

Abstract
Tumor-initiating stem cells (alternatively called cancer stem cells, CSCs) are a subpopulation of tumor cells that plays unique roles in tumor propagation, therapeutic resistance and tumor recurrence. It is becoming increasingly important to understand the molecular signaling that regulates the self-renewal and differentiation of CSCs. Transcription factors are critical for the regulation of normal and neopolastic stem cells. Here, we examined the expression and function of the Krüppel-like family of transcription factors (KLFs) in human glioblastoma (GBM)-drived neurosphere lines and low passage primary GBM-derived neurospheres that are enriched for tumor-initiating stem cells. We identify KLF9 as a relatively unique differentiation-induced transcription factor in GBM-derived neurospheres. KLF9 is shown to induce neurosphere cell differentiation, inhibit neurosphere formation, and inhibit neurosphere-derived xenograft growth in vivo. We also show that KLF9 regulates GBM neurosphere cells by binding to the Notch1 promoter and suppressing Notch1 expression and downstream signaling. Our results show for the first time that KLF9 has differentiating and tumor suppressing functions in tumor-initiating stem cells.
Keywords: cancer stem cell, retinoic acid, differentiation, transcription factor, Notch signaling
Introduction
Human cancers consist of morphological and functionally heterogeneous populations of cells [1, 2]. Only a minority of the heterogeneous tumor cells has the capacity to initiate and sustain tumor xenograft growth when injected into immune-compromised mice [3]. These tumor-initiating cells, alternatively called cancer stem cells (CSCs), display stem-like characteristics including the capacity for long-term self-renewal and multi-lineage differentiation [4-7]. During the past decade, considerable evidence has emerged supporting the existence of CSCs in a wide variety of solid malignancies and their unique roles in tumor propagation, therapeutic resistance and tumor recurrence [8-11]. It is becoming increasingly important to understand the molecular mechanisms that regulate the self-renewal and differentiation of CSCs.
Advances in establishing and maintaining normal embryonic and neural stem cells in culture have been successfully applied for the reliable isolation and growth of CSCs from solid malignancies [6, 12-15]. Under these serum-free culture conditions supplemented with growth factors, the CSCs grow as nonadherent multicellular spheres (e.g. tumor spheres, oncospheres or neurospheres) consisting of self-renewing neoplastic stem-like cells intermixed with their more differentiated progenitor cells that are relatively restricted in self-renewing and tumor-initiating potential. These CSC-enriched culture systems are excellent cancer models by virtue of their ability to recapitulate with high fidelity the molecular, biological, and histopathological characteristics of the parent tumors when implanted to immunodeficient mice [7, 14].
Growing evidence shows that normal and neoplastic stem cells can share signaling pathways that regulate their self-renewal and differentiation. Examples include Bmi1 [16, 17], BMP [18, 19], Notch [20, 21] and Sonic Hedgehog [22, 23]. Therapeutic approaches have been designed to modulate these pathways to successfully inhibit CSCs and their capacity to propagate tumors [19, 21, 23, 24]. Thus, it is promising to address the role of other factors that regulate normal stem cells in the context of CSC. Specific transcription factors are critical for the regulation of normal stem cells. The self-renewal capacity of embryonic stem (ES) cells is regulated by a set of transcription factors including Nanog, Oct4 and Sox2 [25, 26]. In addition, ectopic expression of four transcription factors, KLF4, Sox2, Oct4 and c-Myc, can efficiently reprogram somatic cells to induced pluripotent ES-like (iPS) cells [27, 28]. The likelihood that similar networks of transcription factors regulate CSCs is supported by recent evidence that Sox2 is essential for maintaining the growth and tumor-initiating capacity of CSCs derived from brain tumors [29]. The Krüppel-like family of transcription factors (KLFs) consists of 17 evolutionarily conserved zinc finger containing proteins with diverse regulatory functions [30]. KLFs bind to GC-GT rich sites in gene promoter and enhancer regions and are known to contextually regulate the growth, proliferation, and differentiation of cells including non-neoplastic stem cells and cancer cells [31, 32]. Specific KLF family members are known to influence self-renewal of pluripotent cells. Klf4 supports the reprogramming of somatic cells to iPS cells and the simultaneous depletion of Klf2, Klf4 and Klf5 inhibits ES cell self-renewal and induces cell differentiation [33]. Several KLFs act as tumor suppressors and/or oncogenes under distinct cellular contexts. KLF4 is a transcriptional repressor of p53 and can act as a context-dependent oncogene [34, 35]. KLF6 is a tumor suppressor gene mutated in a significant fraction of human prostate cancer and brain tumors [36, 37]. KLF9, also known as basic transcription element-binding protein 1 (BTEB1), regulates the the transactivation of progesterone-responsive promoters and has been found to be downregulated in endometrial carcinoma and colorectal cancer [32, 38, 39].
Here we use neurospheres derived from human glioblastoma, the most frequent and malignant brain tumors in the adult, to examine the expression and function of KLFs in tumor-initiating CSCs. We found that KLF family members were differentially expressed during the differentiation process of GBM-derived neurospheres. KLF9 was found to be relatively unique in its upregulation in response to two differentiation signals (retinoic acid and serum) and its capacity to induce neurosphere cell differentiation, inhibit neurosphere formation by CSCs and inhibit CSC-initiated xenograft growth in vivo. Finally, we show that KLF9 regulates CSCs by binding to the Notch1 promoter and suppressing Notch1 expression and downstream signaling.
Materials and Methods
Reagents
All reagents were purchased from Sigma-Aldrich unless otherwise stated. All-trans retinoic acid (RA) was prepared as stock solution in DMSO and diluted in cell culture medium to 1μM as working concentration. Doxycycline (Dox) was diluted in cell culture medium to 1 μg/ml as working concentration.
Cell culture
Human GBM neurosphere lines, 0913 (GBM1a) and 0627 (GBM1b), were originally derived by Vescovi and colleagues [14]. The GBM-KK190156 (GBMKK) neurosphere line was derived from a single GBM patient and kindly provided by Dr. Jaroslaw Maciaczyk (University of Freiburg). Cells were cultured in serum-free medium supplemented with EGF/FGF and incubated in 5% CO2/95% air condition at 37°C. U87MG cells (ATCC) were grown in Dulbecco’s Modified Eagle Medium with 10% FBS [14]. The primary neurospheres were derived from GBM tumors at Johns Hopkins University using the same methods and culture conditions as described in Galli et al [14]. Primary neurospheres were used at less than 10 passages.
Human GBM tumors and normal brain tissues were collected at Johns Hopkins University. All human materials were obtained and used in compliance with the Johns Hopkins Medicine Institutional Review Boards.
Lentiviral transduction and cell transfection
Hairpin sequences used for the KLF9 shRNA lentiviral vectors (Open Biosystems) are listed in Supplemental Table 1. N-terminal Flag-tagged KLF9 was constructed with high-fidelity PCR (Roche) and cloned into pTRIPZ vector (Thermo Scientific) with AgeI and MluI. Trans-Lentiviral Packaging System (Thermo Scientific) was used for lentivirus packaging. Cells were infected with lentivirus and selected with puromycin (1μg/ml) for stable cell lines. Nucleofector Transfection System (Lonza) was used for transient cell transfection following manufacturer’s protocol. Human Notch1 Intracellular Domain (NICD) was inserted into pCAGGS vector and kindly provided by Dr. Nicholas Gaiano (Johns Hopkins University).
Neurosphere formation and soft agar clonogenic assays
For neurosphere formation assay, dissociated viable cells (2 × 104 per well) were cultured in 6 well pates. After 5-6 days, neurospheres were fixed in neurosphere culture medium with 1% agarose. For soft agar clonogenic assay, dissociated viable cells (5 × 103) were suspended in neurosphere culture medium containing 0.5% agarose and placed on top of the bottom layer (1% agarose in DMEM) in 6 well plates. Cells were incubated in neurosphere culture medium +/- Dox for 12-14 days. After both assays, colonies were stained with Wright stain solution (1%) and counted by computer-assisted morphometry (MCID software) by measuring the number of neurospheres (>100μm in diameter) in 3 random fields per well.
