

Abstract
Neuropeptide identification in mass spectrometry experiments using database search programs developed for proteins is challenging. Unlike proteins, the detection of the complete sequence using a single spectrum is required to identify neuropeptides or prohormone peptides. This study compared the performance of three open-source programs used to identify proteins, OMSSA, X!Tandem and Crux, to identify prohormone peptides. From a target database of 7850 prohormone peptides, 23550 query spectra were simulated across different scenarios. Crux was the only program that correctly matched all peptides regardless of p-value and at p-value < 1 × 10–2, 33%, 64%, and >75%, of the 5, 6, and ≥7 amino acid-peptides were detected. Crux also had the best performance in the identification of peptides from chimera spectra and in a variety of missing ion scenarios. OMSSA, X!Tandem and Crux correctly detected 98.9% (99.9%), 93.9% (97.4%) and 88.7% (98.3%) of the peptides at E- or p-value < 1 × 10–6 (< 1 × 10–2), respectively. OMSSA and X!Tandem outperformed the other programs in significance level and computational speed, respectively. A consensus approach is not recommended because some prohormone peptides were only identified by one program.
Keywords: peptidomics, database search program, prohormones, Crux, X!Tandem, OMSSA
Introduction
Neuropeptides are a subclass of peptides that can function as neuromodulators, neurotransmitters and hormones and have a critical role in many biological processes such as growth, learning, memory, metabolism and neuronal differentiation and disorders such as depression, Parkinson’s disease, and eating and sleeping disorders.1−5 Most neuropeptides range in length from 3 to 40 amino acids and are produced by a complex post-translational processing. This processing includes removal of a signaling peptide, cleavage of precursor prohormones at basic amino acids (K and R), removal of C-terminal basic amino acids by carboxypeptidases, and post-translational modifications (PTMs).6−9
Mass spectrometry (MS) is a well-established technology to identify proteins and peptides. The shotgun proteomics implementation of the bottom-up approach relies on the direct protease digestion (typically with trypsin) with subsequent separation of the peptides in the first LC separation step for tandem mass spectrometry (MS/MS). In a subsequent step, the resulting digested peptides are subjected to tandem mass spectrometry (MS/MS) to generate MS/MS spectra. Database searching is a common approach to identify peptides and, consequently, proteins from MS/MS spectra. Multiple database search programs are available including SEQUEST,10 X!Tandem,11 and Crux.12 The overall strategy of database searches is to pair observed and theoretical or predicted spectra. The observed spectra arise from MS/MS experiments and the theoretical spectra are the result of in silico prediction based on the known sequence of potential peptides in a database. Most databases include proteins that have been empirically confirmed or predicted from genome sequence assemblies and/or EST libraries.
Database search programs use different algorithms and, thus, differ in the capability to identify peptides.13−15 Comparing the performance of Mascot, SEQUEST, Sonar, X!Tandem and Spectrum Mill, 15% of human serum and plasma MS/MS spectra was identified by at least one program.14 Similarly, on average 34% of the proteins from normal human ovarian epithelium was identified by Mascot, OMSSA, SEQUEST and X!Tandem.16 Out of 1837 human histone MS/MS spectra, 5% was identified by MassMatrix, Mascot, OMSSA and X!Tandem.17 Inconsistencies among programs in peptide identification have been attributed to differences in the matching algorithms although simulated spectra have not been used to assess the performance of the programs.
The same MS/MS database search programs used to identify proteins and peptides in general are also used to identify neuropeptides.2,9,18,19 However, the extrapolation of the performance of these programs to identify neuropeptides is not straightforward. First, the goals are different when characterizing proteins via shotgun proteomics and for a typical neuropeptide measurement. In a proteomics experiment, the proteins are digested into peptides via an enzyme and then the goal is to identify unique peptides from the protein with high enough confidence that the existence of the protein can be inferred. Higher numbers or more unique peptides identified translate to higher evidence supporting the presence of the protein in the sample. On the other hand, neuropeptides already exist in the sample as endogenous peptides prior to sample preparation or enzymatic degradation. Consequently, each individual neuropeptide requires the precise identification of the exact form of the peptide. The second distinctive feature is that the length of neuropeptides tends to be small and, thus, precise short sequence matches tend to be less statistically significant than the matches of potentially many or longer peptide products from shotgun proteomics. Short lengths limit the statistical significance of the match of the peptide to a database and thus the capability to detect peptide matches beyond a user-defined statistical threshold. A third distinctive feature is that neuropeptides may result from cleavages by multiple proteases. Thus, the standard MS digestion model to generate peptides from proteins is not exactly applicable to neuropeptides because many of these may lack additional basic amino acids or result in smaller peptides. Consequently, the digestion model may not identify the correct peptide and fail to distinguish between shorter and longer forms of the same peptide. The fourth distinctive feature is that the proteins and peptides under consideration are synthesized as inactive precursors, that are converted to their mature forms by protein convertases (PCs) resulting in bioactive proteins and peptides.20 Generally, these endoproteases cleave the precursor substrates at the C-terminal side of single, paired, or tetra basic amino acid residues including arginine or lysine.21 Subsequently the C-terminal basic residues of protein/peptide intermediates are eliminated by specialized carboxypeptidases leading to the mature peptides.22 These peptides may undergo additional post-translational modifications including C-terminal amidation, N-terminal acetylation, glycosylation, sulfation, and phosphorylation prior to the formation of the final bioactive peptides.20 This specialized processing is not modeled by protein identification programs used in shotgun proteomics.
