A1. The atRA molecule was obtained from PubChem (Compound Identifier CID 444795), and the protein was energy minimized by the steepest descent approach using the MMFF94 force field (36) in Avogadro (version 1.01). Because the primary site of metabolism is at the C-4 carbon, a time-averaged distance restraint of 3.4 and 7.4 Å was applied between the heme iron and this carbon. The complex was immersed in a simulated water box with 120 Å sides and ∼50,000 waters, corresponding to roughly twice the length of the diagonal of the protein, or ∼50 Å. Next, the complex was energy minimized by the method of steepest descent to remove van der Waals contacts between overlapping waters and the amino acids of the protein. The simulations were run with Berendsen temperature and pressure coupling (37) at 300 K, using the GROMOS 53a6 force field (38, 39) and periodic boundary conditions in all directions. Electrostatics of the system were measured using the particle-mesh Ewald method (40). The molecular dynamics simulation reached an equilibrium at >10 ns, but was continued to 18.75 ns. The root mean square deviation of the backbone over the course of the simulation is shown in supplemental Fig. S1. The structure at the 18.75-ns time point was energy minimized by conjugate gradient energy and steepest descent minimization methods. The secondary structure assignments of this CYP26A1 homology model are shown in supplemental Table S1.
The CYP26A1 homology model with atRA restrained at C-4 was validated as described for other homology models of CYP26A1 (41–43) and the results are presented in supplemental Table S2. The backbone φ and ψ angles of the homology model were evaluated from a Ramachandran plot obtained on the RAMPAGE server. The Ramachandran plot is shown in supplemental Fig. S2. The compatibility of the homology models three-dimensional fold with the amino acid sequence was compared using Verify 3D (44). The program ERRAT was used to analyze the non-bonded interactions between different atom types (45). The quality of the homology model was overall better than previously built homology models (41–43).
Molecular Docking of atRA to CYP3A4
atRA was docked into the active site of CYP3A4 using Autodock Vina 1.1.1 (46). To sample the known CYP3A4 conformations, atRA was docked into the published crystal structures of the enzyme (PDB code 2J0D (47), 3NXU (48), 3TJS (49), 1TQN (50), 3UA1 (49), 2V0M (47), 1W0E (51), 1W0F (51), and 1W0G (51)). AutoDockTools 1.4.6 (The Scripps Research Institute, La Jolla, CA) was used to put Gasteiger charges on the homology model and atRA. During the simulation, the protein was kept rigid and the carboxylic acid group of atRA was allowed to rotate. A box centered 10 Å away from the heme iron with 20 Å sides was searched with the software. The maximum number of potential binding sites was set to 10. Binding sites with ∼100-fold weaker binding affinity (i.e. 3 kcal/mol) than the tightest binding site were excluded as potential binding sites. The dissociation constant (Kd) was calculated for comparative purposes from the free energy (ΔG) in kcal mol−1 using the equation: Kd = e(ΔG)/(RT), where T is the temperature in K and R is the gas constant in kcal mol−1 K−1.
RESULTS
Separation of the 4-OH-RA Enantiomers and Stereoselective Formation of 4-OH-RA by CYP26A1
A method for the separation of 4-OH-RA enantiomers using a Chiralcel OD-RH column was developed, and the separation of (4S)- and (4R)-OH-RA is shown in Fig. 2a. To determine whether formation of 4-OH-RA by CYP26A1 is stereoselective, atRA was incubated with recombinant human CYP26A1 microsomes. A chromatogram of an incubation of atRA with CYP26A1 is shown in Fig. 2b. CYP26A1 formed preferentially a single 4-OH-RA enantiomer. Other hydroxylation products were also detected in the CYP26A1 incubations that eluted after the 4-OH-RA enantiomers (Fig. 2b), as previously reported (11, 25). Based on previous studies (15) these products include 18-OH-RA and 16-OH-RA.
FIGURE 2.

HPLC separation of the 4-OH-RA enantiomers and stereoselective formation of (4S)-OH-RA by CYP26A1. The 4-OH-RA enantiomers were separated from the racemic mixture using a Chiralcel OD-RH column (5 μm, 2.1 × 150 mm) and a water-acetonitrile gradient as described under “Experimental Procedures.” Panel a shows the separation of (4R)- and (4S)-OH-RA in a racemic mixture, and panel b shows the stereoselective formation of (4S)-OH-RA from atRA (200 nm) by CYP26A1 (5 nm). The determination of enantiomeric excess (ee) for the synthetic (−)-(4R)-OH-RA (ee 96.3%, retention time 11.4 min) and (+)-(4S)-OH-RA (ee 95.1%, retention time 9.8 min) and the identification of the two enantiomers is shown in panels c and d.