Western blot
Total cellular protein was extracted with RIPA buffer (Sigma-aldrich) containing protease and phosphatase inhibitor cocktail (Calbiochem). Nuclear protein extraction was performed with NE-PER nuclear protein extraction kit (Pierce Biotechnology). SDS-PAGE was performed with 50μg total cellular protein per lane using 4–20% gradient Tris-glycine gels. Western blot was preformed using Quantitative Western Blot System (LI-COR Biosciences) following manufacturer’s instructions. The primary antibodies were: anti-BTEB1 (Santa Cruz), anti-Histone H3 (Cell Signaling), anti-FLAG-HRP (Sigma), anti-Tuj1 (Millipore), anti-GFAP (DAKO), anti-β-actin (Sigma), anti-Notch1 (D1E11) (Cell Signaling) and anti-cleaved Notch1 (Val1744) (Cell Signaling). Secondary antibodies were labeled with IRDye infrared dyes and protein levels were quantified with Odyssey Infrared Imaging System (LI-COR Biosciences).
Immunofluorescence
Neurosphere cells were collected by cytospin onto glass slides and fixed with 4% paraformaldehyde. The cell or brain slides were immunostained with anti-GFAP (Cell signaling), anti-Tuj1 (Millipore) and anti-human nuclear (Millipore) antibodies following the protocol from Cell Signalling. Secondary antibodies were conjugated with Alexa488. Immunofluorescent images were taken and analyzed using Axiovision software (Zeiss).
Tumor xenografts
For subcutaneous xenografts, female athymic nude mice were injected s.c. in the flank with 5×106 viable cells in 0.1 ml DMEM. When tumors reached about 50mm3, mice were randomly divided into groups for treatment. In RA treatment, mice received RA (1.5μg/kg, i.p. daily) or solvent DMSO as control. For Dox treatment, mice received Dox (2mg/ml in 1% sucrose) in drinking water or 1% sucrose as control. Tumor sizes were determined daily by measuring two dimensions [length (a) and width (b)] and volumes (V) were calculated using the formula V = ab 2 / 2 [40].
For intracranial xenografts, SCID immunodeficient mice received 5,000 viable cells in 2 μl DMEM by stereotactic injection to the right caudate/putamen. Cell viability was determined by trypan blue dye exclusion. Mice were perfused with 4% paraformaldehyde at the indicated times and the brains were removed for histological analysis. Tumor sizes were quantified by measuring maximum tumor volume on H&E-stained brain coronal sections using computer-assisted morphometry (MCID software). All animal protocols were approved by the Johns Hopkins School of Medicine Animal Care and Use Committee.
Quantitative real-time PCR
Quantitative RT-PCR was performed using SYBR Green PCR Master Mix (Applied Biosystems) and IQ5 RT-PCR detection system (Bio-rad). All primer sequences are listed in Supplemental Table 2. Relative expression of each gene was normalized to 18S rRNA.
Microarray analysis
The hybridizations were performed in the Johns Hopkins Microarray core facility using Human Exon 1.0 ST Array (Affymetrix) and data were processed with Partek software. Statistical analysis was performed using the false discovery rate (FDR) method for multiple hypothesis correction. A list of statistically significant differentially expressed genes (FDR=0.2) was generated. Fold change values were calculated on the data set normalized with the robust multiarray average (RMA) method.
Flow cytometric assay
Unfixed cells were stained with CD133/2(293C3)-PE (Miltenyi Biotec) and Notch1-PE (R&D) antibody following manufacturer’s protocol.
Chromatin immunoprecipitation
5×106 GBM1a-KLF9 cells were treated with Dox for 72h and preceded to ChIP using MAGnify chromatin immunoprecipitation system (Invitrogen). Mouse anti-FLAG M2 antibody (Sigma-Aldrich) was coupled with Dynabeads with mouse IgG as the control. ChIP DNA was used in semi-quantitative PCR reactions with primer pairs (ChIP-A and ChIP-B) listed in Table S2.
Luciferase reporter assay
Npro plasmid was kindly provided by Dr. Michael Ruppert and has been used to study the interaction between KLF4 and Notch1 promoter [41]. 3×106 GBM1a-KLF9 cells were treated +/- Dox for 48h and cotransfected with 3 μg Npro and 1 μg pβGal-control plasmids (Clontech). Cells were extracted 48 h later and analyzed for luciferase and β-gal activity using luciferase assay system (Promega) and chemiluminescent β-gal assay (Clontech).
Electrophoretic mobility shift assay (EMSA)
BTE oligonucleotides were synthesized and end-labeled with IRDye 700 (Integrated DNA Technologies). Nuclear protein extraction was performed with NE-PER nuclear protein extraction kit (Pierce Biotechnology). Binding reaction and electrophoresis were performed following the instruction manual from Odyssey Infrared EMSA Kit (LI-COR Biosciences). For supershift assay, mouse anti-FLAG antibody (Sigma) was incubated with nuclear extracts at 4°C for 1 hour before binding reaction.
Statistical analysis
Data were analyzed using Prizm software (GraphPad). All results reported here represent at least three independent replications. Post-hoc tests included the Students t-test and the Tukey multiple comparison tests as appropriate. All data are represented as mean value ± standard error of mean (SEM).
Results
KLF9 is induced during the differentiation of GBM-derived neurospheres
GBM-derived neurosphere lines and low passage primary GBM-derived neurospheres were established and maintained under conditions that enrich for tumor-initiating cells as published extensively by ourselves and others [6, 11, 14, 19, 21, 23, 42]. Changes in the expression of specific Krüppel-like factor family members (KLF1-16) were examined in these cells during their forced differentiation by retinoic acid (RA) or serum-containing medium lacking FGF and EGF. Growth factor withdrawal in the presence of serum is a widely-used method to force CSC differentiation [14, 43]. RA treatment decreased the percentage of CD133+ cells, and induced GFAP and TUJ1, markers of astroglial and neuronal lineage differentiation, respectively (Figure 1A, 1B and 1C), consistent with the results of Campos et al [44]. PCR primers for nine of the sixteen KLFs generated detectable PCR products using cDNAs from the GBM-derived neurosphere line GBM1a as template (data not shown). Quantitative real-time PCR (qRT-PCR) showed that the expression of KLF 3, 4, 6, 11, 13, 15 and 16 was either inhibited or unchanged in response to differentiation conditions for 24-72 h (Figure 1D and 1E; Supplemental Figure 1). KLF10 was induced 3.5-fold in response to serum-induced differentiation (Figure 1E) and was unchanged in response to RA-induced differentiation (Figure 1D and Supplemental Figure 1). Only KLF9 was induced (up to 8-fold) in response to both RA and serum (Figure 1D and 1E). KLF9 expression by neurosphere cells increased as early as 8 hours and persisted for at least 72 hours in response to RA (Figure 1F). KLF9 expression was similarly induced by RA in all GBM-derived neurospheres examined including two additional neurosphere cell lines (GBM1b, GBMKK) and two low passage primary GBM-derived neurosphere isolates (JH276 and JH551) (Figure 1G). By comparison, KLF9 was unchanged by RA treatment in the established U87 glioblastoma cell line maintained as monolayers in serum-free neurosphere culture medium for 48h before RA treatment (Figure 1G). KLF9 induction by RA was confirmed at the protein level by immunoblot analyses. KLF9 protein increased after 72h of RA treatment compared to untreated controls (Figure 1H). To determine if RA induces KLF9 expression in vivo, mice bearing subcutaneous tumor xenografts derived from neurosphere cells were treated with RA (1.5 μg/kg, i.p. daily) or with DMSO as control for 7 days. KLF9 expression in the RA-treated tumors was ~4-fold higher than in DMSO-treated controls as determined by qRT-PCR of tumor-derived total RNA (Figure 1I).
Figure 1. Forced differentiation with either RA or serum induces KLF9 expression in GBM-derived neurospheres.