In summary, the goal in prohormone peptide MS studies is to identify individual and typically small peptides, and not to use the presence of unique peptides to infer the presence of a larger protein. After all, the presence of the protein Angiotensinogen is not as important as detecting the forms of Angiotensin present as each form (whether Angiotensin I, II or III) implies something different. Because of these differences in goals, the need to characterize the exogenous peptides in a sample creates a different set of factors that has a unique and still uncharacterized impact on the performance of the different protein search algorithms to identify neuropeptides.
Sample preparation, instrumental and algorithmic settings need to be tailored to minimize the influence of sample complexity and dynamic range, typically observed in neuropeptide studies. These settings can result in peaks selected for MS/MS representing chimeras that contain more than one peptide with similar mass-to-charge (m/z) values. This situation has been observed in neuropeptides. For example, from a single 875.79 m/z precursor peak, both the ProSAAS big LEN peptide (1744.964 Da with charge state +2) and Rhombex-40 peptide (2623.345 Da with charge state +3) have been identified.23 A least one peptide is expected to be identified in these chimera spectra due to the difference in charge states. A more challenging situation occurs when the peptides in a chimera have similar theoretical mass and identical charge state. Prevalence of chimera events that can lead to inaccurate identification has been estimated to range between 11 and 50% of the MS/MS trypsin-digested spectra.24,25
No large-scale, systematic study of the strengths and weaknesses of database search programs to identify neuropeptides and other potential peptides resulting from prohormone processing have been reported. The unique characteristics of these peptides, compared to peptides resulting from a tryptic digest justify the evaluation and recommendation of database programs and algorithms that best support the identification of neuropeptides. The aims of this study were: (1) to compare the relative advantages of three complementary open-source search methods: OMSSA, X!Tandem and Crux to accurately identify prohormone peptides including neuropeptides; (2) to evaluate the impact of MS factors such as charge on neuropeptide identification; and (3) to offer guidelines to obtain the most comprehensive and accurate survey of the peptides in a sample.
Materials and Methods
A comprehensive set of prohormone and peptide sequences were compiled from our two public repositories, NeuroPred26 (http://neuroproteomics.scs.illinois.edu/neuropred.html) and SwePep27 (http://www.swepep.org), and complemented with information from UniProt28 (http://www.uniprot.org; release 2011_01). NeuroPred was also used to predict the most likely and potentially cleaved peptides from 92 mouse prohormones.21,29−31 The final database consisted of 7850 peptides that ranged in length from 5 to 255 amino acids including experimentally confirmed neuropeptides and predicted peptides (Table 1).
Table 1. Summary of the Peptides Used to Simulate the Query Spectra and Populate the Target Database.
| Number of prohormones | 92 |
| Number of peptides | 7850 |
| Average (minimum, maximum) number of peptides/prohormones | 74.06 (1, 1139) |
| Average (min, max) peptide size (amino acids) | 75.23 (5, 255) |
| Percentage of peptides from UniProt | 3.35% |
| Percentage of peptides not from UniProt | 96.65% |
Peptides from mouse prohormones were used to simulate the observed or query spectra and generate the corresponding search or target database. The rationale for matching the observed data to the same counterpart in the database without the addition of a decoy database is 3-fold. First, a decoy database does not assist in determining if the algorithms can correctly match the spectra to the correct target. Rather a decoy database provides a general measure of confidence among the matches. Second, the simulated data share the same quality and thus the addition of decoy mass spectra does not aid in addressing quality differentials in the present study. Third, neuropeptides tend to be short. A reverse decoy spectra of a short peptide has higher likelihood to be present in nature than that of a longer peptide, thus biasing the objective of these spectra to help assess the probability of a random match. The lack of known spectra with no target database entry prevented the comparison of performance across programs using receiver operating characteristics curves.
Among the database search programs available, three public programs were considered: Crux, X!Tandem, OMSSA.11,12,32 These programs were selected because they are open source and this allows the investigation of the code, computation of matching scores and the algorithmic specifications. These three programs were compiled using the default settings and run on the same computer (a 3.00 GHz Intel X9650 processor).
These three programs compute a score indicator of the similarity (e.g., correlation) between the query and database spectra.14 The score is then used to compute an E-value (Expected value or expected number of database matches by chance with scores equal or higher than the one observed) or p-value (the probability that the match between the query and target sequences is due to chance). The programs differ on the algorithm used to identify and score the matches and statistical significance indicator of each match.
Crux (Version 1.37 released on December 22, 2011) is an alternative implementation of the SEQUEST algorithm.12 Peptide identification relies on searching a collection of spectra against an indexed sequence database, and returning a collection of peptide-spectrum matches (PSMs). Crux option to calculate p-values from a Weibull distribution of the cross-correlation scores was used in this study.13 Although this approach is computationally intensive, this strategy maximizes sensitivity or true positive rate through the ability to identify peptides regardless of the quality of the spectra at the expense of higher rates of false positives or mismatches.