Determination of Absolute Stereochemistry of 4-OH-RA Enantiomers
To determine the absolute stereochemistry of the predominant 4-OH-RA formed by CYP26A1, authentic standards of the (4S)- and (4R)-OH-RA enantiomers were synthesized (Fig. 1) from 4-oxo-RA ester. Diborane (dimethyl sulfide) reduction in the presence of the methylated (S)- and (R)-Corey-Bakshi-Shibata (CBS)-oxazaborolidine catalysts (52), yielded the enantiomeric methyl (4S)- and (4R)-OH-retinoates. The corresponding free acids were obtained by hydrolysis of the methyl esters with aqueous methanolic KOH, followed by acidification with citrate buffer (pH 6). The enantiomeric excess (ee %) of the 4-OH-RA enantiomers was determined using chiral HPLC. The enantiomeric excesses (ee %) of the synthesized (4S)- and (4R)-OH-RA enantiomers were 95 and 96%, respectively (Fig. 2, c and d). The retention times of the synthetic standards show that CYP26A1 forms predominantly the (4S)-OH-RA.
To confirm the absolute stereochemistry of the products, the 4-hydroxyl group of each methyl retinoate enantiomer was coupled to the 4-(dimethylamino)cinnamoyl chromophore (Fig. 1), and the exciton circular dichroism (CD) spectra of the 4-(dimethylamino)cinnamoate esters were obtained for the coupled products (Fig. 3). The CD spectra are mirror images of each other and show exciton split CD Cotton effects. For the (4R)-enantiomer, a negative first (longer wavelength) and positive second Cotton effects were expected, because the 4-(dimethylamino)cinnamoyl and the pentaenacyl chromophores constitute a counterclockwise screw (Fig. 3), whereas for the (4S)-enantiomer the opposite Cotton effects are expected (53). The obtained CD spectra were compared with the CD spectra of related 4-(dimethylamino)cinnamoyl esters of (4S)- and (4R)-OH-retinal (23) and the absolute configuration of the 4-OH-RA enantiomers was confirmed. These data show that CYP26A1 hydroxylates atRA predominantly to the (4S)-OH-RA.
FIGURE 3.

Determination of the absolute configuration of 4-OH-RA enantiomers by exciton CD spectroscopy. CD spectra (top) of the 4-(dimethylamino)cinnamoyl esters of methyl (4S)- and (4R)-OH-retinoate in acetonitrile. The bottom panels show the structures of the 4-(dimethylamino)cinnamoyl esters. The UV visible spectrum of the enantiomers is shown in green in the top panel.
The Michaelis-Menten kinetic values of (4S)-OH-RA formation by CYP26A1 were determined (supplemental Fig. S3). The kcat for (4S)-OH-RA formation was 2.1 ± 0.2 pmol/min/pmol of P450 and Km 10 ± 6 nm. The Km and kcat of (4R)-OH-RA formation could not be determined due to the very low formation of this enantiomer and the lack of concentration dependence in the formation (supplemental Fig. S3).
Stereoselectivity in 4-OH-RA Formation by Other P450 Isoforms
The stereoselective formation of 4-OH-RA enantiomers by CYP3A4, CYP3A5, CYP3A7, and CYP2C8 was determined, as these enzymes have been previously shown to metabolize atRA in human liver. The chromatograms of the separation of the products formed are shown in supplemental Fig. S4. The other P450 enzymes were much less stereoselective in the 4-hydroxylation of atRA than CYP26A1. CYP3A4 formed (4R)-OH-RA and (4S)-OH-RA in a ∼1:1 ratio with equal Km and kcat values (Table 1). CYP3A5 formed (4R)-OH-RA twice as efficiently as (4S)-OH-RA, whereas CYP3A7 formed (4S)-OH-RA nearly twice as efficiently as (4R)-OH-RA (Table 1). The determined kinetic constants summarized in Table 1 show that stereoselective product ratios are mainly due to different kcat values and not differences in Km for formation of the (4S)- and (4R)-OH-RA.
TABLE 1.
Michaelis-Menten kinetic parameters for (4S)-OH-RA and (4R)-OH-RA formation by CYP3A enzymes
| CYP3A4 |
CYP3A5 |
CYP3A7 |
||||
|---|---|---|---|---|---|---|
| (4S)-OH-RA | (4R)-OH-RA | (4S)-OH-RA | (4R)-OH-RA | (4S)-OH-RA | (4R)-OH-RA | |
| Km (μm) | 4.7 ± 0.6 | 4.6 ± 1.1 | 6.0 ± 1.2 | 6.0 ± 1.1 | 3.5 ± 1.3 | 3.1 ± 0.6 |
| kcat (pmol/min/pmol P450) | 5.1 ± 0.2 | 5.3 ± 0.6 | 3.6 ± 0.2 | 7.0 ± 0.4 | 5.1 ± 0.6 | 2.7 ± 0.2 |
| kcat /Km (μl/min/pmol P450) | 1.1 | 1.2 | 0.6 | 1.1 | 1.5 | 0.9 |
Metabolism of 4-OH-RA Enantiomers by CYP26A1
Racemic 4-OH-RA is a substrate of CYP26A1, and several sequential metabolites are formed from 4-OH-RA by CYP26A1 (15). Mass spectrometric analysis of the metabolic products formed from (4S)-OH-RA showed that the dihydroxylated product, 4,16-OH2-RA, was the predominant metabolite formed from (4S)-OH-RA (Fig. 4) and 4-oxo-RA was a minor metabolite. In contrast, 4-oxo-RA was the major product formed from (4R)-OH-RA along with multiple dihydroxylated metabolites including 4,16-OH2-RA, 4,18-OH2-RA, 4-oxo-16-OH-RA, and 4-oxo-18-OH-RA. These products were also detectable from (4S)-OH-RA but were quantitatively minor (Fig. 4). The identification of the metabolites was similar to those shown previously from racemic 4-OH-RA (15).