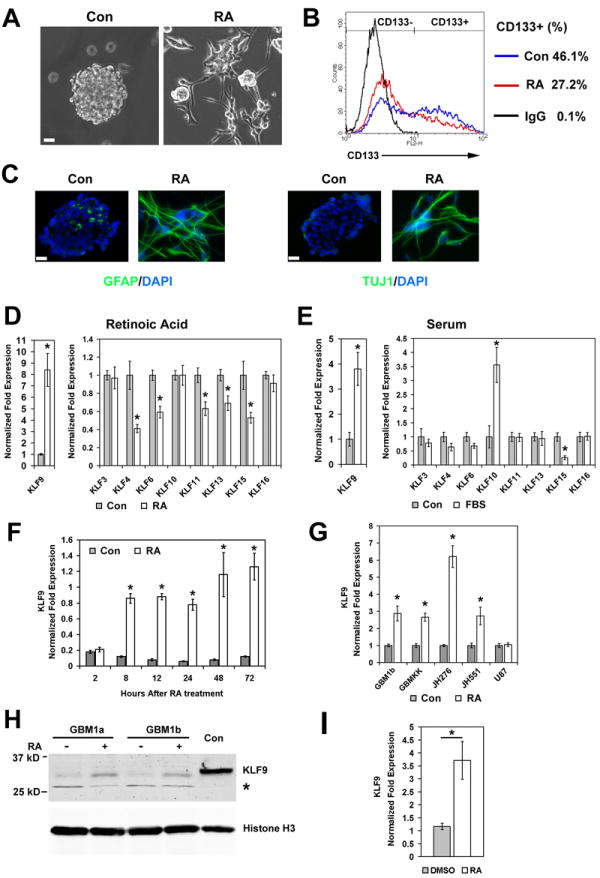
(A, B and C) GBM1a cells were treated +/- retinoic acid (RA, 1μM) for 72h. RA induces neurosphere cell attachment and process formation (A). RA decreases the percentage of CD133+ cells as determined by flow cytometry (B). RA induces GFAP and TUJ1 expression as determined by immunofluorescence staining of sectioned control neurospheres and adherent RA-treated cells (C). Bar = 20μm. (D and E) GBM1a neurospheres were grown in neurosphere medium as control (Con) with either RA or 1% fetal bovine serum (FBS) for 24h. The expression of KLF family members was measured by qRT-PCR. (F) Time course of KLF9 induction in response to RA as measured by qRT-PCR. (G) RA treatment (24 h) induces KLF9 expression as measured by qRT-PCR in GBM-derived neurosphere lines (GBM1b, GBMKK) and in low passage primary GBM-derived neurospheres (JH276 and JH551) but not in U87 cells. (H) GBM1a and GBM1b neurospheres were treated with RA for 72h. Nuclear protein extraction subjected to immunoblot analysis using anti-KLF9 and anti-histone-H3 antibodies reveals induction of nuclear KLF9 protein by RA. Controls (Con) represent protein extraction from 293T cells transfected with FLAG-tagged KLF9; * indicates a nonspecific band. (I) Mice bearing subcutaneous xenografts derived from GBMKK neurspheres were treated with RA (1.5 μg/kg i.p. daily) or with DMSO as control for 7 days. KLF9 expression in tumor xenografts was measured with qRT-PCR. Data represents Mean ± SEM; * indicates p<0.01.
KLF9 knockdown rescues neurospheres from RA-induced growth inhibition and differentiation
KLF9 knockdown GBM1a neurospheres (designated GBM1a-KLF9KD) were generated using lentiviral vectors expressing two distinct KLF9 shRNAs. These cells were used to determine if KLF9 induction is essential for the neurosphere cell responses to RA. GBM1a-KLF9KD cells and control cells with non-silencing shRNA were treated +/- RA for 24h. Both KLF9 shRNAs inhibited KLF9 induction by RA at both RNA and protein levels (Figure 2A). RA induced GBM-derived neurosphere cells to attach and extend processes on the cell culture substrata reflecting morphologic differentiation (Figure 1A). KLF9 knockdown inhibited RA-induced cell adhesion and process formation (Figure 2B), consistent with the involvement of KLF9 in RA-induced differentiation. The capacity to form renewable neurospheres is a biomarker of GBM cell stemness and correlates with tumor-initiating capacity [45]. RA potently inhibited neurosphere formation (Figure 2C). KLF9 knockdown did not affect basal neurosphere formation and significantly reversed by ~40% the capacity of RA to inhibit neurosphere formation (Figure 2C). KLF9 knockdown also inhibited GFAP and TUJ1 induction by RA (Figure 2D). Thus, the induction of KLF9 is required for these RA-induced responses in GBM-derived neurospheres.
Figure 2. KLF9 knockdown rescues neurospheres from RA-induced effects.
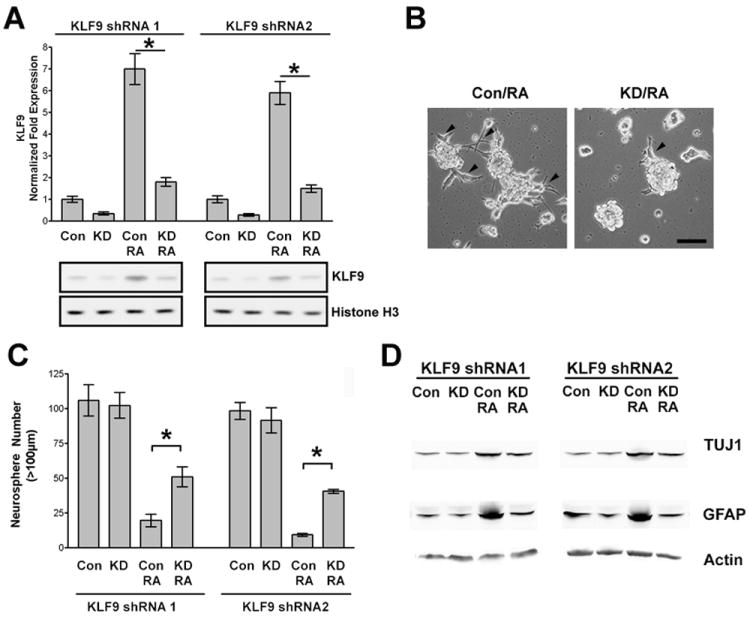
(A) GBM1a neurospheres were infected with lentivirus coding for either KLF9 shRNA1 or KLF9 shRNA2 and control shRNA to establish stable cell lines. Knockdown (KD) and control (Con) cells were treated +/- RA (1μM) for 24h. KLF9 expression was quantified by qRT-PCR (upper panel) and by immunoblot analysis of nuclear protein using anti-KLF9 antibody (lower panal). (B) KLF9 Knockdown and control neurosphere cells were treated with RA for 48h. Control neurosphere cells attach to culture substratum and extend processes (arrowheads) in response to RA. KLF9 knockdown reduced neurosphere cell attachment and process formation. Bar = 100μm. (C and D) KLF9 knockdown and control neurospheres were treated +/- RA for 6 days. Neurospheres (>100μm diameter) were counted by computer-based image analysis (C). KLF9 knockdown reversed the inhibition of neurosphere formation by RA. Whole cell extracts were subjected to immunoblot analysis with anti-GFAP and anti-TUJ1 antibody (D). KLF9 knockdown inhibited RA-induced upregulation of GFAP and TUJ1. Data represents Mean ± SEM (n=5); * indicates p<0.01.
KLF9 expression inhibits neurosphere-forming capacity, induces neurosphere differentiation and depletes neurospheres of CD133+ stem-like cells
Three independent neurosphere lines engineered to express a Dox-inducible N-terminal FLAG tagged KLF9 transgene (designated as GBM1a-KLF9, GBM1b-KLF9 and GBMKK-KLF9) were used to determine if KLF9 induction phenocopies neurosphere cell responses to RA (for lentiviral vector map see Supplemental Figure 2A). Dox treatment for 48h induced transgenic KLF9 expression (Figure 3A). After Dox removal, KLF9 expression levels rapidly returned to baseline (Figure 3B). KLF9 was induced 15-fold and 29-fold in response to RA and Dox, respectively, in GBM1a-KLF9 cells (Supplemental Figure 2B). We examined neurosphere cell growth during Dox induction of KLF9 (KLF9 induction) and following Dox withdrawal (KLF9 pre-induction). Cell growth was inhibited only during KLF9 induction and normalized under conditions of KLF9 pre-induction (Figure 3C). KLF9 did not alter cell viability under either condition (Figure 3D). We examined the effect of KLF9 on the neurosphere forming capacity of GBM-derived stem cells. KLF9 induction for 6 days significantly decreased neurosphere formation by 66-95% in all three neurosphere lines tested (Figure 3E). Expressing an alternative KLF family member (KLF6) under identical conditions had no effect on neurosphere formation, indicating that neurosphere cell responses to KLF9 were both KLF family member specific and not a nonspecific response to Dox alone (Supplemental Figure 3A and 3B). KLF9 preinduction also inhibited neurosphere formation by ~55% (Figure 3F). We also examined the effects of KLF9 induction on the capacity of neurosphere cells to form colonies in soft agar, an in vitro biomarker that correlates with tumor initiating capacity. KLF9 induction for 10-12 days significantly decreased colony formation by 35-60% (Figure 3G).
Figure 3. Forced KLF9 expression inhibits neurosphere formation and colony formation.
