

Background: Antimicrobial peptides are new antibiotics avoiding resistance problems.
Results: Eurocin is a new antimicrobial peptide featuring a cysteine-stabilized αβ-fold. Eurocin binds the cell wall precursor lipid II but does not disrupt cell membranes.
Conclusion: Eurocin acts by inhibiting cell wall synthesis. Its structure is typical for invertebrate defensins.
Significance: Knowing the mode of action and structure is a prerequisite for pharmaceutical application of an antibiotic.
Keywords: Antibiotics Action, Antimicrobial Peptides, Cell Wall, Defensins, Nuclear Magnetic Resonance, Vesicles
Abstract
Antimicrobial peptides are a new class of antibiotics that are promising for pharmaceutical applications because they have retained efficacy throughout evolution. One class of antimicrobial peptides are the defensins, which have been found in different species. Here we describe a new fungal defensin, eurocin. Eurocin acts against a range of Gram-positive human pathogens but not against Gram-negative bacteria. Eurocin consists of 42 amino acids, forming a cysteine-stabilized α/β-fold. The thermal denaturation data point shows the disulfide bridges being responsible for the stability of the fold. Eurocin does not form pores in cell membranes at physiologically relevant concentrations; it does, however, lead to limited leakage of a fluorophore from small unilamellar vesicles. Eurocin interacts with detergent micelles, and it inhibits the synthesis of cell walls by binding equimolarly to the cell wall precursor lipid II.
Introduction
Throughout evolution antimicrobial peptides (AMP)4 are found to be an important defensive weapon in virtually all multicellular organisms (1). AMPs have been recognized as an important part of the innate immune system and have remained effective against bacterial, fungal, and viral infections to this day (2). Today antimicrobial peptides are of great interest in medicine as these peptides have bactericidal effects and are active against a broad range of pathogens. The sustained effectiveness throughout evolution suggests that antimicrobial peptides limit the opportunity for the development of bacterial resistance and that they could be the means to overcome the increasing problem with bacterial resistance to commonly used antibiotics (3, 4).
AMPs show enormous sequence diversity, and also different classes of structures of AMPs are known. A large group of AMPs have an amphipathic structure and are cationic due to a high content of arginine and lysine residues. The positive charge promotes the binding to membranes of microbes that are generally negatively charged (3). AMPs have been loosely classified into four classes according to their sequence and structure. These are: amphipathic α-helices, loops due to a single disulfide bond, extended molecules, and β-sheet molecules stabilized by two or more disulfide bonds (3).
Two predominant classes of AMPs in vertebrates are cathelicidins and defensins, both containing three conserved disulfide bonds. Cathelicidins are characterized by a highly conserved signal sequence and pro-regions, whereas the C-terminal domain, encoding the mature peptide, displays much diversity in sequence and structure (5). Defensins are characterized by a β-sheet-rich fold stabilized by three conserved intramolecular cysteine disulfide bridges (6). Defensins are divided into α-, β-, and θ-defensins. α- and β-defensins differ in peptide length and the pairing of cysteines in disulfide bonds. The θ-defensin is a circular peptide isolated from rhesus monkeys (7, 8).
Defensin-like peptides from higher plants and insects have a β-defensin like structure (2, 9). Several defensins have been isolated, and surprisingly, defensins from closely related species show less homology than defensins from species of different phyla (10). Many invertebrate (11) and fungal (12) defensins have a cysteine-stabilized α-helix/β-sheet structure similar to the structure of β-defensins from vertebrates. Among others, structures have been solved for Phormia defensin (13), Sarcophaga defensin (14), drosomycin (15), heliomycin (16), terramycin (17), MGD-1 (18), lucifensin (19), and plectasin (20). They all form an α-helix and two anti-parallel β-strands (αββ-scaffold) with the α-helix stabilized by two disulfide bridges to one strand of the β-sheet. Drosomycin and heliomycin are further characterized by an additional N-terminal β-strand (βαββ-scaffold) identical to the scaffold of some antifungal plant defensins (11).
AMPs make attractive candidates for therapeutic use for several reasons. They kill their target rapidly (compared with conventional antibiotics the development of antimicrobial resistance is unlikely), and they have additional adjuvant effects on the immune system (21). In this paper we present the structure and mechanism of action of the new defensin eurocin isolated from the fungus Eurotium amstelodami.
The gene encoding eurocin was discovered in a cDNA library of the ascomycete E. amstelodami (US2006/0223751). The cDNA clone encoded a putative defensin-like peptide that was named eurocin. The gene encodes 90 amino acids (signal peptide (amino acids (aa) 1–20), propeptide (aa 21–48), and defensin peptide (aa 49–90)). The mature peptide consists of 42 amino acids (Fig. 1) and has a molecular mass of 4.3 kDa. Eurocin has a high content (>10%) of both glycine (24%) and cysteine (14%) residues. The peptide encloses six cysteine residues, allowing for the formation of three disulfide bonds. This indicates that eurocin belongs to the group of antimicrobial peptides with β-sheets stabilized by two or more disulfide bonds.
FIGURE 1.

A, multiple alignment of eurocin, plectasin from P. nigrella (20) (gi:82407586), and six amino acid sequences of defensins found by a BLASTP on the amino acid sequence of eurocin are shown. The multiple alignment is performed with ClustalX Version 2.0 (64) using a BLOSUM matrix with a gap cost of 10 and a gap extension cost of 0.2. The six defensins are defensin from southern hawker (gi:118430) (13), amercin from lone star tick (gi:114438982) (65) (the N terminus is not shown), defensin-1 from American oyster (gi:118582050) (66), MGD-1 from Mediterranean mussel (gi:12084380) (67), micasin from M. canis (gi:343481536), and hemocyte defensin Defh2 from Pacific Oyster (gi:89275885) (47) (the N terminus is not shown). -, gaps; *, fully conserved amino acid; :, conserved region and ·, semi-conserved region. B, shown is a neighbor-joint tree based upon sequence alignment of eurocin. The tree was constructed in MEGA Version 5.05 (68) with 1000 bootstrap replications. The percentage of bootstrap appearance is indicated at the branches.
Bacterial cell wall biosynthesis represents a most relevant target pathway for antibiotic intervention. Numerous antibiotic classes like glycopeptides, e.g. vancomycin (22) and the lantibiotics (23, 24), have been shown to act via the disruption of this essential biosynthesis pathway through interaction with the cell wall precursor lipid II. Recently, it was shown that eurocin along with other fungal and metazoan defensins specifically forms a complex with the central cell wall precursor lipid II (25). It was also demonstrated that human β-defensin 3 (26) and human neutrophil peptide-1 (27) bind lipid II. It was, therefore, obvious to further investigate the structure and the mode of action of eurocin.
In the following, we describe the structural characterization of eurocin by solution-state NMR spectroscopy, its antimicrobial effect in vivo and in vitro, and the investigation of its mode of action by a broad range of experiments.
EXPERIMENTAL PROCEDURES
Materials
Dihexanoylphosphatidylcholine and dioleoylphosphatidylcholine were from Avanti Phospholipids (Alabaster, AL). Deuterium-labeled DPC was purchased from Cambridge Isotope Laboratories. All other chemicals used were purchased from Sigma.
Peptide Production
The gene encoding eurocin was cloned in the Aspergillus oryzae proprietary production host as previously described (20). By a three-step purification procedure, we were able to recover grams of purified peptide. More specifically, cells were removed from the fermentation broth by centrifugation at 6000 rpm. The supernatant was filtered (Fast PES bottle top filters, 0.22 μm; Nalgene 595-4520) and adjusted to pH 7.0 with NaOH. The peptide was then captured on a single-step MEP HyperCel capture column connected to an ÄKTA explorer 100. The column was equilibrated with 50 mm phosphate buffer, pH 7.0, and peptides were eluted with 50 mm formic acid, pH 4.0. The fractions containing the peptide was pooled, and the pH was adjusted to 5.0 with NaOH and loaded onto an ion exchange column equilibrated and washed with 50 mm malonic acid, pH 5.0. The peptide was eluted with a gradient of 50 mm malonic acid, 2 m NaCl, pH 5.0, 0–100% for 50 min. Finally, the sample was adjusted to pH 7.0 and loaded onto a 100-ml MEP HyperCel column equilibrated with 50 mm phosphate, pH 7.0. The column was washed with phosphate buffer and eluted with 50 mm acetate, pH 4.0. At this point, the purity exceeded 95%.