FIGURE 4.

Identification of the metabolites formed from (4S)-OH-RA (a) and (4R)-OH-RA (b) by CYP26A1. The negative ion MS/MS transitions monitored are shown next to each trace and the identified metabolites are indicated for each peak. Incubations and analysis were carried out as described under “Experimental Procedures” and metabolic products were identified as described previously (15).
To determine the affinity of (4S)-OH-RA and (4R)-OH-RA to CYP26A1, both enantiomers were incubated with CYP26A1 and the formation of 4-oxo-RA from (4R)-OH-RA and 4,16-OH2-RA from (4S)-OH-RA was measured as a function of 4OH-RA concentration (Fig. 5, a and b). Although the Km value for (4S)-OH-RA (37.4 ± 9.7 nm) was lower than that for (4R)-OH-RA (45.8 ± 12.3 nm), the Km values were not significantly different from each other (p > 0.25). The kcat value for 4-oxo-RA formation from (4R)-OH-RA was 11.3 ± 1.3 pmol/min/pmol of P450.
FIGURE 5.

Metabolism of (4S)-OH-RA (left panel) and (4R)-OH-RA (right panel) by CYP26A1. The formation of specific metabolites was measured using (4S)-OH-RA (panel a) and (4R)-OH-RA (panel b) as substrates, and the Km value was determined according to Equation 2. The relative formation rate of 4,16-OH2-RA was quantified from the peak height ratio, whereas 4-oxo-RA formation was quantified using a standard curve. Error bars show the S.D. of triplicate measurements for each point. Panels c and d show the depletion of (4S)-OH-RA and (4R)-OH-RA by CYP26A1 at 2.5 nm initial concentration (closed circles). The closed squares show the depletion of 4OH-RA enantiomers in no-enzyme control incubations. No NADPH controls were also done and they were identical to the no-enzyme controls (not shown). Error bars show the S.D. of triplicate incubations. Representative determinations of the concentration-dependent depletion of (4S)-OH-RA and (4R)-OH-RA by CYP26A1 are shown in panels e and f, respectively. The depletion rates were measured at six different concentrations and the depletion rate constants were determined (insets in panels e and f). All depletion experiments were conducted on multiple days and in triplicate on each day. S.D. for the individual measurements are shown by error bars. Equation 1 was fitted to the data of depletion constants as a function of 4-OH-RA concentration to determine the Km and kdep,max of (4S)-OH-RA and (4R)-OH-RA depletion by CYP26A1. The kinetic values for (4S)-OH-RA were Km = 5.2 ± 1.3 nm and kdep,max = 2.9 ± 0.3 min−1. For (4R)-OH-RA the values were Km = 11.0 ± 5.2 nm and kdep,max = 1.5 ± 0.2 min−1.
To determine the catalytic efficiency of CYP26A1 in metabolizing 4-OH-RA enantiomers substrate depletion experiments were conducted. When (4S)-OH-RA and (4R)-OH-RA were incubated with CYP26A1 at 2.5 nm concentrations (≪Km) the depletion of (4S)-OH-RA was significantly faster (p < 0.005, kdep = 1.8 ± 0.5 1/min) than that of (4R)-OH-RA (kdep = 0.6 ± 0.02 1/min) (Fig. 5). To further confirm the different kinetics of (4R)-OH-RA and (4S)-OH-RA depletion by CYP26A1 the depletion rates were analyzed at different concentrations. The depletion curves as well as the relationship between kdep and the 4-OH-RA concentration are shown in Fig. 5, e and f. The (4S)-OH-RA had significantly higher (p < 0.005) intrinsic clearance (1.4 ± 0.1 ml/min/pmol P450) than (4R)-OH-RA (0.7 ± 0.1 ml/min/pmol of P450). The affinity of (4S)-OH-RA (Km = 5.2 ± 1.3 nm) was not significantly different from that of (4R)-OH-RA (Km = 11.0 ± 5.2 nm).
Homology Model of atRA-bound CYP26A1 and Molecular Docking of atRA to CYP3A4
To explore the stereoselectivity in 4-OH-RA formation by CYP26A1 in comparison to CYP3A4, a homology model of CYP26A1 with atRA in the active site was constructed and energy minimized. atRA was also docked to the CYP3A4 active site. Fig. 6, a and b, shows the orientation of atRA in the CYP26A1 active site.
FIGURE 6.