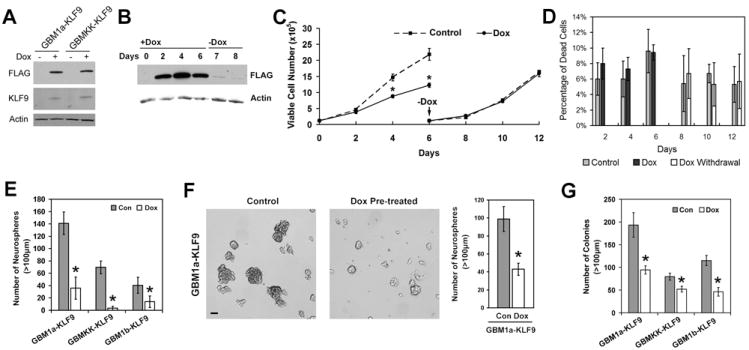
(A) GBM-derived neurosphere cells infected with lentivirus harboring doxycycline (Dox)-inducible Flag-tagged KLF9 cDNA were cultured in neurosphere growth medium +/- Dox for 48h. Whole cell lysates were subjected to immunoblot analysis using anti-FLAG and anti-KLF9 antibody to show KLF9 induction by Dox. (B) GBM1a-KLF9 cells were treated with Dox for 6 days and then passaged to Dox-free medium. Aliquotes of cells were collected on the days shown and whole cell lysates subjected to anti-FLAG immunoblot analysis. Flag-tagged KLF9 rapidly returns to baseline after Dox withdrawal. (C and D) GBM1a-KLF9 cells were treated +/- Dox for 6 days. Neurospheres were then passaged to Dox-free medium only and cultured for an additional 6 days. All cells within cultures were counted on the days shown to assess cell growth and viability using trypan blue staining (n=3). Cell growth was inhibited during the period of Dox-induced KLF9 expression and normalized after Dox withdrawal (C). Dox-induced KLF9 did not alter cell viability (D). (E) GBM-derived neurosphere cells were treated +/- Dox for 6 days and the numbers of neurospheres (>100 μm diameter) were counted (n=5). KLF9 induction inhibits neurosphere formation. (F) GBM1a-KLF9 cells were treated +/- Dox for 6 days and then equal numbers of viable cells were passaged to Dox-free medium for 6 additional days. Photomicrographs of representative microscopic fields are shown (left panel; Bar = 100μm). Neurospheres (>100μm diameter) were counted. Transient KLF9 induction inhibited neurosphere-forming capacity. (G) GBM neurosphere cells were dissociated, suspended in soft agar, and cultured in neurosphere growth medium +/- Dox for 12-14 days. The numbers of colonies (>100μm diameter) were counted (n=5). KLF9 induction inhibits colony formation. Data represents Mean ± SEM; * indicates p<0.01.
The persistent inhibition of neurosphere formation under both KLF9 induction and pre-induction conditions suggests the relative depletion of the neurosphere-forming stem cell population in response to KLF9 expression. The effects of KLF9 on the differentiation of GBM-derived neurospheres and their stem-like cells were examined in more detail. CD133 expression has been shown to correlate with the tumor-initiating capacity of GBM-derived cells and is used as a biomarker of GBM-derived stem-like cells [6, 46]. KLF9 induction for 6 days decreased the percentage of CD133+ cells from 42.8% to 30.3% (Figure 4A). After removing Dox for 3 days to turn off KLF9 induction, the percentage of CD133+ cells was still decreased from 45.6% to 31.0% (Figure 4B). Expression of Olig2, another stem cell-associated marker in human glioma [47] was also inhibited significantly by KLF9 (Figure 4C). KLF9 induction increased neurosphere expression of both GFAP and TUJ1 and increased the percentage of GFAP+ and TUJ1+ cells (Figure 4D and 4E).
Figure 4. Forced KLF9 expression induces cell differentiation and reduces stem-cell marker expression.
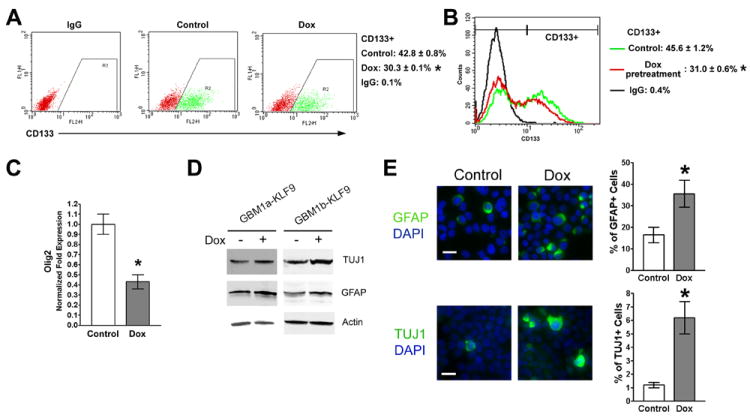
(A) GBM1a-KLF9 cells were treated +/- Dox for 6 days and analyzed by flow cytometry using CD133 antibodies and isotype IgG control (n=3). Representative dot plots and the percentage of CD133+ cells are shown. (B) GBM1a-KLF9 cells were treated +/- Dox for 6 days. Equal numbers of viable cells were passaged to Dox-free medium for an additional 3 days and analyzed by flow cytometry using CD133 antibodies and isotype IgG control (n=3). Representative histogram and the percentage of CD133+ cells are shown. (C) GBM1a-KLF9 cells were treated +/- Dox for 6 days and expression of Olig2 was quantified by qRT-PCR. KLF9 induction significantly inhibits Olig2 expression. (D and E) GBM1a-KLF9 and GBM1b-KLF9 cells were treated +/- Dox for 6 days. Immunoblot analysis of whole cell extracts shows increased expression of GFAP and TUJ1 in Dox-treated cells (D). Immunostaining and quantification of neurosphere cells collected by cytospin show increased percentage of GFAP+ and TUJ1+ cells in response to KLF9 expression (E). Bar = 10μm. Data represents Mean ± SEM; * indicates p<0.01.
KLF9 expression inhibits the growth of tumor xenografts established from GBM-derived neurospheres
We examined the effects of KLF9 on the growth of subcutaneous (s.c.) and intracranial (i.c.) xenografts established from GBM-derived neurospheres. Mice bearing pre-established s.c. xenografts derived from GBMKK-KLF9 cells received Dox (or sucrose as control) in drinking water beginning on post-implantation day (PID) 5. Tumors were measured from PID 8 through PID 14 to monitor the effects of KLF9 induction (Figure 5A). The average tumor size increased from 96 mm3 to 270 mm3 in control mice and from 83 mm3 to 144 mm3 in Dox-treated mice. By PID 14, KLF9 induction had inhibited tumor growth by 47%. Growth inhibition was confirmed following tumor dissection from sacrificed mice (Figure 5B). Immunoblot of whole tumor protein extracts using anti-FLAG antibody confirmed KLF9 induction in vivo in comparison to untreated mice (Figure 5C).
Figure 5. KLF9 induction inhibits the in growth of xenografts derived from GBM neurospheres.
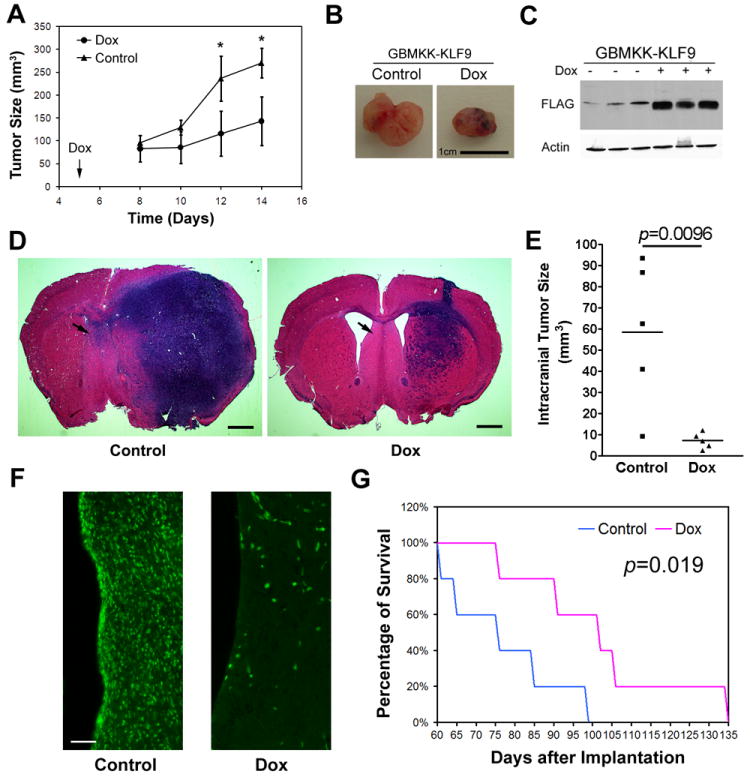
(A and B) Mice (n=6) were implanted s.c. with GBMKK-KLF9 cells and then received doxycycline (Dox) in drinking water beginning on post implantation day (PID) 5. Tumor sizes were measured as described in Methods (A). KLF9 induction inhibited xenograft growth. Representative xenografts dissected from control and Dox-fed mice on PID 14 are shown (B, Bar = 1cm). (C) Immunoblot analysis of xenograft protein extractions using anti-FLAG antibody confirms Dox-induced KLF9 expression in vivo. (D and E) GBM1a-KLF9 cells were pretreated +/- Dox and equal numbers of viable cells were transplanted into the brains of SCID mice (n=5). Hematoxyline and eosin stained coronal brain sections (20 μm) obtained from post-implantation day 60 animals are shown (D, Bar = 1mm). Arrows indicate regions examined for contralateral tumor cell infiltration (see F). Quantification of xenograft tumor volume shows that KLF9 induction reduces xenograft growth in vivo (E). (F) Brain sections stained with anti-human nuclei antibody show human tumor cells that had invaded contralateral to hemisphere of cell implantation (region shown indicated by arrows in D). KLF9 expression reduced the number of distantly invading tumor cells (Bar = 50μm). (G) Mice were implanted with neurosphere cells pretreated +/- Dox as in (D) and monitored for survival. Kaplan-Meier survival curve demonstrate that mice bearing xenografts derived from Dox-pretreated cells have significantly prolonged survival when compared to controls (p=0.019, log-rank analysis, n=5). Data represents Mean ± SEM; * indicates p<0.01, t-test.