Bacterial Strains and Growth Conditions
Staphylococcus simulans 22 and Staphylococcus carnosus TM 300 were maintained on tryptic soy agar and grown at 37 °C.
Micrococcus Luteus
DSM 1790 was used for membrane preparations and was grown at 30 °C in Tryptone soy broth.
Susceptibility Testing
Determination of minimal inhibitory concentration (MIC) was performed in 96-well polypropylene microtiter plates (Nunc brand) by standard broth microdilution in cation-adjusted Mueller-Hinton broth (Oxoid) according to the general guidelines provided by Clinical and Laboratory Standards Institute/National Committee for Clinical Laboratory Standards (M7-A5). A bacterial collection of ATCC susceptibility reference strains as well as 183 clinical important strains, mainly Gram-positive bacteria, were tested against eurocin and appropriate reference antibiotics. The bacteria tested here cover 15 different genera of aerobic or facultative anaerobic strains of human clinical origin including Staphylococcus aureus, Streptococcus pneumoniae, and Enterococcus faecalis. 15 Gram-negative strains were tested representing Enterobacteriaceae as well as Aeromonas hydrophila, Pseudomonas aeruginosa, Stenotrophomonas maltophilia, Burkholderia cepacia, and Moraxella catarrhalis.
In Vivo Efficacy
The in vivo anti-infective activity of eurocin was evaluated in a mouse model of systemic bacteremia peritoneal infection. The peritonitis model was performed with a S. pneumoniae strain serotypes 2 (D39) ATCC33400 where a 106 cfu inoculum was introduced into the peritoneum. The effect of treatment was evaluated as a reduction in peritoneal bacterial counts where inoculated mice were treated 1 h later with either eurocin (10 mg/kg, intravenously) or vancomycin (70 mg per kg, subcutaneously) as compared with untreated control mice, and peritoneal bacterial counts were performed at 0, 2, and 5 h after treatment (20).
To evaluate the effective dose (ED50) 38 NMRI mice were infected intraperitoneally with 106 cfu of S. pneumoniae D39. 1 h post-inoculation the mice were treated (intravenously) once with 1 of 8 doses (0.06–14 mg/kg). Four hours later, bacterial counts were obtained from both blood and peritoneal fluid, and ED50 values were calculated analyzing a sigmoid dose-response with variable slope.
NMR Structure Determination
Eurocin samples for structure determination by NMR contained 1.68 mm eurocin and 95 mm formic acid in 95% MilliQ H2O, 5% D2O (v/v). The samples had a pH of 4.5.
All NMR experiments were carried out using a BRUKER DRX600 spectrometer operating at a field strength of 14.1 tesla with a 5-mm triple-axis gradient TXI(H/C/N) probe. TopSpin v. 1.3 was used for recording and processing NMR data. Spectra were referenced relative to internal 4,4-dimethyl-4-silapentane-1-sulfonic acid. All experiments were performed at 298.1 K unless indicated otherwise. Spectra recorded were double quantum filtered-COSY, [1H,1H]-clean-TOCSY (28) with 80 ms spin-lock of 15 kHz and WATERGATE (29) water suppression, and [1H,1H]-NOESY with a 75-ms mixing time and WATERGATE water suppression. In addition, natural abundance [1H,15N]-HSQC without water saturation (30) and [13C,1H]-HSQC spectra for the aliphatic and the aromatic region were obtained. Assignment of the NMR resonances was performed by the “sequential walk” method (31). Interpretation and assignment were carried out using the program CARA 1.5.5 (32). Backbone torsion angle restraints were obtained from secondary chemical shifts using the program TALOS+ (33). TALOS+-derived angle constraints were only accepted for residues, where all the 10 best database hits were situated within the same region of the Ramachandran plot. A deviation from the TALOS+-derived value of ±30° was allowed. The CALIBA (34) subroutine in CYANA was used to convert cross-peak intensities from NOESY spectra into distance constraints. The CYANA subroutine FOUND (35) was used to add angle restraints. On the basis of this, input the structure was calculated using the torsion angle dynamics program CYANA 2.1 (36). Structure calculations were started from 100 conformers with random torsion angle values. The 20 conformers with the lowest final CYANA target function values were energy-minimized with YASARA (37) through two steps. First, in vacuo with the NOVA force field (37) and then, second, with water as explicit solvent using the particle mesh Ewald method (38) and the YASARA force field (39). No further refinement was performed. The structures were checked by PROCHECK_NMR (40).
Structures were initially calculated without any assumptions on disulfide bridge topology. Disulfide bridge topology was derived from the calculated structures, and distance constraints defining the disulfide bridge topology were added for another round of structure calculations.
Secondary Structure and Thermal Stability
CD wavelength scans were performed on a 45 μm eurocin solution at pH values from 2.0 to 12.0 in steps of 1 pH unit. The buffers used were phosphate (pH 2, 3, 7, 8, and 12), acetate (pH 4 and 5), MES (pH 6), glycine (pH 9 and 10), CAPS (pH 11); all buffers were 50 mm in concentration. Spectra were recorded from 250 to 200 nm at 298.1 K on a Jasco J-810 spectropolarimeter with a Jasco PTC-423S temperature control unit and using Spectra Manager v. 1.53.01 software. At each pH value, thermal scans were recorded at a wavelength of 215 nm. The scans were measured from 298.1 to 378.1 K with a data pitch of 0.2 K and a temperature slope of 60 K h −1. The melting temperature was calculated by fitting data to Equation 1,
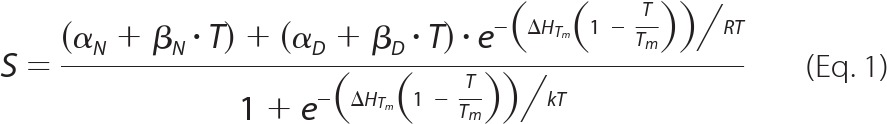 |
where S is the molar ellipticity (deg cm2 dmol−1), ΔHTm is the temperature-dependent enthalpy change (J mol−1), T is the temperature (K), Tm refers to the melting temperature (K), and R is the ideal gas constant 8.3144 (J mol−1 K−1). The parameters αN, βN, αD, and βD refer to the values (α) and temperature dependence (β) of the ellipticity in the native (N) and denatured (D) states, respectively.
A CD thermal scan at pH 4.5 was repeated in the presence of 1 and 20 mm DTT to assess the importance of the disulfide bridges on the structural stability. For this scan, the wavelength of 220 nm was monitored.
Interaction with Lipid and Detergents
CD wavelength scans were repeated in the presence of 0, 1, 2, 4, 8, 16, or 64 mm DPC and 0, 2, 6, 10, 20, 41, or 100 mm dihexanoylphosphatidylcholine. CD thermal scans were repeated as described above in the presence of 32 mm DPC between pH 2.0 and 12.0 in steps of 1 pH unit.
NMR -[1H,1H]-clean-TOCSY with a 20-ms spin-lock of 15 kHz and WATERGATE water suppression were recorded at 310.1 K on samples containing 0.84 mm eurocin, 95 mm formic acid, to which DPC was added to concentrations of 0.33, 0.66, 1, 2, 3, 4, 6, 8, 10, 12, 14, 16, 20, 22, and 50 mm. The NMR chemical shift changes of Hα and HN atoms of eurocin upon titration with DPC are given as an absolute change in chemical shift, Δδabs, calculated by Equation 2
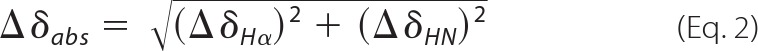 |
Chemical shift changes of eurocin were used to calculate the concentration of free and DPC-bound eurocin, respectively. A Langmuir isotherm (Equation 3) was then used to model the binding of eurocin to the DPC micelle (41).
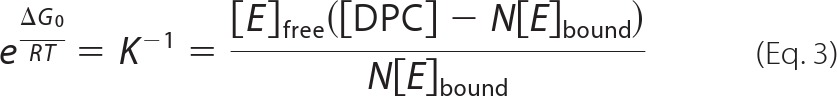 |
where [E]free and [E]bound are the concentrations of free and bound peptide, respectively, N is the number of DPC molecules interacting per molecule eurocin, K is the affinity constant of eurocin for DPC, and ΔG0 is the Gibb's free energy of the interaction.
Fluorescence measurements were conducted on an Eclipse fluorimeter (Cary-Varian, Palo Alto, CA). Fluorescence emission was scanned from 310 to 400 nm with an excitation wavelength of 295 nm to monitor tryptophan fluorescence of eurocin at pH 6 in the absence and presence of 50 mm DPC.