The binding of atRA to CYP26A1 (a and b) and CYP3A4 (c and d). The orientation of atRA within the energy minimized CYP26A1 homology model is shown from two different angles in panels a and b. In panels c and d, only the 1TQN x-ray crystal structure of CYP3A4 is shown for clarity. The atRA molecule bound to the 1TQN x-ray crystal structure is highlighted, because it is roughly average from all the bound orientations, which are shown semi-transparent. Stick models of atRA (orange), interacting residues (green), and the heme (red) with a schematic (gray) representation of protein structures in the background. Only the interacting amino acid side chains are shown.
According to the model of atRA bound with CYP26A1, the β-ionone ring is positioned over the heme allowing oxidation of C-4 as well as C-18 and C-16. The residues surrounding the β-ionone ring (Phe-299, Trp-112, Leu-120, Trp-304, and Ser-126, Fig. 6, a and b) position the (4S)-hydrogen 1.5 Å closer to the heme than (4R)-hydrogen (4S-hydrogen distance 4.6 Å, (4R)-hydrogen distance 6.1 Å). The distance between C-4 and the heme iron was 5.6 Å. Overall the CYP26A1 active site shown in the model is small and consists of a network of hydrophobic residues that surround the nonatetraene chain of atRA. The nonatetraene chain of atRA directly interacts with several residues, including Phe-84, Trp-112, Pro-113, Val-116, Phe-374, and the hydrophobic parts of Glu-303 and Val-370. The terminal carboxylic acid group of atRA interacts with Arg-64, the positively charged residue in the active site.
To investigate the potential binding orientations of atRA within the CYP3A4 active site, atRA was docked in the published CYP3A4 crystal structures (supplemental Fig. S5). atRA was found in a number of orientations within the CYP3A4 active site as shown in Fig. 6, c and d. The distances between the heme iron and the hydrogen atoms at C-4 varied between 3.1 and 16.5 Å. The average distance was not different for (4S) and (4R) positions (Table 2), in agreement with similar rates of formation of (4R)- and (4S)-OH-RA by CYP3A4. Consistent with a large flexible active site of CYP3A4, the predicted affinity of the preferred orientation of atRA within the CYP3A4 active site varied 50-fold among the known crystal structures. The average affinity of 2.5 μm is similar to the experimentally measured Km of 3 μm.
TABLE 2.
Kd and distance parameters for docking of atRA to the available crystal structures of CYP3A4
| PDB ID | Kd | Distance of 4R-H from heme | Distance of 4S-H from heme |
|---|---|---|---|
| μm | Å | Å | |
| 1TQN | 0.93 | 7.3 | 8.2 |
| 1W0E | 0.93 | 5.3 | 6.3 |
| 1W0F | 0.93 | 4.3 | 4.0 |
| 1W0G | 3.00 | 4.0 | 5.3 |
| 2J0D | 2.15 | 11.3 | 9.9 |
| 2V0M | 0.30 | 16.5 | 16.4 |
| 3TJS | 0.40 | 12.0 | 11.9 |
| 3UA1 | 13.52 | 3.1 | 3.5 |
| 3NXU | 0.67 | 9.1 | 9.5 |
| Average | 2.54 ± 3.97 | 8.1 ± 4.2 | 8.3 ± 3.9 |
The orientation of atRA docked in the 1TQN crystal structure of CYP3A4 (Fig. 6, c and d) corresponds best to the average orientation of atRA in the different CYP3A4 structures. In this orientation, the β-ionone ring is located between residues Arg-105, Phe-108, Ile-120, Phe-215, and Ser-119 positioning the ring in an angle that allows hydrogen atom abstraction from both the pro-S and pro-R position. The orientation of the β-ionone ring within the CYP3A4 active site in all structures (supplemental Fig. S5) has the C-16 located away from the heme, which is consistent with the lack of formation of the 16-OH-RA by CYP3A4. The nonatetrene chain of atRA interacts with hydrophobic residues, Phe-215, Met-371, Leu-482, Leu-483, and Ile-369, in addition to the hydrophobic regions of Arg-212 and Thr-309 in the atRA docked 1TQN structure. The carboxylic acid group of atRA hydrogen bonds with Ser-312 and Glu-308. Repulsion from the hydrophilic residues within the active site might explain the large differences in the atRA orientations between the docked structures (Table 2).
DISCUSSION
The physiological effects of RA demonstrate high stereoselectivity, as atRA is believed to be the biologically active geometric isomer with less activity residing in the 13-cis-RA, 9-cis-RA, and 9,13-dicis-RA stereoisomers. Generally, atRA has a higher affinity for many proteins such as RARs, CYP26A1, and cellular retinoic acid-binding proteins than the other RA isomers (16, 26, 54). However, the stereoselective metabolism of atRA and the role of stereoselectivity in the activity and biological fate of the metabolites have not been previously reported. This study shows for the first time that the hydroxylation of atRA by CYP26A1 is highly stereoselective. In contrast, the P450 enzymes that are not believed to be critical in atRA metabolism, CYP3A4, CYP3A5, CYP3A7, and CYP2C8, showed limited stereoselectivity suggesting that this is an inherent property of the enzymes that evolved to mediate atRA homeostasis.