We examined the effect of KLF9 induction on the growth of orthotopic intracranial xenografts derived from GBM neurospheres. GBM1a-KLF9 neurosphere cells were treated +/- Dox for 6 days and then 5,000 viable cells per animal were implanted into the brains of SCID mice. Five animals for each group were randomly selected and sacrificed 60 days later and coronal histological brain sections were examined for tumor size and growth pattern. Another 5 mice from each group were monitored for survival. As shown in Figure 5D and 5E, control neurosphere cells generated much larger tumors (58.6±34.6 mm3 max tumor volume) than Dox pre-treated cells (7.2±3.7 mm3 max tumor volume). Substantially fewer tumor cells, identified by anti-human nuclei immunofluorescence, were found to have invaded the contralateral hemisphere in the animals implanted with KLF9 pretreated cells (Figure 5F). Mice bearing intracranial xenografts derived from Dox pretreated cells lived significantly longer than controls (median survival: 76 days vs 102 days, p=0.019; Figure 5G).
KLF9 suppresses Notch1 transcription in GBM-derived neurospheres
KLF9 is known to function as a transcription repressor by binding to GC-rich basic transcriptional elements (BTE) in gene promoter regions [48]. To elucidate the transcriptional effects of KLF9 in GBM-derived neurospheres, we treated GBM1a-KLF9 neurospheres +/- Dox for 48h and transcriptional profiles were analyzed by Affymetrix gene expression microarray. This analysis revealed an ~45% reduction in Notch1 expression in response to KLF9 (fold change −1.74, p=0.0002), which was further validated using qRT-PCR. After 48h of Dox treatment, Notch1 expression was reduced by 93% and 76% in GBM1a-KLF9 and GBM1b-KLF9 cells, respectively (Figure 6A). Inducing another KLF family member (KLF6) under identical conditions had no effect on Notch1 expression (Supplemental Figure 3C). Immunoblot analysis for Notch1 and the Notch1 intracellular domain (NICD) proteins confirmed the qRT-PCR results. KLF9 induction for 72h reduced total cellular Notch1 levels by ~50% and NICD protein levels by ~56% in both neurosphere lines (Figures 6B and 6C). KLF9 induction for 72h decreased the percentage of Notch1+ neurosphere cells from ~88% to ~59% in Dox-treated cells, as determined by flow cytometry (Figure 6D). We examined the effects of KLF9 induction on the expression of Notch pathway target genes. KLF9 induction also decreased the expression of the Notch pathway target genes, HES1 and HES5 expression by ~50-65% and HEY2 expression by ~80% (Figure 6E).
Figure 6. KLF9 suppresses Notch1 transcription in GBM-derived neurospheres.
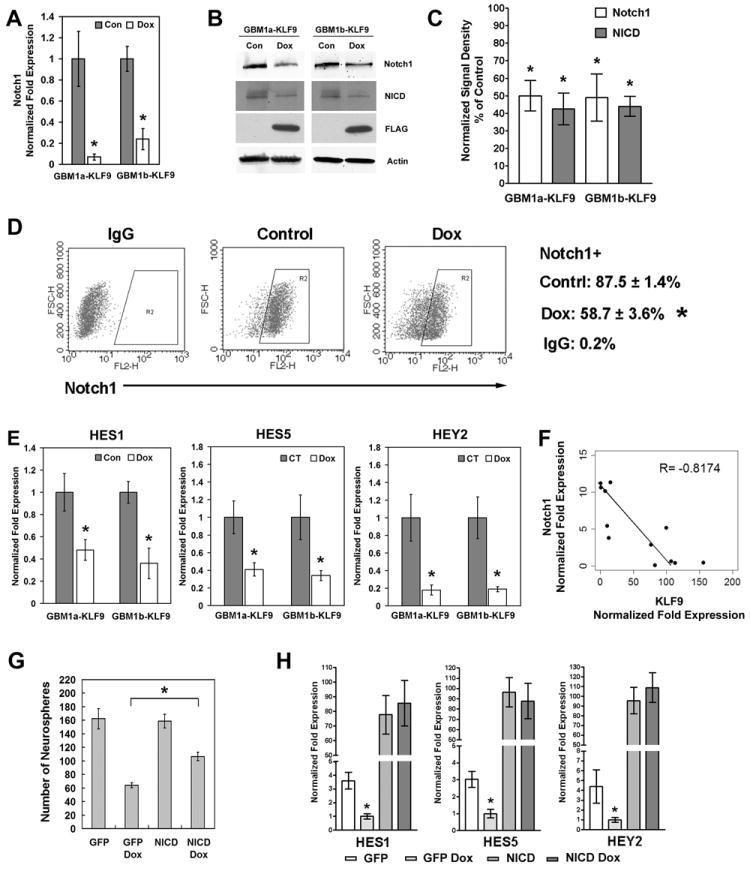
(A) GBM1a-KLF9 and GBM1b-KLF9 cells were cultured in neurosphere medium +/- Dox for 48h and Notch1 expression measured by qRT-PCR. KLF9 induction inhibited Notch1 expression. (B and C) GBM1a-KLF9 and GBM1b-KLF9 cells were treated +/- Dox for 72h. Whole cell protein was subjected to immunoblot using antibodies against Notch1, NICD, FLAG and β-Actin. Expression levels of Notch1 and NICD normalized to β-Actin are shown in C. KLF9 induction inhibited the expression of Notch1 and NICD proteins. (D) GBM1a-KLF9 cells were treated +/- Dox as in (B) and live cells were subject to flow cytometry using anti-Notch1 or isotype IgG control antibodies. Representative dot plots and the percentages of Notch1+ cells are shown. KLF9 induction decreased the percentage of Notch1+ cells. (E) GBM1a-KLF9 and GBM1b-KLF9 cells were treated +/- Dox for 48h. KLF9 induction decreased the expression of HES1, HES5 and HEY2 as determined by qRT-PCR. (F) Notch1 and KLF9 expression were quantified and normalized to 18s rRNA in 12 independent low passage primary GBM neurosphere isolates using qRT-PCR. Notch1 expression inversely correlates with KLF9 expression as shown by the scatter plot and linear regression analysis. (G) GBM1a-KLF9 cells were transfected with the active form of Notch1 receptor (NICD) or GFP as the control and treated +/- Dox for 6 days. The number of neurospheres (>100μm diameter) was counted. Constitutive activation of Notch signaling by NICD partially reversed the inhibition of neurosphere formation by KLF9 (n=5). (H) GBM1a-KLF9 cells were transfected with either NICD or GFP as in (H) and treated +/- Dox for 48h. The expression of HES1, HES5 and HEY2 was analyzed using qRT-PCR. Constitutive activation of Notch signaling abrogates the inhibition of HES1, HES5 and HEY2 expression by KLF9. Data represents Mean ± SEM; * indicates p<0.01.