Intracellular Accumulation of the Final Soluble Cell Wall Precursor UDP-N-acetyl-muramyl Pentapeptide
Analysis of the cytoplasmic peptidoglycan nucleotide precursor pool was examined using the method of Kohlrausch and Höltje (42) with slight modifications. S. simulans 22 was grown in Mueller-Hinton broth to an A600 of 0.5 and supplemented with 130 pg/ml chloramphenicol. After 15 min of incubation, vancomycin and eurocin were added at 10 × MIC (5 μg/ml and 3 μg/ml, respectively) and incubated for another 30 min. Subsequently, cells were rapidly cooled on ice and spun down (15,000 × g, 5 min, 4 °C), resuspended in cold water, and under stirring, extracted with boiling water. Cell debris was removed (48,000 × g, 30 min), and the supernatant was lyophilized. UDP-linked cell wall precursors were analyzed by reversed-phase HPLC, and the identities were confirmed by mass spectrometry.
Potassium Release from Whole Cells
Cells of S. carnosus TM300 were harvested at an A600 of 1.0–1.5, washed with cold choline buffer (300 mm choline chloride, 30 mm MES, 20 mm Tris, pH 6.5) and resuspended to an A600 of 30. The concentrated cell suspension was kept on ice and used within 30 min. For each measurement the cells were diluted in choline buffer supplemented with 10 mm glucose (25 °C) to an A600 of about 3. Peptide-induced leakage was monitored relative to the total amount of potassium release after the addition of 1 μm lantibiotic nisin (positive control) over 300 s using a potassium-sensitive electrode. Eurocin was added at 1, 3, and 10 × MIC (corresponding to 0.069, 0.21, and 0.69 μm, respectively). The fungal defensin plectasin (10 × MIC (2 μm)) was used as the negative control.
In Vitro Peptidoglycan Synthesis with Isolated Membranes
In vitro lipid II synthesis was performed using membranes of M. luteus as previously described (43). Briefly, synthesis was performed in a total volume of 50 μl containing 150–200 μg of membrane protein, 5 nmol of undecaprenyl phosphate (C55-P), 50 nmol of UDP-MurNAc-pentapeptide, 50 nmol of UDP-GlcNAc in 60 mm Tris-HCl, 5 mm MgCl2, pH 8, and 0.5% (w/v) Triton X-100. For quantitative analysis, [14C]UDP-GlcNAc (0.25 nmol) was added to the reaction mixture. After incubation for 1 h at 30 °C, lipid II synthesized was extracted from the reaction mixture and separated by TLC.
Bactoprenol-containing products were extracted with butanol-pyridine acetate (2:1; v/v; pH 4.2) and analyzed by thin layer chromatography (TLC; silica plates, 60F254, Merck). Radiolabeled spots were visualized by iodine vapor, excised, and counted. Eurocin was added to the reaction mixture in molar ratios as indicated referring to the total amount of C55-P. For purification of milligram quantities of lipid II, the analytical procedure was scaled up by a factor of 500 and purified as described (43). Radiolabeled lipid II was synthesized using [14C]UDP-GlcNAc as substrate.
In Vitro PBP2 Catalyzed Transglycosylation Using Purified Enzyme and Substrates
Cloning, expression, and purification of recombinant PBP2-His6 was performed as described (25). Enzymatic activity of PBP2 was determined by incubating 2.5 nmol of [14C]lipid II in 100 mm MES, 10 mm MgCl2, pH 5.5, and 0.1% Triton X-100 in a total volume of 50 μl. The reaction was initiated by the addition of 7.5 μg of PBP2-His6 and incubated for 1.5–2 h at 30 °C. Eurocin was added in molar ratios with respect to the lipid II substrate. Analysis of lipid II polymerization catalyzed by PBP2 was carried out by applying reaction mixtures directly onto TLC plates developed in solvent B (butanol-acetic acid-water-pyridine; 15:3:12:10, v/v/v/v) and subsequent quantification of residual radiolabeled free lipid II using phosphorimaging.
Complex Formation of Eurocin with Lipid II as Analyzed by TLC
Purified [14C]lipid II (2.5 nmol) was incubated in 10 mm Tris/HCl, pH 7.5, in the presence of increasing eurocin concentrations (eurocin-lipid II molar ratios ranging from 0.25 to 1:1) in a total volume of 30 μl. After incubation for 30 min at 30 °C, the mixture was analyzed by TLC using solvent B (butanol-acetic acid-water-pyridine; 15:3:12:10, v/v/v/v). Analysis was carried out by phosphorimaging (STORM).
Preparation of Liposomes
Small unilamellar vesicles containing calcein were prepared using stock solutions of dioleoylphosphatidylcholine that were first dissolved in methanol and dried in a desiccator for 5 h. Lipids were then resuspended by vortexing in an aqueous buffer-free solution of 70 mm calcein (sodium salt) to a final concentration of 10 g/liter (∼14 mm). The suspension was exposed to at least 7 cycles of freezing in liquid nitrogen followed by thawing in a 50 °C water bath before extrusion through a 200-nm pore filter 12 times using a 10-ml thermo barrel extruder (Northern Lipids, Vancouver, Canada). The lipid solutions were run on a PD10 column pre-equilibrated with a 50 mm NaCl solution. Eluent fractions were gathered and tested by fluorescence measurements with and without the addition of Triton X-100 to test for calcein release. Those with the highest signal-to-background ratio were selected for further use. All extruded vesicles were used the same day they were made.
Calcein Release Assay Measured by Fluorescence
All measurements were conducted on an Eclipse fluorimeter (Cary-Varian). To monitor the release of free calcein from the vesicles and the concomitant rise in fluorescence, the solution was excited at 490 nm, measuring emission at 515 nm with intervals of 0.1 s using a slit width of 2.5 nm for both monochromators. The vesicles were diluted in the respective buffers to a concentration of ∼0.005 g/liter or 5 μm. A 10-mm quartz cuvette with magnetic stirring was used, and the vesicle solution was allowed to equilibrate for a minute before starting to record fluorescence. During the recording, protein was injected after 1 min (defined as time t = 0), and the fluorescence was followed until it reached a plateau. Spectra were normalized with regard to maximum fluorescence, i.e. the fluorescence level achieved when 1% of Triton X-100 was added using Equation 4,
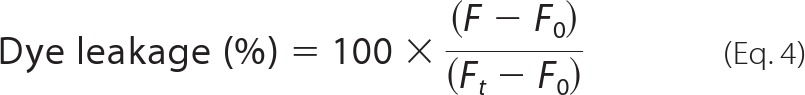 |
where F is the fluorescence intensity achieved by the peptides, and F0 and Ft are fluorescence intensities without the peptides and with Triton X-100, respectively.
RESULTS
Antimicrobial Activity
MIC was tested for 183 bacterial strains, the majority being Gram-positive human pathogens of species Staphylococcus and Streptococcus. The more susceptible species were Streptococcus pneumonia, Streptococcus pyogenes, and Streptococcus agalactiae; they showed MIC values of 0.06–1 μg/ml. A larger variation was seen among other species; MIC for Staphylococcus ssp. were found to be in the range of 0.5–128 μg/ml and for Enterococcus, 0.25–128 μg/ml. As an example, the MIC for the susceptibility reference strains of important human Gram-positive pathogen bacteria were: S. aureus (ATCC29213), 16 μg/ml; Staphylococcus epidermidis (ATCC12228), 16 μg/ml; Enterococcus faecium (ATCC49624), 16 μg/ml; E. faecalis (ATCC29212), 2 μg/ml; S. pneumonia (ATCC49619), 0.25 μg/ml.
No Activity Was Seen among the Gram-negative Bacteria Tested Here (MIC > 32 μg/ml)
Fig. 2A depicts the MIC90 against a wide range of clinically relevant isolates of Streptococci. Antimicrobial activities indicated that eurocin is active at low concentrations under complex ionic conditions as applied in the National Committee for Clinical Laboratory Standards/Clinical and Laboratory Standards Institute MIC microbroth dilution assay.
FIGURE 2.

In vitro and in vivo antimicrobial activity of eurocin.
A, distribution of MIC values of eurocin (*), penicillin (□), and vancomycin (▿) against 57 isolates of Streptococcus. B, shown is antibacterial action of eurocin (●) and vancomycin (▵) against S. pneumoniae in a mouse sepsis model. The y axis shows the number of cfu/ml in the peritoneal fluid of infected mice. Values observed from the negative control experiment (vehicle alone) are shown by filled squares (■). C, shown is a dose-response curve of eurocin and its action against S. pneumoniae in a mouse sepsis model. The number of cfu/ml is given for blood (□) and peritoneal fluid ( ).