Despite the fact that CYP26A1 forms preferentially (4S)-OH-RA, both 4-OH-RA enantiomers are good substrates of CYP26A1. This is plausible as the (4R)-OH-RA can be formed by the fetal liver CYP3A7, and by CYP3A4, CYP3A5, and CYP2C8 in the adult liver, and hence an elimination pathway for this enantiomer is also needed. However, the metabolism of 4-OH-RA by CYP26A1 was steresoselective with (4S)-OH-RA having significantly higher (p < 0.005) intrinsic clearance than (4R)-OH-RA. Although the Km value for (4S)-OH-RA depletion was consistently lower than that of (4R)-OH-RA the Km values between the enantiomers were not significantly different. As such, the greater CLint of (4S)-OH-RA is likely due to higher kcat for the formation of the diol metabolite observed from (4S)-OH-RA than for the formation of 4-oxo-RA from the (4R)-OH-enantiomer. Interestingly, as shown before for metabolism of atRA by CYP26A1, the Km values measured via product formation were higher than those measured via substrate depletion. This could, in part be due to the fact that the determination of the affinity via product formation allows correction for the depletion of free substrate due to binding to CYP26A1. The CLint and Km values obtained for the 4-OH-RA enantiomers were similar to the CLint values obtained for 4-OH-RA formation and atRA depletion by CYP26A1 (15, 25).
Different metabolites were formed from (4S)- and (4R)-OH-RA by CYP26A1, suggesting that the binding orientations of the two enantiomers in the CYP26A1 active site are different. The profile of metabolites formed from (4S)-OH-RA by CYP26A1 is similar to that observed following incubations of atRA with CYP26A1, whereas the products formed from (4R)-OH-RA do not agree well with the product profile from atRA (15). These results support the finding that CYP26A1 metabolizes atRA predominantly to (4S)-OH-RA, which is sequentially metabolized to the 4,16-OH2-RA product. In addition, the fact that only 4,16-OH2-RA was formed from (4S)-OH-RA suggests that the other diols observed in atRA incubations with CYP26A1 are formed by 4-hydroxylation of the primary metabolites 18-OH-RA and 16-OH-RA. These data also suggest that in vivo 4-oxo-RA is formed via different enzymatic pathway from atRA, and not by CYP26A1. The high binding affinity of (4S)-OH-RA determined here also partially explains the observed nondissociative metabolism of atRA by CYP26A1 (25). Whether metabolism of 4-OH-RA by UGT enzymes and enzymes forming 4-oxo-RA is also stereoselective is currently not known.
The stereoselectivity of hydroxylation of atRA by CYP26A1 offers a unique opportunity to enhance our understanding of the molecular interactions between atRA and CYP26A1, and to refine the understanding of the structure of the CYP26A1 active site. The stereoselective formation of (4S)-OH-RA by CYP26A1 likely requires that the hydroxyl group be delivered from the si-face of the molecule to give (4S)-OH-RA. This is well illustrated in the homology model of CYP26A1, which shows a very well defined niche for atRA within the CYP26A1 active site and a distinct orientation of the β-ionone ring above the plane of the heme. The orientation of atRA within the CYP26A1 active site is consistent with stereoselective hydrogen atom abstraction from the pro-(S)-position and subsequent hydroxyl radical rebound. P450-mediated hydroxylations usually occur with retention of the stereochemistry at the reacting carbon, although occasionally products arising from both retention and inversion are reported (55).
In terms of the active site residues, the presented homology model is in good agreement with previously reported homology models of CYP26A1 (41–43). The previous models are also consistent with the stereoselective 4S-hydroxylation of atRA. The presented model also explains both the stereoselectivity of 4-hydroxylation of atRA and the observed lack of regioselectivity in hydroxylation (15, 26). It shows that the hydrogen atoms in C-16 and C-18 lie in a plane above the heme together with the pro-S hydrogen atom at C-4. As such, the model suggests that hydroxylation at the C-16 position would also be stereoselective as only one of the gem-methyl groups is close to the heme. The inferred limited mobility of atRA within the CYP26A1 active site is in contrast with the apparent greater mobility within the CYP3A4 active site. The multiple binding modes found for atRA with the CYP3A4 structures together with the similar average distances of the pro-S and pro-R hydrogen atoms is in agreement with the observed equal rates of (4S)-OH-RA and (4R)-OH-RA products formed by CYP3A4.
In summary, this study reports for the first time the synthesis and determination of the absolute configuration of 4-OH-RA enantiomers and shows that CYP26A1 forms the (4S)-OH-RA stereoselectively. The observed product stereoselectivity and metabolism of both 4-OH-RA enantiomers by CYP26A1 will be useful in better modeling of CYP26A1 and aid in the structural design of novel CYP26A1 inhibitors as therapeutic agents. These results show that CYP26A1 accepts various biologically active retinoids as substrates, but has a conserved preference for the (4S)-OH-RA enantiomer over (4R)-OH-RA. These results show that the two 4-OH-RA enantiomers are physiologically distinct due to their different formation and elimination kinetics resulting in likely different cellular exposures to these enantiomers and formation of different downstream active metabolites such as 4-oxo-RA. Further studies are needed to determine the role of stereoselectivity in the activity of the 4-OH-RA enantiomers and in the formation of 4-oxo-RA from the individual stereoisomers.