The relationship between KLF9 and Notch1 expression was examined in 12 independent low passage primary GBM neurosphere isolates derived from fresh clinical pathological tissues. Figure 6F shows a scatter plot representing the levels of Notch1 and KLF9 for each neurosphere isolate as determined by qRT-PCR. The correlation coefficient (-0.8174) indicates a strong negative relationship between Notch1 and KLF9 expression in primary GBM-derived neurospheres, consistent with the repressive effect of KLF9 on Notch1 expression shown earlier. To determine if the inhibition of Notch1 signaling mediates the neurosphere response to KLF9 induction, we transfected GBM1a-KLF9 cells with an active form of Notch1 receptor (NICD) or GFP as control followed by Dox treatment to induce KLF9 expression (Figure 6G and Supplemental Figure 4). In control neurospheres expressing GFP, Dox-induced KLF9 inhibited neurosphere formation by 60%. In NICD transfected neurospheres, Dox-induced KLF9 only inhibited neurosphere formation by 33%. NICD transfection also reversed the downregulation of HES1, HES5 and HEY2 induced by KLF9 induction (Figure 6H). These results support a mechanism by which Notch1 signaling inhibition mediates the effects of KLF9 on GBM derived neurosphere-forming cells.
KLF9 binds the Notch1 BTE site and inhibits Notch1 promoter activity
We asked if KLF9 inhibits Notch1 expression through indirect mechanisms or by directly repressing the Notch1 promoter. The human Notch1 promoter sequence was found to contain two BTE sites located at -307 and −292 bps relative to the ATG start codon (Figure 7A). These BTE sites were also conserved in mouse and rat Notch1 promoters. We used chromatin immunoprecipitation (ChIP) to determine if KLF9 binds to the human Notch1 promoter. GBM1a-KLF9 cells were treated with Dox to induce KLF9 expression. Anti-FLAG antibody specifically precipitated FLAG-tagged KLF9 (Figure 7B). PCR analysis including sequencing of the amplified product revealed that anti-FLAG antibody co-precipitated promoter sequences flanking the two human Notch1 promoter BTE sites but not control sequences lacking a BTE site (Figures 7A and 7C). As control, non-immune IgG failed to precipitate either fragment. Electrophoretic mobility shift and supershift assays confirmed that transgenic FLAG-KLF9 is targeted to nuclei and binds BTE sequences in GBM neurosphere cells (Supplemental Figure 7). Thus, KLF9 directly binds to Notch1 promoter BTE sites in GBM-derived neurosphere cells.
Figure 7. KLF9 binds to the Notch1 promoter basic transcriptional element (BTE) sites and inhibits promoter activity.
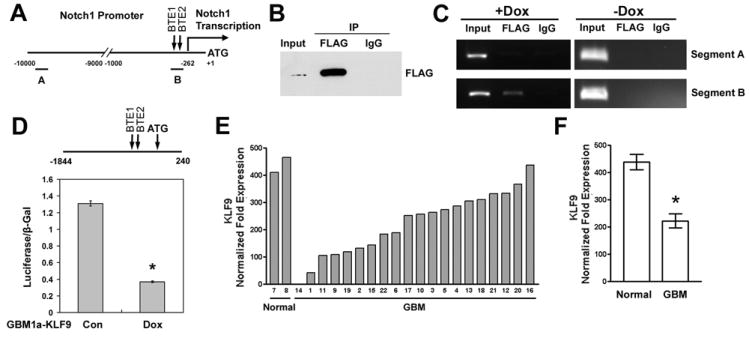
(A) Schematic of the human Notch1 promotor (-10000 to +1 bp relative to the ATG site; BTE1: GGGGCGGGGC; BTE2: GGGGCGGAGC; Notch1 transcription start site: -262 bp). Segment A and segment B indicate the locations of amplified sequences used for analysis of chromatin immunoprecipitation (ChIP) as described in (C). (B) GBM1a-KLF9 cells were treated with Dox for 72h. Anti-FLAG antibody but not nonimmune IgG specifically precipitated FLAG-tagged KLF9 from the fragmented DNA-protein complexes. (C) GBM1a-KLF9 cells were treated +/- Dox for 72h. DNA fragments were precipitated and analyzed with PCR primer pairs designed to amplify BTE-free and BTE-containing Notch promoter sequences (see A). In Dox-treated but not untreated cells, anti-FLAG antibody co-precipitated BTE-containing (segment B) but not BTE-free (segment A) promoter regions. Control IgG failed to precipitate either fragment. (D) Human Notch1 promoter sequences (-1844 to +240 relative to ATG site, shown on top of the panel) were inserted into the luciferase reporter pGL3-Basic to generate Npro vector. GBM1a-KLF9 cells were treated +/- Dox for 48h and cotransfected with Npro and β-Gal plasmids. 48h after transfection, luciferase activity was measured and normalized to β-Gal activity. KLF9 induction inhibited the luciferase activity driven by the Notch1 promoter. * indicates p<0.01. (E and F) KLF9 expression in 2 normal brains and 20 GBMs was measured by qRT-PCR and normalized to 18S rRNA. KLF9 expression was significantly lower in GBM compared with normal brains (* indicates p=0.0176, t test). Data represents Mean ± SEM.
Next, we used a Notch1 promoter-reporter assay to determine the effect of KLF9 on Notch1 promoter function. A BTE-containing fragment of the human Notch1 promoter (-1844 to +240) was cloned into a luciferase reporter vector to generate the Npro vector (Figure 7D). GBM1a-KLF9 cells were treated +/- Dox for 48h and co-transfected with Npro and β-Gal vectors. As shown in Figure 7D, KLF9 induction inhibited Notch1 promoter-driven luciferase activity by ~72% when compared with controls. This combined with the ChIP results shows that KLF9 suppresses Notch1 transcription by directly acting upon the Notch1 promoter.
KLF9 is down regulated in human GBM tumors
The ability of KLF9 to inhibit GBM neurosphere formation, deplete neurospheres of CD133+ stem-like cells, and inhibit the growth and invassiveness of neurosphere-drived tumor xenografts is consistent with a tumor suppressive function within tumor-initiating stem cells. Therefore, we asked if KLF9 expression differs between non-neoplastic (normal) human brain and clinical GBM specimens. Total RNA from 20 human GBM tumors and 2 non-neoplastc human temporal lobe specimens, were examined for KLF9 expression using qRT-PCR (Figure 7E). Normalized KLF9 expression was found to be ~2-fold higher in normal brains compared to GBM tumors (Figure 7F, 438 ± 28 vs 222 ± 25, Mean ± SEM, p<0.05). Serial analysis of gene expression (SAGE) provides an additional sensitive and reliable method for comparing transcript levels between different sources, including transcription factor transcripts that are commonly expressed at relatively low levels. We analyzed the SAGE dataset from Parsons et al. containing clinical GBM specimens, human GBM xenografts, and normal human brains [49] for KLF9 transcript levels. KLF9 expression was found to be significantly decreased in both human GBM tumors and GBM xenografts when compared with normal brains (Supplemental Figure 5).
Discussion
We identified changes in the expression of KLF transcription factors as GBM-derived neurospheres, enriched for tumor-initiating stem cells, transit from conditions of growth/self-renewal to conditions that induce differentiation. KLF9 was found to be the only KLF that was upregulated in response to both differentiating stimuli examined, retinoic acid and serum. KLF9 induction was absent in the established adherent GBM cell line (U87MG), suggesting that KLF9 may regulate transcriptional mechanisms that are specific to neurosphere-forming cells with the capacity for multiple lineage differentiation. KLF9, also known as basic transcription element-binding protein 1 (BTEB1) is currently best known as a regulator of steroid hormone signaling [48, 50]. KLF9 interacts with the progesterone receptor (PGR) B to promote the PGR-B-dependent transactivation of progesterone-responsive promoters [51, 52]. KLF9 is also known to function as either a negative or a positive regulator of estrogen receptor alpha signaling in a cell context dependent manner [53, 54]. Recent evidence suggests an association between KLF9 and human endometrial tumor pathology, with significantly higher KLF9 transcript levels in normal endometrium and stage I endometrial tumors when compared to more aggressive stage II-IV tumors [32]. Similarly in colorectal cancer, both KLF9 mRNA and protein levels were down regulated in tumor tissues relative to matched normal tissues [38]. Despite the relative absence of information linking KLF9 mechanistically to neoplastic processes, these two studies suggest a possible tumor suppressive role for KLF9 in certain tissues. To our knowledge, KLF9 has not been studied previously within the context of normal or neoplastic stem cell biology.