).
In Vivo Efficacy
When 10 mg/kg eurocin was administered, the concentration of viable S. pneumoniae in the peritoneum of mice dropped by approximately 3 orders of magnitude after 2 h (Fig. 2B). This was comparable to the effect of vancomycin when administered at 70 mg/kg. This reduction is paralleled by a similar reduction in the blood (data not shown). In the untreated controls, the concentration of viable pneumococci in the peritoneum increased >10-fold during 5 h (data not shown).
Eurocin is effective at low dosage concentrations against an intraperitoneal infection with S. pneumoniae D39. Four hours after administration, the bacterial counts from both blood and peritoneal fluid showed an effect at concentrations of ∼0.5 mg/kg. The effective dose 50 (ED50) was 2 mg/kg (Fig. 2C).
At the same time, eurocin was found non-cytotoxic up to a concentration of 1024 μg/ml (hemolytic effect of 0.5–4%; data not shown). Mammalian in vitro toxicity was examined using both L929 mouse fibroblasts and freshly prepared human erythrocytes. Both methods showed low cytotoxicity of eurocin even at the highest concentrations tested (1 mg/ml, data not shown).
Eurocin NMR Solution Structure
All 1H NMR resonances were assigned except for Gly-1 HN, Phe-2 Hζ, Trp-31 Hϵ3, Hζ3, Hη2, and Hζ2, and Leu-33 HN. In addition to that, natural abundance HSQC spectra allowed the unambiguous assignment of ∼50% of 13C and 15N resonances, respectively. The chemical shift assignments have been deposited in the BioMagResBank (accession number 18463).
The three-dimensional structure of eurocin was solved. Supplemental Table S1 contains the classification of NOEs, the number of angle constraints used, and criteria for quality assessment of the structure (CYANA target function, root mean square deviation, and number of violations). Eurocin consists of a moderately flexible N terminus followed by an α-helix (α1, residues A8-L18). After a turn, a β strand (β1, residues Thr-23–Cys-27) followed by a flexible loop (residues Ala-28–Thr-37), leads to another β-strand (β2, residue Cys-38–Ser-41) forming an antiparallel β-sheet. The structure is illustrated in Fig. 3. The structure of eurocin is thus highly homologous to other fungal and invertebrate defensin structures published. The three-dimensional structure of eurocin has been deposited to the Protein Data Bank, accession code 2LT8.
FIGURE 3.

Structural features of eurocin. A, shown is a schematic of the structure with the lowest final force field energy; the helical region is shown in red, the β-strand regions are in cyan. N and C denote the N and C termini, respectively. B, shown is a bundle of 20 conformers after energy refinement; the helical region is shown in red, the β-sheet region is in cyan, and disulfide bridges are in yellow. C, shown is a schematic drawing of the structure of the conformer with the lowest force field energy showing the acidic (Glu, Asp) side chains in red, the basic (Lys, Arg) side chains in blue, and weakly polar and apolar (Ala, Val, Ile, Leu, Pro, Phe, Tyr, and Trp) side chains in gray. D is the same as C but turned 180° around the vertical axis as indicated. E and F, shown is a comparison of micelle binding site in eurocin (panel E) and plectasin (PDB ID 1ZFU, panel F). The residues changing chemical shifts upon interaction with a DPC micelle are shown as stick models in blue, whereas the remaining residues are only shown with the same colors as in B. The structure models shown here are those with the lowest force-field energy. The figures were produced with YASARA (37).
Of the three proline residues, the peptide bond from Cys-4 to Pro-5 could be assigned a cis conformation based on the chemical shifts of Cβ and Cγ (36), and Pro-30 could be assigned a trans conformation, whereas the chemical shifts did not allow any prediction of the conformation of Pro-36.
The presence of the helix α1 is supported by a row of dαN(i,i+3) NOEs, some dαβ(i,i+3) and sequential dNN(i,i+1). The β-sheet topology is supported by interstrand NOEs. The relative orientation of the β-sheet on one side and the N-terminal residues plus the α-helix on the other side with respect to each other is defined by a number of long range NOEs.
The structure calculated without any assumptions regarding disulfide bridge topology only allowed one possible combination of cysteines to disulfide bridges: 4–27, 11–38, and 15–40. Among others, an NOE between Cys-15 Hβ2 and Cys-40 Hα and an NOE between Pro-5 Hα and Cys-27 Hβ2 was observed. This is also in agreement with the disulfide bridge topology of homologous peptides.
Thermal Stability
Fig. 4 shows the pH stability profile of eurocin. Eurocin has the lowest denaturation temperature tm of 56.5 ± 1.1 °C at pH 7. In the acidic range, tm increased steadily under more acidic conditions with the highest tm of 71.7 ± 0.9 °C at pH 2. With increasingly alkaline pH values, tm increased, reaching a maximum tm of 70 ± 2 °C between pH 10 and 11. At pH 12, tm dropped again. The addition of 1 mm DTT caused the melting temperature at pH 4.5 to drop by 3.3 °C in the presence of 20 mm DTT tm dropped by 8 °C (data not shown). However, the protein unfolded in a cooperative fashion, indicating that intact disulfide bonds are not essential for the protein ability to fold into a stable structure.
FIGURE 4.

Thermal stability profile of eurocin at different pH values in the absence (○, solid line) and presence (●, dashed line) of DPC (32 mm) as measured by CD. Bars indicate the error from data fitted to Equation 1. The lines solely serve as a guide to the eye.
Interaction with Lipids and Detergents
The CD spectrum of eurocin does not change upon interaction with either DPC or dihexanoylphosphatidylcholine (data not shown). However, NMR data reveal a specific interaction between DPC and eurocin above the critical micelle concentration of DPC. Fig. 5A shows Δδabs as calculated by Equation 2 plotted versus the sequence of eurocin. The amino acids in the flexible loop between β1 and β2 as well as some adjacent amino acids in the N terminus are affected by the presence of DPC, whereas there are only very small changes in chemical shift throughout the remaining part of the protein. The changes occur only at concentrations of DPC above its critical micelle concentration (1.1 mm (44)), not below. Based on the data and our structure, the SAMPLEX program (45) suggests residues 6 and 31–35 as the core interaction site, which matches the most perturbed residues except for Trp-31, where no data could be obtained. However, fluorescence data clearly indicate that Trp-31 changes from a hydrophilic to a hydrophobic environment upon DPC binding (Fig. 5B). DPC titration data can be fitted to a Langmuir isotherm yielding an affinity constant of 5268 ± 1388, which corresponds to a ΔG0 of −21.2 ± 0.8 kJ mol−1, and n = 11.1 ± 1.1 (Fig. 5C).
FIGURE 5.

Eurocin binding to DPC micelles. A, shown is accumulated chemical shift changes of Hα and HN atoms of eurocin as calculated by Equation 2 upon the addition of 50 mm DPC. For Pro-30, only the Hα chemical shift change is shown. No data could be obtained from Pro-5 (Hα obscured by H2O), Trp-31 (cross-peak invisible in spectra at 50 mm DPC), and Pro-36 (Hα vanishes at DPC > 20 mm). B, shown is a fluorescence emission scan (excitation at 295 nm) of eurocin at pH 6 in the absence (dashed line) and presence (solid line) of DPC. a.u., absorbance units. C, shown is a binding isotherm of eurocin to DPC micelles. The dashed line shows the fitted model, whereas circles show individual data points. The inset shows the same data, only zoomed closer to the origin.
Based on the clear evidence for interactions between eurocin and DPC, we investigated whether DPC affected the thermal stability of eurocin. Fig. 4 shows the tm values for eurocin in the presence of 0 and 32 mm DPC. Interestingly, binding to DPC micelles strongly increased the thermal stability in the acidic pH range, whereas thermal stability in the alkaline pH range is not significantly affected. The stability minimum shifts by ∼1 pH unit toward the alkaline region.
Fluorescence Measurements of Peptide-induced Vesicle Disruption
To observe whether the interaction between eurocin and lipid vesicles can lead to pore formation or membrane disruption, we prepared zwitterionic (100% dioleoylphosphatidylcholine) calcein-loaded lipid vesicles and followed the calcein release as a function of eurocin concentration.