Acknowledgments
We thank Dr. Alex Zelter and Dr. Jayne E. Thatcher for their assistance in enzyme expression, preparation of microsomes, and conduct of preliminary experiments, Dr. David R. Goodlett for use of the Linux cluster for the molecular dynamics simulations, and Dr. Justin Lutz for assistance in obtaining the accurate mass measurements.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-GM081569, R01-GM081569-S1, and GM054995-16.

This article contains supplemental Data, Tables S1 and S2, and Figs. S1–S5.
- atRA
- all-trans-retinoic acid
- RAR
- retinoic acid receptor.
REFERENCES
- 1. Blomhoff R., Blomhoff H. K. (2006) Overview of retinoid metabolism and function. J. Neurobiol. 66, 606–630 [DOI] [PubMed] [Google Scholar]
- 2. Jacobs S., Lie D. C., DeCicco K. L., Shi Y., DeLuca L. M., Gage F. H., Evans R. M. (2006) Retinoic acid is required early during adult neurogenesis in the dentate gyrus. Proc. Natl. Acad. Sci. U.S.A. 103, 3902–3907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Manolescu D. C., Sima A., Bhat P. V. (2010) All-trans retinoic acid lowers serum retinol-binding protein 4 concentrations and increases insulin sensitivity in diabetic mice. J. Nutr. 140, 311–316 [DOI] [PubMed] [Google Scholar]
- 4. Altucci L., Leibowitz M. D., Ogilvie K. M., de Lera A. R., Gronemeyer H. (2007) RAR and RXR modulation in cancer and metabolic disease. Nat. Rev. Drug. Discov. 6, 793–810 [DOI] [PubMed] [Google Scholar]
- 5. Samokyszyn V. M., Gall W. E., Zawada G., Freyaldenhoven M. A., Chen G., Mackenzie P. I., Tephly T. R., Radominska-Pandya A. (2000) 4-Hydroxyretinoic acid, a novel substrate for human liver microsomal UDP-glucuronosyltransferase(s) and recombinant UGT2B7. J. Biol. Chem. 275, 6908–6914 [DOI] [PubMed] [Google Scholar]
- 6. Nadin L., Murray M. (1999) Participation of CYP2C8 in retinoic acid 4-hydroxylation in human hepatic microsomes. Biochem. Pharmacol. 58, 1201–1208 [DOI] [PubMed] [Google Scholar]
- 7. McSorley L. C., Daly A. K. (2000) Identification of human cytochrome P450 isoforms that contribute to all-trans-retinoic acid 4-hydroxylation. Biochem. Pharmacol. 60, 517–526 [DOI] [PubMed] [Google Scholar]
- 8. Marill J., Cresteil T., Lanotte M., Chabot G. G. (2000) Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol. Pharmacol. 58, 1341–1348 [DOI] [PubMed] [Google Scholar]
- 9. Eckhoff C., Nau H. (1990) Identification and quantitation of all-trans- and 13-cis-retinoic acid and 13-cis-4-oxoretinoic acid in human plasma. J. Lipid Res. 31, 1445–1454 [PubMed] [Google Scholar]
- 10. Arnold S. L., Amory J. K., Walsh T. J., Isoherranen N. (2012) A sensitive and specific method for measurement of multiple retinoids in human serum with UHPLC-MS/MS. J. Lipid Res. 53, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. White J. A., Beckett-Jones B., Guo Y. D., Dilworth F. J., Bonasoro J., Jones G., Petkovich M. (1997) cDNA cloning of human retinoic acid-metabolizing enzyme (hP450RAI) identifies a novel family of cytochromes P450. J. Biol. Chem. 272, 18538–18541 [DOI] [PubMed] [Google Scholar]
- 12. White J. A., Ramshaw H., Taimi M., Stangle W., Zhang A., Everingham S., Creighton S., Tam S. P., Jones G., Petkovich M. (2000) Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc. Natl. Acad. Sci. U.S.A. 97, 6403–6408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thatcher J. E., Zelter A., Isoherranen N. (2010) The relative importance of CYP26A1 in hepatic clearance of all-trans-retinoic acid. Biochem. Pharmacol. 80, 903–912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Duester G. (2008) Retinoic acid synthesis and signaling during early organogenesis. Cell 134, 921–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Topletz A. R., Thatcher J. E., Zelter A., Lutz J. D., Tay S., Nelson W. L., Isoherranen N. (2012) Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem. Pharmacol. 83, 149–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Idres N., Marill J., Flexor M. A., Chabot G. G. (2002) Activation of retinoic acid receptor-dependent transcription by all-trans-retinoic acid metabolites and isomers. J. Biol. Chem. 277, 31491–31498 [DOI] [PubMed] [Google Scholar]
- 17. Reynolds N. J., Fisher G. J., Griffiths C. E., Tavakkol A., Talwar H. S., Rowse P. E., Hamilton T. A., Voorhees J. J. (1993) Retinoic acid metabolites exhibit biological activity in human keratinocytes, mouse melanoma cells and hairless mouse skin in vivo. J. Pharmacol. Exp. Ther. 266, 1636–1642 [PubMed] [Google Scholar]