We found that KLF9 induction is essential for the complete response of CSCs to forced differentiation. Upregulating KLF9 expression was found to inhibit GBM neurosphere self-renewal and to phenocopy the effects of forced differentiation based on multiple experimental endpoints. These include the inhibition of neurosphere formation, the induction of differentiation–associated markers such as GFAP and TUJ1, and the attenuation of stemness markers, including CD133 and Olig2. Conversely, KLF9 knockdown inhibited GBM neurosphere differentiation induced by RA and also rescued cells from the RA-induced inhibition of neurosphere formation, a marker of self-renewal capacity. These results demonstrate that KLF9 functions downstream in the RA-induced CSC differentiation response. Another KLF family member, KLF6, which has been reported to inhibit the proliferation of the established U87 GBM cell line [36], had no inhibitory effects on GBM neurospheres. Thus, KLF9 has a selective role in GBM CSCs, which is consistent with its relatively selective upregulation in response to differentiating signals. It is notable that KLF9 knockdown alone had no obvious effects on neurosphere formation or neurosphere cell proliferation under basal growth conditions devoid of differentiation signals. This context-dependent effect of KLF9 knockdown is best explained by the fact that KLF9 expression levels are low under basal growth conditions and that differentiation signals induce KLF9 expression leading to new KLF9-dependent transcriptional events that differ from those under basal growth conditions. We show that one of these differentiation-induced KLF9 transcriptional events acts on the Notch1 promoter.
We show the first time that KLF9 suppresses Notch1 expression and downstream signaling and depletes GBM neurospheres of their stem-like phenotype through these mechanisms. Notch signaling normally supports development and tissue homeostasis by mediating communication between neighboring cells [55, 56]. Notch signaling is known to be essential for maintaining a variety of adult stem cells, including mammary [57, 58], intestinal [59, 60], and neural stem cells [61, 62], by promoting self-renewal and inhibiting differentiation. Abnormal Notch activity has been found in a wide range of human tumors, such as leukemia [63] and breast cancer [64, 65]. In GBM, Notch1 protein was found to be overexpressed in three of four tumor cell lines, and in four of five primary clinical samples [66]. Recent evidence shows that Notch inhibition by γ-secretase inhibitors depletes CD133+ GBM CSCs, inhibits tumor initiation, and sensitizes CSCs to DNA damage [21, 67]. We used multiple complementary criteria to show that KLF9 induction inhibits Notch1 signaling. These include reductions in Notch1 mRNA and protein, cell surface Notch1, activated Notch1 cleavage product (NICD), and the downregulation of HES1, HES5, and HEY2. KLF9 was found to suppress the Notch1 promoter in a direct manner as indicated by the chromatin immunoprecipitation and luciferase reporter assays. A repressive role for KLF9 on Notch1 expression in tumor initiating cells was further supported by the strong negative correlation between Notch1 and KLF9 expression in twelve primary low passage human neurosphere isolates. Our finding that KLF4, a KLF family member that supports normal stem cells and the generation of induced pluripotent stem (iPS) cells, was down regulated during forced GBM neurosphere differentiation is interesting in light of the findings of Liu et al. showing that KLF4 also directly binds the Notch1 promoter, and activates Notch1 expression in epithelial cells [41]. Consistent with published evidence showing that KLF family members can share promoter binding sites and thereby coordinately regulate gene expression [30, 31, 33], it is possible that KLF4 and KLF9 may regulate the balance between stemness and differentiation in normal and neoplastic cells via Notch signaling.
Conclusion
We identify KLF9 as a differentiation-induced transcription factor in GBM neurosphere cultures that are enriched in tumor-initiating stem cells. KLF9 is shown to function as a suppressor of neoplastic cell stemness and an activator of differentiation by directly suppressing Notch1 signaling. These findings point to a suppressive function of KLF9 on brain tumor-initiating stem cells. This conclusion is further supported by KLF9’s downregulation in clinical GBM specimens relative to normal brain [49], in endometrial carcinoma [32, 39] and in colorectal cancer [38]. Our findings expand the role of KLF family members in stem cell and cancer biology, support KLF9 as a regulator of GBM malignancy, and as a potential biomarker and target for cancer therapeutics.
Supplementary Material
Acknowledgments
This research was funded by NIH R01 NS43987 (JL), NIH 5K08 NS055851 (AQH), the James S. McDonnell Foundation (JL, ALV, CGB and AQH) and the Maryland Stem Cell Research Fund (MY, SX and HGC). We thank Jonathan Pevsner for microarray analysis.
Footnotes
Author contributions: M.Y.: Conception and design, Collection and assembly of data, Data analysis and interpretation, Manuscript writing, Final approval of Manuscript; Y.S., Y.L., S.X.: Collection and assembly of data, Final approval of Manuscript; H.G., A.Q., A.L.V., C.G.E.: Providing material, Final approval of Manuscript; J.L.: Conception and design, Financial support, Administrative support, Data analysis and interpretation, Manuscript writing, Final approval of Manuscript.
Disclosure of Potential Conflicts of Interest
The authors indicate no potential conflicts of interest.
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Park CH, Bergsagel DE, McCulloch EA. Mouse myeloma tumor stem cells: a primary cell culture assay. J Natl Cancer Inst. 1971;46:411–422. [PubMed] [Google Scholar]
- 3.Reya T, Morrison SJ, Clarke MF, et al. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 4.Lobo NA, Shimono Y, Qian D, et al. The biology of cancer stem cells. Annu Rev Cell Dev Biol. 2007;23:675–699. doi: 10.1146/annurev.cellbio.22.010305.104154. [DOI] [PubMed] [Google Scholar]
- 5.Ward RJ, Dirks PB. Cancer stem cells: at the headwaters of tumor development. Annu Rev Pathol. 2007;2:175–189. doi: 10.1146/annurev.pathol.2.010506.091847. [DOI] [PubMed] [Google Scholar]
- 6.Singh SK, Hawkins C, Clarke ID, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 7.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 8.Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008;8:755–768. doi: 10.1038/nrc2499. [DOI] [PubMed] [Google Scholar]
- 9.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer. 2006;6:425–436. doi: 10.1038/nrc1889. [DOI] [PubMed] [Google Scholar]
- 10.Stiles CD, Rowitch DH. Glioma stem cells: a midterm exam. Neuron. 2008;58:832–846. doi: 10.1016/j.neuron.2008.05.031. [DOI] [PubMed] [Google Scholar]
- 11.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 12.Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao S, Wu Q, Sathornsumetee S, et al. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006;66:7843–7848. doi: 10.1158/0008-5472.CAN-06-1010. [DOI] [PubMed] [Google Scholar]
- 14.Galli R, Binda E, Orfanelli U, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 15.Hemmati HD, Nakano I, Lazareff JA, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci U S A. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park IK, Qian D, Kiel M, et al. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 17.Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423:255–260. doi: 10.1038/nature01572. [DOI] [PubMed] [Google Scholar]
- 18.Panchision DM, McKay RD. The control of neural stem cells by morphogenic signals. Curr Opin Genet Dev. 2002;12:478–487. doi: 10.1016/s0959-437x(02)00329-5. [DOI] [PubMed] [Google Scholar]
- 19.Piccirillo SG, Reynolds BA, Zanetti N, et al. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature. 2006;444:761–765. doi: 10.1038/nature05349. [DOI] [PubMed] [Google Scholar]
- 20.Androutsellis-Theotokis A, Leker RR, Soldner F, et al. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–826. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- 21.Fan X, Khaki L, Zhu TS, et al. NOTCH pathway blockade depletes CD133-positive glioblastoma cells and inhibits growth of tumor neurospheres and xenografts. Stem Cells. 2010;28:5–16. doi: 10.1002/stem.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heo JS, Lee MY, Han HJ. Sonic hedgehog stimulates mouse embryonic stem cell proliferation by cooperation of Ca2+/protein kinase C and epidermal growth factor receptor as well as Gli1 activation. Stem Cells. 2007;25:3069–3080. doi: 10.1634/stemcells.2007-0550. [DOI] [PubMed] [Google Scholar]