Typical calcein release data after the addition of eurocin to lipid vesicles can be seen in Fig. 6. In all cases, the final amount of calcein released is well below 100% (as defined by the amount of calcein released by addition of Triton X-100), suggesting that the calcein leakage is due to neither pore formation nor total vesicle disruption. In support of this statement, we could not observe any peptide-induced disruption of the vesicle membrane integrity by scanning laser confocal microscopy (data not shown).
FIGURE 6.

Eurocin-induced fluorescence release from dioleoylphosphatidylcholine vesicles. Fluorescence intensity on the y axis is normalized to the fluorescence release obtained with Triton X-100, defined as 100. The eurocin concentrations shown correspond to 30, 62, 125, and 250 × MIC for S. simulans 22. a.u., absorbance units.
Effect of Eurocin on Whole Cells
The reported binding of eurocin to lipid II suggests a mode of action of eurocin through interference with peptidoglycan biosynthesis. To investigate this in further detail, we first determined the cytoplasmic levels of UDP-muramic acid-pentapeptide (Fig. 7). Antibiotics such as vancomycin, which interfere with the late stages of peptidoglycan synthesis, trigger an accumulation of this ultimate soluble peptidoglycan precursor in the cytoplasm.
FIGURE 7.

Intracellular accumulation of the soluble cell wall precursor UDP-MurNAc-pentapeptide in vancomycin-treated (dotted line) and eurocin-treated (dashed line) cells of S. simulans 22. Cells were treated for 30 min with peptides at 10× MIC. Treated cells were extracted with boiling water, and the intracellular nucleotide pool was analyzed by reversed-phase HPLC. Untreated cells of S. simulans 22 were used as control (solid line).
Treatment of S. simulans 22 with eurocin (10× MIC) led to significant accumulation of the soluble cell wall precursor UDP-MurNAc-pentapeptide, similar to the extent of accumulation seen with vancomycin-treated controls (Fig. 7). HPLC analysis and subsequent mass spectrometry confirmed the identity of UDP-MurNAc-pentapeptide (m/z 1148.3). The continuous biosynthesis of UDP-MurNAc-pentapeptide and subsequent accumulation in the cytoplasm of treated cells further suggests that eurocin does not impair or depolarize the cytoplasmic membrane, as the precursor was retained in the cytoplasm and did not leak from treated cells.
Membrane damage or pore formation should further result in a release of potassium from whole cells; however, release of potassium was not observed after the addition of eurocin at 1, 3, and 10 × MIC (corresponding to 0.069, 0.21, and 0.69 μm, respectively), compared with the pore forming lantibiotic nisin (data not shown), which rules out pore formation as mechanism of action.
Impact of Eurocin on in Vitro Cell Wall Biosynthesis
Cell wall peptidoglycan is synthesized (46) starting from UDP-activated N-acetylmuramic acid-pentapeptide, which is linked to a membrane carrier, bactoprenolphosphate (C55-P) by the glycosyltransferase MraY, yielding lipid I. Lipid I is further glycosylated by MurG-catalyzed addition of an N-acetylglucosamine moiety to form the cell wall building block lipid II that is translocated across the cytoplasmic membrane to be assembled into the growing peptidoglycan network, this step being catalyzed by enzymes collectively designated as PBPs. In staphylococci, lipid II is further modified by FemXAB-catalyzed addition of a pentaglycine chain to the pentapeptide (43), where the product is translocated to the outside of the cell to form the peptidoglycan layer, releasing the C55-P molecule again.
Membrane preparations of M. luteus catalyze the membrane-associated synthesis (MraY and MurG) of lipid II in vitro in the presence of defined amounts of the soluble precursors UDP-MurNAc-pentapeptide and UDP-GlcNAc and of the bactoprenol carrier C55-P. The addition of increasing concentrations of eurocin to this test system resulted in an inhibition of the overall lipid II synthesis, as observed by TLC (Fig. 8). In the positive control, where no inhibitor was present, the complete conversion of C55-P to lipid II was achieved (Fig. 8A, lane 1). Quantitative analysis using radiolabeled precursors showed that increasing concentrations of eurocin led to enhanced inhibition of the lipid II synthesis, and an almost complete inhibition was achieved at equimolar concentrations of antibiotic to lipid carrier (Fig. 9A).
FIGURE 8.

Overall lipid II synthesis using M. luteus membranes. The analytical assay was performed in a total volume of 50 μl containing 200 μg of membrane protein, 5 nmol of C55-P, 50 nmol of UDP-N-acetylmuramic acid pentapeptide, 50 nmol UDP-N-acetylglucosamine in 60 mm Tris-HCl, 5 mm MgCl2, pH 8, 0.5% (w/v) Triton X-100. Eurocin was added to the reaction mixture in molar ratios as indicated referring to the total amount of 5 nmol C55-P. After incubation of 1 h at 30 °C lipid II, synthesized was extracted from the reaction mixture and separated by TLC. A, TLC of the reaction mixture at different molar ratios of C55-P and eurocin is shown. B, amount synthesized lipid II at different C55-P:eurocin ratios, normalized to the amount obtained in the absence of eurocin is shown.
FIGURE 9.

A, impact of eurocin on the PBP2 catalyzed transglycosylation of lipid II. Enzymatic activity of PBP2 was determined by incubating 2. 5 nmol of [14C]lipid II in 100 mm MES, 10 mm MgCl2, pH 5.5, and 0.1% Triton X-100 in a total volume of 50 μl. The reaction was initiated by the addition of 7.5 μg of PBP2-His6 and incubated for 2 h at 30 °C. Analysis of the lipid II conversion catalyzed by PBP2 was carried out by applying reaction mixtures directly onto TLC plates that were then developed in butanol-acetic acid-water-pyridine (15:3:12:10, v/v/v/v). B, complex formation of eurocin with lipid II was analyzed by TLC. [14C]Lipid II was incubated in the presence of increasing eurocin concentrations (eurocin-lipid II molar ratios ranging from 0.25–1:1). After incubation, the mixture was analyzed by TLC. Nisin was used as a positive control.
To further characterize the inhibitory nature of eurocin, we tested its impact on the individual PBP2-catalyzed transglycosylation/transpeptidation reaction, which occurs at the extracytoplasmic site of the membrane and, therefore, is the likely target reaction of eurocin. Reconstitution of the PBP2-catalyzed lipid II polymerization in vitro and subsequent TLC analysis and quantification of residual [14C]lipid II revealed that eurocin completely inhibited lipid II conversion at equimolar ratio of eurocin to lipid II, suggesting a 1:1 complex of peptide:lipid II (rather than inhibition of the enzyme). Binding stoichiometry of eurocin and lipid II was further validated by incubating purified [14C]lipid II together with eurocin in various molar ratios. Subsequent TLC was used to analyze the migration behavior (Fig. 9B). Free lipid II migrated to a defined position on the chromatogram (lane 1), whereas free eurocin was not detectable. However, in complex with eurocin, lipid II remained close to the starting point (lanes 3–5), as observed with the positive control nisin (lane 2). As has been observed in the cell-free assays, only at an equimolar ratio, no free lipid II was detectable, substantiating the formation of a 1:1 stoichiometric eurocin-lipid II complex.
DISCUSSION
Structure
The three-dimensional structure of eurocin can be classified as a cysteine-stabilized αβ fold. This class of proteins comprises several defensins from insects, invertebrates, and fungi, like insect defensin A (13), lucifensin (19), Mediterranean mussel defensin MGD-1 (18), oyster defensin from Crassostrea gigas (47), micasin from the dermatophytic fungus Microsporum canis (48), and plectasin from the saprophytic fungus Pseudoplectania nigrella (20). The members of the cysteine-stabilized αβ family show the common feature of being toxic to cells; however, they achieve this toxicity through different functions. Scorpion toxins have been found to inhibit potassium channels (49, 50). Micasin was suggested to act intracellularly through interference with protein folding (48). Plant γ-thionins are a group of peptides with a variety of antibacterial and antifungal activities (51). In some cases the mode of action is known, e.g. Cp-thionin inhibiting proteases (52) and γ-hordothionin inhibiting α-amylase (53). Apart from plectasin and micasin, eurocin is the only fungal defensin with a known three-dimensional structure. Mammalian defensins belong to a different structural family (54, 55), e.g. human β-defensin-2, which consists of a helical segment and a three-stranded β-sheet (54).