- 18. van der Leede B. M., van den Brink C. E., Pijnappel W. W., Sonneveld E., van der Saag P. T., van der Burg B. (1997) Autoinduction of retinoic acid metabolism to polar derivatives with decreased biological activity in retinoic acid-sensitive, but not in retinoic acid-resistant human breast cancer cells. J. Biol. Chem. 272, 17921–17928 [DOI] [PubMed] [Google Scholar]
- 19. Sonneveld E., van den Brink C. E., Tertoolen L. G., van der Burg B., van der Saag P. T. (1999) Retinoic acid hydroxylase (CYP26) is a key enzyme in neuronal differentiation of embryonal carcinoma cells. Dev. Biol. 213, 390–404 [DOI] [PubMed] [Google Scholar]
- 20. Langton S., Gudas L. J. (2008) CYP26A1 knockout embryonic stem cells exhibit reduced differentiation and growth arrest in response to retinoic acid. Dev. Biol. 315, 331–354 [DOI] [PubMed] [Google Scholar]
- 21. Niederreither K., Abu-Abed S., Schuhbaur B., Petkovich M., Chambon P., Dollé P. (2002) Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat. Genet. 31, 84–88 [DOI] [PubMed] [Google Scholar]
- 22. Achkar C. C., Derguini F., Blumberg B., Langston A., Levin A. A., Speck J., Evans R. M., Bolado J., Jr., Nakanishi K., Buck J., Gudas L. J. (1996) 4-Oxoretinol, a new natural ligand and transactivator of the retinoic acid receptors. Proc. Natl. Acad. Sci. U.S.A. 93, 4879–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Katsuta Y., Ito M., Yoshihara K., Nakanishi K., Kikkawa T., Fujiwara T. (1994) Retinoids and related compounds. Part 16. Synthesis of (+)-(4S)- and (−)-(4R)-(11Z)-4-hydroxyretinals and determination of the absolutestereochemistry of a visual pigment chromaphore in the firefly squid, Watasenia scintillans. J. Org. Chem. 59, 6917–6921 [Google Scholar]
- 24. Moise A. R., Domínguez M., Alvarez S., Alvarez R., Schupp M., Cristancho A. G., Kiser P. D., de Lera A. R., Lazar M. A., Palczewski K. (2008) Stereospecificity of retinol saturase. Absolute configuration, synthesis, and biological evaluation of dihydroretinoids. J. Am. Chem. Soc. 130, 1154–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lutz J. D., Dixit V., Yeung C. K., Dickmann L. J., Zelter A., Thatcher J. E., Nelson W. L., Isoherranen N. (2009) Expression and functional characterization of cytochrome P450 26A1, a retinoic acid hydroxylase. Biochem. Pharmacol. 77, 258–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thatcher J. E., Buttrick B., Shaffer S. A., Shimshoni J. A., Goodlett D. R., Nelson W. L., Isoherranen N. (2011) Substrate specificity and ligand interactions of CYP26A1, the human liver retinoic acid hydroxylase. Mol. Pharmacol. 80, 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Obach R. S., Reed-Hagen A. E. (2002) Measurement of Michaelis constants for cytochrome P450-mediated biotransformation reactions using a substrate depletion approach. Drug. Metab. Dispos. 30, 831–837 [DOI] [PubMed] [Google Scholar]
- 28. Takakuwa T., Konno T., Meguro H. (1985) A new standard substance for calibration of circular dichroism. Ammonium d10 camphorsulfonate. Anal. Sci. 1, 215–218 [Google Scholar]
- 29. Eswar N., Eramian D., Webb B., Shen M. Y., Sali A. (2008) Protein structure modeling with MODELLER. Methods Mol. Biol. 426, 145–159 [DOI] [PubMed] [Google Scholar]
- 30. Fiser A., Sali A. (2003) Modeller. Generation and refinement of homology-based protein structure models. Methods Enzymol. 374, 461–491 [DOI] [PubMed] [Google Scholar]
- 31. Kühnel K., Ke N., Cryle M. J., Sligar S. G., Schuler M. A., Schlichting I. (2008) Crystal structures of substrate-free and retinoic acid-bound cyanobacterial cytochrome P450 CYP120A1. Biochemistry 47, 6552–6559 [DOI] [PubMed] [Google Scholar]
- 32. Schoch G. A., Yano J. K., Sansen S., Dansette P. M., Stout C. D., Johnson E. F. (2008) Determinants of cytochrome P450 2C8 substrate binding. Structures of complexes with montelukast, troglitazone, felodipine, and 9-cis-retinoic acid. J. Biol. Chem. 283, 17227–17237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Berendsen H. J. C., van der Spoel D., Drunen R. (1995) GROMACS. A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 91, 43–56 [Google Scholar]
- 34. Autenrieth F., Tajkhorshid E., Baudry J., Luthey-Schulten Z. (2004) Classical force field parameters for the heme prosthetic group of cytochrome c. J. Comput. Chem. 25, 1613–1622 [DOI] [PubMed] [Google Scholar]