- 23.Bar EE, Chaudhry A, Lin A, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–2533. doi: 10.1634/stemcells.2007-0166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abdouh M, Facchino S, Chatoo W, et al. BMI1 sustains human glioblastoma multiforme stem cell renewal. J Neurosci. 2009;29:8884–8896. doi: 10.1523/JNEUROSCI.0968-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boyer LA, Lee TI, Cole MF, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122:947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang J, Rao S, Chu J, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–368. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- 27.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 28.Wernig M, Meissner A, Foreman R, et al. In vitro reprogramming of fibroblasts into a pluripotent ES-cell-like state. Nature. 2007;448:318–324. doi: 10.1038/nature05944. [DOI] [PubMed] [Google Scholar]
- 29.Gangemi RM, Griffero F, Marubbi D, et al. SOX2 silencing in glioblastoma tumor-initiating cells causes stop of proliferation and loss of tumorigenicity. Stem Cells. 2009;27:40–48. doi: 10.1634/stemcells.2008-0493. [DOI] [PubMed] [Google Scholar]
- 30.Kaczynski J, Cook T, Urrutia R. Sp1- and Kruppel-like transcription factors. Genome Biol. 2003;4:206. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pearson R, Fleetwood J, Eaton S, et al. Kruppel-like transcription factors: a functional family. Int J Biochem Cell Biol. 2008;40:1996–2001. doi: 10.1016/j.biocel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 32.Simmen RC, Pabona JM, Velarde MC, et al. The emerging role of Kruppel-like factors in endocrine-responsive cancers of female reproductive tissues. J Endocrinol. 2010;204:223–231. doi: 10.1677/JOE-09-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang J, Chan YS, Loh YH, et al. A core Klf circuitry regulates self-renewal of embryonic stem cells. Nat Cell Biol. 2008;10:353–360. doi: 10.1038/ncb1698. [DOI] [PubMed] [Google Scholar]
- 34.Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005;7:1074–1082. doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- 35.Zhang W, Geiman DE, Shields JM, et al. The gut-enriched Kruppel-like factor (Kruppel-like factor 4) mediates the transactivating effect of p53 on the p21WAF1/Cip1 promoter. J Biol Chem. 2000;275:18391–18398. doi: 10.1074/jbc.C000062200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimmelman AC, Qiao RF, Narla G, et al. Suppression of glioblastoma tumorigenicity by the Kruppel-like transcription factor KLF6. Oncogene. 2004;23:5077–5083. doi: 10.1038/sj.onc.1207662. [DOI] [PubMed] [Google Scholar]
- 37.Narla G, Heath KE, Reeves HL, et al. KLF6, a candidate tumor suppressor gene mutated in prostate cancer. Science. 2001;294:2563–2566. doi: 10.1126/science.1066326. [DOI] [PubMed] [Google Scholar]
- 38.Kang L, Lu B, Xu J, et al. Downregulation of Kruppel-like factor 9 in human colorectal cancer. Pathol Int. 2008;58:334–338. doi: 10.1111/j.1440-1827.2008.02233.x. [DOI] [PubMed] [Google Scholar]
- 39.Simmen FA, Su Y, Xiao R, et al. The Kruppel-like factor 9 (KLF9) network in HEC-1-A endometrial carcinoma cells suggests the carcinogenic potential of dys-regulated KLF9 expression. Reprod Biol Endocrinol. 2008;6:41. doi: 10.1186/1477-7827-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lal B, Xia S, Abounader R, et al. Targeting the c-Met pathway potentiates glioblastoma responses to gamma-radiation. Clin Cancer Res. 2005;11:4479–4486. doi: 10.1158/1078-0432.CCR-05-0166. [DOI] [PubMed] [Google Scholar]
- 41.Liu Z, Teng L, Bailey SK, et al. Epithelial transformation by KLF4 requires Notch1 but not canonical Notch1 signaling. Cancer Biol Ther. 2009;8:1840–1851. doi: 10.4161/cbt.8.19.9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sun P, Xia S, Lal B, et al. DNER, an epigenetically modulated gene, regulates glioblastoma-derived neurosphere cell differentiation and tumor propagation. Stem Cells. 2009;27:1473–1486. doi: 10.1002/stem.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pollard SM, Yoshikawa K, Clarke ID, et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- 44.Campos B, Wan F, Farhadi M, et al. Differentiation therapy exerts antitumor effects on stem-like glioma cells. Clin Cancer Res. 16:2715–2728. doi: 10.1158/1078-0432.CCR-09-1800. [DOI] [PubMed] [Google Scholar]
- 45.Laks DR, Masterman-Smith M, Visnyei K, et al. Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells. 2009;27:980–987. doi: 10.1002/stem.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bidlingmaier S, Zhu X, Liu B. The utility and limitations of glycosylated human CD133 epitopes in defining cancer stem cells. J Mol Med. 2008;86:1025–1032. doi: 10.1007/s00109-008-0357-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ligon KL, Huillard E, Mehta S, et al. Olig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron. 2007;53:503–517. doi: 10.1016/j.neuron.2007.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Imataka H, Sogawa K, Yasumoto K, et al. Two regulatory proteins that bind to the basic transcription element (BTE), a GC box sequence in the promoter region of the rat P-4501A1 gene. Embo J. 1992;11:3663–3671. doi: 10.1002/j.1460-2075.1992.tb05451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kobayashi A, Sogawa K, Imataka H, et al. Analysis of functional domains of a GC box-binding protein, BTEB. J Biochem. 1995;117:91–95. doi: 10.1093/oxfordjournals.jbchem.a124727. [DOI] [PubMed] [Google Scholar]
- 51.Zhang D, Zhang XL, Michel FJ, et al. Direct interaction of the Kruppel-like family (KLF) member, BTEB1, and PR mediates progesterone-responsive gene expression in endometrial epithelial cells. Endocrinology. 2002;143:62–73. doi: 10.1210/endo.143.1.8590. [DOI] [PubMed] [Google Scholar]
- 52.Zhang XL, Zhang D, Michel FJ, et al. Selective interactions of Kruppel-like factor 9/basic transcription element-binding protein with progesterone receptor isoforms A and B determine transcriptional activity of progesterone-responsive genes in endometrial epithelial cells. J Biol Chem. 2003;278:21474–21482. doi: 10.1074/jbc.M212098200. [DOI] [PubMed] [Google Scholar]
- 53.Pabona JM, Velarde MC, Zeng Z, et al. Nuclear receptor co-regulator Kruppel-like factor 9 and prohibitin 2 expression in estrogen-induced epithelial cell proliferation in the mouse uterus. J Endocrinol. 2009;200:63–73. doi: 10.1677/JOE-08-0383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Velarde MC, Zeng Z, McQuown JR, et al. Kruppel-like factor 9 is a negative regulator of ligand-dependent estrogen receptor alpha signaling in Ishikawa endometrial adenocarcinoma cells. Mol Endocrinol. 2007;21:2988–3001. doi: 10.1210/me.2007-0242. [DOI] [PubMed] [Google Scholar]
- 55.Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131:965–973. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- 56.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 57.Dontu G, Jackson KW, McNicholas E, et al. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004;6:R605–615. doi: 10.1186/bcr920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farnie G, Clarke RB. Mammary stem cells and breast cancer--role of Notch signalling. Stem Cell Rev. 2007;3:169–175. doi: 10.1007/s12015-007-0023-5. [DOI] [PubMed] [Google Scholar]
- 59.Fre S, Huyghe M, Mourikis P, et al. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964–968. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 60.van Es JH, van Gijn ME, Riccio O, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 61.Hitoshi S, Alexson T, Tropepe V, et al. Notch pathway molecules are essential for the maintenance, but not the generation, of mammalian neural stem cells. Genes Dev. 2002;16:846–858. doi: 10.1101/gad.975202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakamura Y, Sakakibara S, Miyata T, et al. The bHLH gene hes1 as a repressor of the neuronal commitment of CNS stem cells. J Neurosci. 2000;20:283–293. doi: 10.1523/JNEUROSCI.20-01-00283.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weng AP, Ferrando AA, Lee W, et al. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science. 2004;306:269–271. doi: 10.1126/science.1102160. [DOI] [PubMed] [Google Scholar]
- 64.Pece S, Serresi M, Santolini E, et al. Loss of negative regulation by Numb over Notch is relevant to human breast carcinogenesis. J Cell Biol. 2004;167:215–221. doi: 10.1083/jcb.200406140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Politi K, Feirt N, Kitajewski J. Notch in mammary gland development and breast cancer. Semin Cancer Biol. 2004;14:341–347. doi: 10.1016/j.semcancer.2004.04.013. [DOI] [PubMed] [Google Scholar]
- 66.Kanamori M, Kawaguchi T, Nigro JM, et al. Contribution of Notch signaling activation to human glioblastoma multiforme. J Neurosurg. 2007;106:417–427. doi: 10.3171/jns.2007.106.3.417. [DOI] [PubMed] [Google Scholar]
- 67.Wang J, Wakeman TP, Lathia JD, et al. Notch promotes radioresistance of glioma stem cells. Stem Cells. 2010;28:17–28. doi: 10.1002/stem.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.