Like MGD-1, eurocin also exhibits a cis-proline at position 4. Because cis-prolines rarely succeed cysteine residues (56), there must be a special reason for this peculiar geometry. Both the cysteine and the proline residue are conserved, the disulfide-forming cysteine even to 100% (see Fig. 1) in the sequences described so far, further corroborating the essential role of the proline residue at this position in the sequence. The only exception is plectasin, where two extra amino acids are inserted between the cysteine and the proline and where proline is in the trans-conformation. The remaining two proline residues of eurocin, Pro-30 and Pro-36, which are not conserved in homologous proteins, show a trans-conformation. Given their location on both termini of the flexible loop connecting the two β-strands, they probably act as secondary structure breakers.
A striking feature of the structure is the absence of a hydrophobic core. Fig. 3 shows the location of the hydrophobic residues of eurocin. They are scattered around the surface with a concentration of three hydrophobic residues in a row in the flexible loop, which was seen to interact with DPC micelles. Fig. 3 shows the location of charged residues and demonstrates that salt bridges are not involved in keeping the tertiary structure of the protein intact. Neither could any H-bonds be found between residues in the helix and residues in the β-sheet. The fold seems to be kept intact solely by the three disulfide bridges. This is corroborated by the marked decrease of the denaturation temperature, tm, in the presence of DTT.
Interaction with Micelles and Vesicles
The majority of antimicrobial peptides have been found to interact with membrane lipids or lipid mimics like detergents. Eurocin is no exception in that it was found to interact with DPC micelles in a well defined way. The interaction site was found around the three hydrophobic residues Trp-31, Tyr-32, and Leu-33 in the flexible loop and to a minor extent in the neighboring N-terminal loop (Gly-6). The resonances of Trp-31, Tyr-32, and Leu-33 were considerably weaker than other resonances in the absence of DPC, possibly as a consequence of increased molecular mobility. After binding to DPC micelles, the resonances became approximately as strong as other residues. This hints at a reduced mobility of the loop after interaction with the micelle. NMR signals of the DPC bound form of eurocin were generally broader and weaker than the signals of the free form, as can be expected due to slower molecular tumbling. No high resolution structure of the DPC-bound form was calculated. However, the limited changes occurring in the NOESY spectra of the bound form clearly demonstrate that no overall change in structure took place as a consequence of interaction with the micelles. This no doubt reflects rigidity in the structure imparted by the three disulfide bonds.
The data suggest a binding of eurocin to the surface of the micelle. However, the peptide does not seem to penetrate the micelle deeply. This makes pure membrane disruption a very unlikely mechanism of action for eurocin. This was further substantiated by the data obtained on real phospholipids by vesicle disruption experiments; although the eurocin concentrations tested were above a protein-to-lipid ratio of 5 and between 30 and 240 × MIC, we could not observe complete release of vesicle contents in contrast to the high efficiency reported for antimicrobial peptides such as Ac-RRWWRF-NH2 (57), Hnp-2 (58), gramicidin A, gramicidin S, amphotericin (59), and melittin (60) that are proposed to work by either barrel-stave, torroidal pore, or carpet mechanism. The synthetic antimicrobial peptide novispirin (61, 62) effects full release of calcein from 5 μm lipid at 0.2 μm peptide, which is around 2 orders of magnitude more efficient than that of eurocin (63). Thus eurocin is clearly a very inefficient membrane permeabilizer, and membrane permeabilization is not the primary mode of its action. It is, however, worth noting that eurocin binds to lipid aggregates even if there is no lipid II, its natural ligand, present.
It is interesting to compare the DPC binding sites on the surfaces of eurocin and plectasin: Fig. 3, E and F, show the two peptides with their respective binding sites. DPC binding occurs at the same end of the peptide molecule, where there are two loops, one connecting the N terminus with helix 1 and the other connecting the two β-strands. In plectasin, both loops are approximately equally long, the N-terminal loop being slightly longer. In eurocin, the N-terminal loop is much shorter than the loop connecting the two β-strands. Looking at the sequence alignment (Fig. 1) in the loop regions, it can be seen that the extended N-terminal loop is a unique feature of plectasin (residues Asn-5–Trp-8 inserted), whereas the loop connecting the two β-strands seems to be extended by three amino acids (Gly-34–Pro-36) in eurocin compared with other invertebrate defensins. The free energy of binding of eurocin to DPC micelles was found to be −21.2 ± 0.8 kJ/mol. This is weaker than the −27 ± 1 kJ/mol found for plectasin. This probably reflects the smaller buried solvent-accessible surface area of 1040 Å2 for eurocin (calculated as the solvent accessible surface area of residues Gly-6, Trp-31, Tyr-32, Leu-33, Gly-34, and His-35) compared with 1145 Å2 for plectasin (residues Gly-6, Trp-8, Asp-9, Ala-31, Lys-32, Gly-33, Gly-34, Phe-35, Val-36, and Cys-37).
Antimicrobial Effect
Like plectasin, eurocin has a much stronger effect on Gram-positive bacteria than on Gram-negative bacteria. This is contrasted by micasin, which shows good antimicrobial activity against the Gram-negative P. aeruginosa and Agrobacterium tumefaciens. This difference is a further indication that eurocin and plectasin on the one hand and micasin on the other hand have different mechanisms of action. Eurocin is shown here to efficiently combat a range of Gram-positive infections both in vitro and in vivo.
Mechanism of Action
The experimental results obtained on the impact of eurocin on cell wall synthesis allows a clear picture of the eurocin mode of action. Using a combination of in vivo and in vitro test systems we found that the defensin eurocin selectively inhibits peptidoglycan biosynthesis through complex formation with the cell wall precursor lipid II. Sequestration of the central cell wall precursor lipid II prevents the incorporation in the growing peptidoglycan network and the formation of a vital cell wall, as revealed by inhibition of the PBP-catalyzed polymerization of lipid II in vitro. In this system eurocin was found to almost completely inhibit the lipid II consuming reaction at equimolar ratio (peptide:lipid), strongly suggesting complex formation rather than inhibition of the enzyme PBP2. The TLC experiment further confirmed the formation of a specific eurocin-lipid II complex observed in the cell-free assays. This is essentially the same mechanism, like that of the fungal defensin plectasin (25). Our observation that eurocin binds to and is stabilized by phospholipid-like micelles and interacts with phospholipid vesicles demonstrates eurocin affinity for phospholipids. This affinity helps prepositioning the peptide at the cell surface where its ligand lipid II occurs.
In line with the accumulation of the ultimate soluble cell wall precursor UDP-MurNAc-pentapeptide in the cytoplasm upon treatment with eurocin, we could not observe eurocin-induced potassium release from whole cells, indicating that eurocin does not impair membrane integrity or induce the formation of pores in the cytoplasmic membrane.
Conclusion
Eurocin is a fungal defensin with a cysteine-stabilized αβ fold as seen for other defensins. Eurocin binds to lipid aggregates, but its primary mode of action is not the formation of pores in cell membranes.
In vivo and in vitro analysis of the eurocin mechanism of action revealed that the peptide inhibits peptidoglycan biosynthesis of Gram-positive bacteria without comprising membrane integrity. Antimicrobial assays carried out both in vivo and in vitro showed that eurocin is a fast and effective antibiotic against Streptococci even at low concentrations, whereas it does not show good activity against Gram-negative pathogens.
Acknowledgments
We thank Steen Nielbo and Birthe Kragelund for access to NMR facilities, Allan Stensballe for assistance with MS experiments, and Kell Andersen for technical assistance.
This work was supported by the Obel and SparNord Foundations.

This article contains supplemental Table S1.
The atomic coordinates and structure factors (code 2LT8) have been deposited in the Protein Data Bank (http://wwpdb.org/).
The chemical shift assignments are available from the BioMagResBank under BMRB number 18463.
- AMP
- antimicrobial peptide
- CAPS
- N-cyclohexyl-3-aminopropanesulfonic acid
- DPC
- dodecylphosphocholine
- HSQC
- heteronuclear single-quantum coherence
- MIC
- minimal growth inhibitory concentration
- MurNAc
- N-acetylmuramyl
- PBP
- penicillin-binding protein
- NOE
- nuclear Overhauser effect
- TOCSY
- total correlation spectroscopy
- C55-P
- undecaprenylphosphate
- MES
- 4-morpholineethanesulfonic acid.