- 35. Oda A., Yamaotsu N., Hirono S. (2005) New AMBER force field parameters of heme iron for cytochrome P450s determined by quantum chemical calculations of simplified models. J. Comput. Chem. 26, 818–826 [DOI] [PubMed] [Google Scholar]
- 36. Halgren T. A. (1996) Merck molecular force field. I. Basis, form, scope, parameterization and performance of MMFF94. J. Comput. Chem. 17, 490–519 [Google Scholar]
- 37. Berendsen H. J. C., Postma J. P. M., van Gunsteren W., DiNola A., Haak J. R. (1984) Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 38. Oostenbrink C., Soares T. A., van der Vegt N. F., van Gunsteren W. F. (2005) Validation of the 53A6 GROMOS force field. Eur. Biophys. J. 34, 273–284 [DOI] [PubMed] [Google Scholar]
- 39. Oostenbrink C., Villa A., Mark A. E., van Gunsteren W. F. (2004) A biomolecular force field based on the free enthalpy of hydration and solvation. The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 25, 1656–1676 [DOI] [PubMed] [Google Scholar]
- 40. Darden T., York D., Pedersen L. (1993) Paraticle mesh Ewald. An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 41. Gomaa M. S., Yee S. W., Milbourne C. E., Barbera M. C., Simons C., Brancale A. (2006) Homology model of human retinoic acid metabolising enzyme cytochrome P450 26A1 (CYP26A1). Active site architecture and ligand binding. J. Enzyme Inhib. Med. Chem. 21, 361–369 [DOI] [PubMed] [Google Scholar]
- 42. Karlsson M., Strid A., Sirsjo A., Eriksson L. A. (2008) Homology models and molecular modeling of human retinoic acid metabolizing enzymes cytochrome P450 26A1 (CYP26A1) and P450 26B1 (CYP26B1). J. Chem. Theory Comput. 4, 1021–1027 [DOI] [PubMed] [Google Scholar]
- 43. Ren J. H., Xiong X. Q., Sha Y., Yan M. C., Lin B., Wang J., Jing Y. K., Zhao D. M., Cheng M. S. (2008) Structure predication and R115866 binding study of human CYP26A1. Homology modeling, fold recognition, molecular docking and MD simulations. Mol. Simul. 34, 337–346 [Google Scholar]
- 44. Eisenberg D., Lüthy R., Bowie J. U. (1997) VERIFY3D. Assessment of protein models with three-dimensional profiles. Methods Enzymol. 277, 396–404 [DOI] [PubMed] [Google Scholar]
- 45. Colovos C., Yeates T. O. (1993) Verification of protein structures. Patterns of nonbonded atomic interactions. Protein Sci. 2, 1511–1519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Trott O., Olson A. J. (2010) AutoDock Vina. Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multi-threading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ekroos M., Sjögren T. (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U.S.A. 103, 13682–13687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sevrioukova I. F., Poulos T. L. (2012) Structural and mechanistic insights into the interaction of cytochrome P4503A4 with bromoergocryptine, a type I ligand. J. Biol. Chem. 287, 3510–3517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sevrioukova I. F., Poulos T. L. (2010) Structure and mechanism of the complex between cytochrome P4503A4 and ritonavir. Proc. Natl. Acad. Sci. U.S.A. 107, 18422–18427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yano J. K., Wester M. R., Schoch G. A., Griffin K. J., Stout C. D., Johnson E. F. (2004) The structure of human microsomal cytochrome P450 3A4 determined by x-ray crystallography to 2.05-Å resolution. J. Biol. Chem. 279, 38091–38094 [DOI] [PubMed] [Google Scholar]
- 51. Williams P. A., Cosme J., Vinkovic D. M., Ward A., Angove H. C., Day P. J., Vonrhein C., Tickle I. J., Jhoti H. (2004) Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science 305, 683–686 [DOI] [PubMed] [Google Scholar]
- 52. Corey E. J., Bakshi R. K., Shibata S. (1987) Highly enantioselective borane reduction of ketones catalyzed by chiral oxazaborolidines. Mechanism and synthetic implications. J. Am. Chem. Soc. 109, 5551–5553 [Google Scholar]
- 53. Cai G., Bozhkova N., Odingo J., Berova N., Nakanishi K. (1993) Circular dichroism exciton chirality method. New red-shifted chromophores for hydroxyl groups. J. Am. Chem. Soc. 115, 7192–7198 [Google Scholar]
- 54. Fiorella P. D., Napoli J. L. (1991) Expression of cellular retinoic acid-binding protein (CRABP) in Escherichia coli. Characterization and evidence that holo-CRABP is a substrate in retinoic acid metabolism. J. Biol. Chem. 266, 16572–16579 [PubMed] [Google Scholar]
- 55. Ortiz de Montellano P. R. (2010) Hydrocarbon hydroxylation by cytochrome P450 enzymes. Chem. Rev. 110, 932–948 [DOI] [PMC free article] [PubMed] [Google Scholar]