REFERENCES
- 1. Zasloff M. (2002) Antimicrobial peptides of multicellular organisms. Nature 415, 389–395 [DOI] [PubMed] [Google Scholar]
- 2. Ganz T. (2003) Defensins. Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3, 710–720 [DOI] [PubMed] [Google Scholar]
- 3. Hancock R. E. (2001) Cationic peptides. Effectors in innate immunity and novel antimicrobials. Lancet Infect. Dis. 1, 156–164 [DOI] [PubMed] [Google Scholar]
- 4. Hancock R. E., Sahl H. G. (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 24, 1551–1557 [DOI] [PubMed] [Google Scholar]
- 5. Zanetti M., Gennaro R., Romeo D. (1995) Cathelicidins. A novel protein family with a common proregion and a variable C-terminal antimicrobial domain. FEBS Lett. 374, 1–5 [DOI] [PubMed] [Google Scholar]
- 6. Lehrer R. I. (2004) Primate defensins. Nat. Rev. Microbiol. 2, 727–738 [DOI] [PubMed] [Google Scholar]
- 7. Tran D., Tran P. A., Tang Y. Q., Yuan J., Cole T., Selsted M. E. (2002) Homodimeric θ-defensins from rhesus macaque leukocytes. Isolation, synthesis, antimicrobial activities, and bacterial binding properties of the cyclic peptides. J. Biol. Chem. 277, 3079–3084 [DOI] [PubMed] [Google Scholar]
- 8. Tang Y. Q., Yuan J., Osapay G., Osapay K., Tran D., Miller C. J., Ouellette A. J., Selsted M. E. (1999) A cyclic antimicrobial peptide produced in primate leukocytes by the ligation of two truncated α-defensins. Science 286, 498–502 [DOI] [PubMed] [Google Scholar]
- 9. García-Olmedo F., Molina A., Alamillo J. M., Rodríguez-Palenzuéla P. (1998) Plant defense peptides. Biopolymers 47, 479–491 [DOI] [PubMed] [Google Scholar]
- 10. Lowenberger C. (2001) Innate immune response of Aedes aegypti. Insect Biochem. Mol. Biol. 31, 219–229 [DOI] [PubMed] [Google Scholar]
- 11. Bulet P., Stöcklin R., Menin L. (2004) Anti-microbial peptides. From invertebrates to vertebrates. Immunol. Rev. 198, 169–184 [DOI] [PubMed] [Google Scholar]
- 12. Zhu S. (2008) Discovery of six families of fungal defensin-like peptides provides insights into origin and evolution of the CSαβ defensins. Mol. Immunol. 45, 828–838 [DOI] [PubMed] [Google Scholar]
- 13. Cornet B., Bonmatin J. M., Hetru C., Hoffmann J. A., Ptak M., Vovelle F. (1995) Refined three-dimensional solution structure of insect defensin A. Structure 3, 435–448 [DOI] [PubMed] [Google Scholar]
- 14. Hanzawa H., Shimada I., Kuzuhara T., Komano H., Kohda D., Inagaki F., Natori S., Arata Y. (1990) 1H nuclear magnetic resonance study of the solution conformation of an antibacterial protein, sapecin. FEBS Lett. 269, 413–420 [DOI] [PubMed] [Google Scholar]
- 15. Landon C., Sodano P., Hetru C., Hoffmann J., Ptak M. (1997) Solution structure of drosomycin, the first inducible antifungal protein from insects. Protein Sci. 6, 1878–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lamberty M., Caille A., Landon C., Tassin-Moindrot S., Hetru C., Bulet P., Vovelle F. (2001) Solution structures of the antifungal heliomycin and a selected variant with both antibacterial and antifungal activities. Biochemistry 40, 11995–12003 [DOI] [PubMed] [Google Scholar]
- 17. Da Silva P., Jouvensal L., Lamberty M., Bulet P., Caille A., Vovelle F. (2003) Solution structure of termicin, an antimicrobial peptide from the termite Pseudacanthotermes spiniger. Protein Sci. 12, 438–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang Y. S., Mitta G., Chavanieu A., Calas B., Sanchez J. F., Roch P., Aumelas A. (2000) Solution structure and activity of the synthetic four-disulfide bond Mediterranean mussel defensin (MGD-1). Biochemistry 39, 14436–14447 [DOI] [PubMed] [Google Scholar]
- 19. Nygaard M. K., Andersen A. S., Kristensen H.-H., Krogfelt K. A., Fojan P., Wimmer R. (2012) The insect defensin lucifensin from Lucilia sericata. J. Biomol. NMR 52, 277–282 [DOI] [PubMed] [Google Scholar]
- 20. Mygind P. H., Fischer R. L., Schnorr K. M., Hansen M. T., Sönksen C. P., Ludvigsen S., Raventós D., Buskov S., Christensen B., De Maria L., Taboureau O., Yaver D., Elvig-Jørgensen S. G., Sørensen M. V., Christensen B. E., Kjaerulff S., Frimodt-Moller N., Lehrer R. I., Zasloff M., Kristensen H.-H. (2005) Plectasin is a peptide antibiotic with therapeutic potential from a saprophytic fungus. Nature 437, 975–980 [DOI] [PubMed] [Google Scholar]
- 21. Izadpanah A., Gallo R. L. (2005) Antimicrobial peptides. J. Am. Acad. Dermatol. 52, 381–390 [DOI] [PubMed] [Google Scholar]
- 22. Reynolds P. E. (1989) Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 8, 943–950 [DOI] [PubMed] [Google Scholar]
- 23. Brötz H., Bierbaum G., Leopold K., Reynolds P. E., Sahl H.-G. (1998) The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother. 42, 154–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Willey J. M., van der Donk W. A. (2007) Lantibiotics. Peptides of diverse structure and function. Annu. Rev. Microbiol. 61, 477–501 [DOI] [PubMed] [Google Scholar]
- 25. Schneider T., Kruse T., Wimmer R., Wiedemann I., Sass V., Pag U., Jansen A., Nielsen A. K., Mygind P. H., Raventós D. S., Neve S., Ravn B., Bonvin A. M., De Maria L., Andersen A. S., Gammelgaard L. K., Sahl H.-G., Kristensen H.-H. (2010) Plectasin, a Fungal Defensin, Targets the Bacterial Cell Wall Precursor Lipid II. Science 328, 1168–1172 [DOI] [PubMed] [Google Scholar]
- 26. Sass V., Schneider T., Wilmes M., Körner C., Tossi A., Novikova N., Shamova O., Sahl H.-G. (2010) Human β-defensin 3 inhibits cell wall biosynthesis in Staphylococci. Infect. Immun. 78, 2793–2800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. de Leeuw E., Li C., Zeng P., Li C., Diepeveen-de Buin M., Lu W. Y., Breukink E., Lu W. (2010) Functional interaction of human neutrophil peptide-1 with the cell wall precursor lipid II. FEBS Lett. 584, 1543–1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Griesinger C., Otting G., Wüthrich K., Ernst R. R. (1988) Clean TOCSY for 1H spin system identification in macromolecules. J. Am. Chem. Soc. 110, 7870–7872 [Google Scholar]
- 29. Piotto M., Saudek V., Sklenár V. (1992) Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J. Biomol. NMR 2, 661–665 [DOI] [PubMed] [Google Scholar]
- 30. Mori S., Abeygunawardana C., Johnson M. O., van Zijl P. C. (1995) Improved sensitivity of HSQC spectra of exchanging protons at short interscan delays using a new fast HSQC (FHSQC) detection scheme that avoids water saturation. J. Magn. Reson. B 108, 94–98 [DOI] [PubMed] [Google Scholar]
- 31. Wüthrich K. (1986) NMR of Proteins and Nucleic Acids, John Wiley & Sons, Inc., New York [Google Scholar]
- 32. Keller R. (2004) The Computer-aided Resonance Assignment Tutorial, 1st Ed., CANTINA Verlag, Goldau, Switzerland [Google Scholar]
- 33. Shen Y., Delaglio F., Cornilescu G., Bax A. (2009) TALOS+, a hybrid method for predicting protein backbone torsion angles from NMR chemical shifts. J. Biomol. NMR 44, 213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Güntert P., Braun W., Wüthrich K. (1991) Efficient computation of three-dimensional protein structures in solution from nuclear magnetic resonance data using the program DIANA and the supporting programs CALIBA, HABAS, and GLOMSA. J. Mol. Biol. 217, 517–530 [DOI] [PubMed] [Google Scholar]
- 35. Güntert P., Billeter M., Ohlenschläger O., Brown L. R., Wüthrich K. (1998) Conformational analysis of protein and nucleic acid fragments with the new grid search algorithm FOUND. J. Biomol. NMR 12, 543–548 [DOI] [PubMed] [Google Scholar]
- 36. Güntert P., Mumenthaler C., Wüthrich K. (1997) Torsion angle dynamics for NMR structure calculation with the new program DYANA. J. Mol. Biol. 273, 283–298 [DOI] [PubMed] [Google Scholar]
- 37. Krieger E., Koraimann G., Vriend G. (2002) Increasing the precision of comparative models with YASARA NOVA, a self-parameterizing force field. Proteins 47, 393–402 [DOI] [PubMed] [Google Scholar]
- 38. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577 [Google Scholar]
- 39. Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K. (2009) Improving physical realism, stereochemistry, and side-chain accuracy in homology modeling. Four approaches that performed well in CASP8. Proteins 77, 114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., Thornton J. M. (1996) AQUA and PROCHECK-NMR. Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 [DOI] [PubMed] [Google Scholar]
- 41. Shenkarev Z. O., Nadezhdin K. D., Sobol V. A., Sobol A. G., Skjeldal L., Arseniev A. S. (2006) Conformation and mode of membrane interaction in cyclotides. Spatial structure of kalata B1 bound to a dodecylphosphocholine micelle. FEBS J. 273, 2658–2672 [DOI] [PubMed] [Google Scholar]
- 42. Kohlrausch U., Höltje J. V. (1991) Analysis of murein and murein precursors during antibiotic-induced lysis of Escherichia coli. J. Bacteriol. 173, 3425–3431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schneider T., Senn M. M., Berger-Bächi B., Tossi A., Sahl H. G., Wiedemann I. (2004) In vitro assembly of a complete, pentaglycine interpeptide bridge containing cell wall precursor (lipid II-Gly5) of Staphylococcus aureus. Mol. Microbiol. 53, 675–685 [DOI] [PubMed] [Google Scholar]
- 44. Stafford R. E., Fanni T., Dennis E. A. (1989) Interfacial properties and critical micelle concentration of lysophospholipids. Biochemistry 28, 5113–5120 [DOI] [PubMed] [Google Scholar]
- 45. Krzeminski M., Loth K., Boelens R., Bonvin A. M. (2010) SAMPLEX. Automatic mapping of perturbed and unperturbed regions of proteins and complexes. BMC Bioinformatics 11, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. van Heijenoort J. (2007) Lipid intermediates in the biosynthesis of bacterial peptidoglycan. Microbiol. Mol. Biol. Rev. 71, 620–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gueguen Y., Herpin A., Aumelas A., Garnier J., Fievet J., Escoubas J. M., Bulet P., Gonzalez M., Lelong C., Favrel P., Bachère E. (2006) Characterization of a defensin from the oyster Crassostrea gigas. Recombinant production, folding, solution structure, antimicrobial activities, and gene expression. J. Biol. Chem. 281, 313–323 [DOI] [PubMed] [Google Scholar]
- 48. Zhu S., Gao B., Harvey P. J., Craik D. J. (2012) Dermatophytic defensin with antiinfective potential. Proc. Natl. Acad. Sci. U.S.A. 109, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Delepierre M., Prochnicka-Chalufour A., Boisbouvier J., Possani L. D. (1999) Pi7, an orphan peptide from the scorpion Pandinus imperator. A 1H NMR analysis using a nano-NMR probe. Biochemistry 38, 16756–16765 [DOI] [PubMed] [Google Scholar]
- 50. Savarin P., Romi-Lebrun R., Zinn-Justin S., Lebrun B., Nakajima T., Gilquin B., Menez A. (1999) Structural and functional consequences of the presence of a fourth disulfide bridge in the scorpion short toxins. Solution structure of the potassium channel inhibitor HsTX1. Protein Sci. 8, 2672–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pelegrini P. B., Franco O. L. (2005) Plant γ-thionins. Novel insights on the mechanism of action of a multifunctional class of defense proteins. Int. J. Biochem. Cell Biol. 37, 2239–2253 [DOI] [PubMed] [Google Scholar]
- 52. Melo F. R., Rigden D. J., Franco O. L., Mello L. V., Ary M. B., Grossi de Sá M. F., Bloch C., Jr. (2002) Inhibition of trypsin by cowpea thionin. Characterization, molecular modeling, and docking. Proteins 48, 311–319 [DOI] [PubMed] [Google Scholar]
- 53. Mendez E., Moreno A., Colilla F., Pelaez F., Limas G. G., Mendez R., Soriano F., Salinas M., de Haro C. (1990) Primary structure and inhibition of protein synthesis in eukaryotic cell-free system of a novel thionin, γ-hordothionin, from barley endosperm. Eur. J. Biochem. 194, 533–539 [DOI] [PubMed] [Google Scholar]
- 54. Pazgier M., Hoover D. M., Yang D., Lu W., Lubkowski J. (2006) Human β-defensins. Cell. Mol. Life Sci. 63, 1294–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. De Smet K., Contreras R. (2005) Human antimicrobial peptides. Defensins, cathelicidins, and histatins. Biotechnol. Lett. 27, 1337–1347 [DOI] [PubMed] [Google Scholar]
- 56. Pal D., Chakrabarti P. (1999) Cis peptide bonds in proteins. Residues involved, their conformations, interactions, and locations. J. Mol. Biol. 294, 271–288 [DOI] [PubMed] [Google Scholar]
- 57. Jing W., Hunter H. N., Hagel J., Vogel H. J. (2003) The structure of the antimicrobial peptide Ac-RRWWRF-NH2 bound to micelles and its interactions with phospholipid bilayers. J. Pept. Res. 61, 219–229 [DOI] [PubMed] [Google Scholar]
- 58. Wimley W. C., Selsted M. E., White S. H. (1994) Interactions between human defensins and lipid bilayers. Evidence for formation of multimeric pores. Protein Sci. 3, 1362–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Katsu T., Imamura T., Komagoe K., Masuda K., Mizushima T. (2007) Simultaneous measurements of K+ and calcein release from liposomes and the determination of pore size formed in a membrane. Anal. Sci. 23, 517–522 [DOI] [PubMed] [Google Scholar]
- 60. Popplewell J. F., Swann M. J., Freeman N. J., McDonnell C., Ford R. C. (2007) Quantifying the effects of melittin on liposomes. Biochim. Biophys. Acta 1768, 13–20 [DOI] [PubMed] [Google Scholar]
- 61. Sawai M. V., Waring A. J., Kearney W. R., McCray P. B., Jr., Forsyth W. R., Lehrer R. I., Tack B. F. (2002) Impact of single-residue mutations on the structure and function of ovispirin/novispirin antimicrobial peptides. Protein Eng. 15, 225–232 [DOI] [PubMed] [Google Scholar]
- 62. Wimmer R., Andersen K. K., Vad B., Davidsen M., Mølgaard S., Nesgaard L. W., Kristensen H. H., Otzen D. E. (2006) Versatile interactions of the antimicrobial peptide novispirin with detergents and lipids. Biochemistry 45, 481–497 [DOI] [PubMed] [Google Scholar]
- 63. Vad B., Thomsen L. A., Bertelsen K., Franzmann M., Pedersen J. M., Nielsen S. B., Vosegaard T., Valnickova Z., Skrydstrup T., Enghild J. J., Wimmer R., Nielsen N. C., Otzen D. E. (2010) Divorcing folding from function. How acylation affects the membrane-perturbing properties of an antimicrobial peptide. Biochim. Biophys. Acta 1804, 806–820 [DOI] [PubMed] [Google Scholar]
- 64. Thompson J. D., Gibson T. J., Plewniak F., Jeanmougin F., Higgins D. G. (1997) The CLUSTAL_X windows interface. Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Todd S. M., Sonenshine D. E., Hynes W. L. (2007) Tissue and life-stage distribution of a defensin gene in the Lone Star tick, Amblyomma americanum. Med. Vet. Entomol. 21, 141–147 [DOI] [PubMed] [Google Scholar]
- 66. Seo J.-K., Crawford J. M., Stone K. L., Noga E. J. (2005) Purification of a novel arthropod defensin from the American oyster, Crassostrea virginica. Biochem. Biophys. Res. Commun. 338, 1998–2004 [DOI] [PubMed] [Google Scholar]
- 67. Hubert F., Noel T., Roch P. (1996) A member of the arthropod defensin family from edible Mediterranean mussels (Mytilus galloprovincialis). Eur. J. Biochem. 240, 302–306 [DOI] [PubMed] [Google Scholar]
- 68. Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011) MEGA5. Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 [DOI] [PMC free article] [PubMed] [Google Scholar]