X!Tandem (http://www.thegpm.org/tandem; Version 2010.12.01.1 released on December 01, 2010) was developed to optimize speed and to minimize the computational requirements.11 The algorithm preprocesses the observed spectra to remove noise and technical artifacts, processes database peptide sequences with cleavage reagents, post-translational and chemical modifications and scores the peptide matches between the observed and predicted spectra.13 The scores are converted to hyperscores and the distribution of hyperscores of all matches is used to translate the hyperscore of each match into an E-value.
The Open Mass Spectrometry Search Algorithm (OMSSA; Version 2.1.7 released on June 15, 2010; http://pubchem.ncbi.nlm.nih.gov/omssa) centers on optimizing the speed of database searching approach.32 The scoring of each match assumes that the number of matches between observed and predicted peaks for a peptide sequence follows a Poisson distribution. The lambda (or average) parameter of the Poisson distribution is calculated as a function of the fragment ion tolerance, the number of predicted and observed peaks and the neutral mass of the precursor ion. OMSSA provides E- and p-values based on the dimensions of the target database.
Simulated spectra were used to compare the performance of the three database search programs. There are three advantages of simulating the observed peptides to be queried against a database. First, the use of simulated mass spectra overcomes the limited number of neuropeptides with mass spectra information of comparable quality obtained using the same or similar technologies. Second, the analysis of simulated mass spectra that share the same quality level allows benchmarking the database search programs irrespectively of sample or data quality issues including low mass accuracy, noise and low signal-to-noise ratio. Third, simulated mass spectra offers an absolute control of the peptides that should be detected and accurate evaluation of the number of true positives (detected and correctly identified peptides), false positives (detected but incorrectly identified peptides) and false negatives (missed peptides).
Ideal uniform spectra that have either +1, +2, and +3 peptide charge states were simulated for each peptide precursor ion in the target database. For each peptide charge status, only +1 charged b- and y-product ions (b- and y-ion series) were simulated with equal intensity. Neutral losses of a water (−18 Da) and/or ammonia (−17 Da) were simulated when the ion contained either one of four water losing amino acids (Ser, Thr, Glu, Asp) or ammonia losing amino acids (Arg, Lys, Gln, Asn). Neutral losses from b- and y-ion series occurred regardless of position of these amino acids in the ions.
Complementary scenarios of neutral mass loss and ion availability conditions were simulated across the three peptide charge states and searched against the database to investigate the impact of these situations on the identification of neuropeptides. The simulated query scenarios included:
-
1)
All b- and y-ion series including all neutral mass losses due to water and ammonia. This scenario constitutes the baseline for comparison.
-
2)
Only the possible b- and y-ion series excluding neutral mass losses.
-
3)
Only the possible b-ion series including all neutral mass losses.
-
4)
Only the possible y-ion series including all neutral mass losses.
-
5)
Random 50% of b- and y-ion series including all neutral mass losses.
-
6)
Random 25% of b- and y-ion series including all neutral mass losses.
-
7)
Only scoring the b-ion series from b- and y-ion spectra including all neutral mass losses.
-
8)
Only scoring the y-ion series from b- and y-ion spectra including all neutral mass losses.
The performance of the database search programs to identify peptides from chimera spectra was investigated. Chimera representation was achieved by combining the simulated spectra from peptides that have similar theoretical mass values and have the same charge state. Only peptides that were correctly identified by the three algorithms at E- or p-value < 0.01 with a single peptide simulated with all neutral losses and ions were used. The resulting peptides were grouped such that the maximum difference in the theoretical mass of each group was within ±0.4 Da. Individual spectrum was simulated for each peptide including all the b- and y-ion series, neutral losses and +1 peptide charge state. Chimera spectra were generated for each mass group by merging these individual simulated peptide spectra into a single spectrum using the average theoretical precursor mass for the precursor ion m/z value.
The peptide identification search programs OMSSA, X!Tandem and Crux were evaluated using comparable algorithmic specifications and excluding PTMs. The default values of the programs were used in addition to the following specifications: (1) precursor ion tolerance: 1.5 Da; (2) product or fragment ion tolerance: 0.3 Da; (3) no fixed or variable modifications; (4) “whole protein” (OMSSA) or “enzyme: custom cleavage site” (X!Tandem and Crux) to prevent cleavage since the detection of neuropeptides does not involve protease digestion; (5) peptide length: 5–255 residues; (6) peptide ion charge: +1, +2, +3; (7) product ion charge: default values; (8) no complete or partial modifications; and (9) peptide mass: monoisotopic.
For comparison purposes, Crux probability scores (ranging from 0 to 1), X!Tandem E-values (ranging from 1 × 10–45 to 1 × 10+3) and OMSSA E-values (ranging from 1 × 10–15 to 1 × 10+4) were transformed using a base 10 logarithm. The match or hit with lowest E- or p-value among all hits per input spectrum was analyzed. The 1 × 10–6 threshold based on a 1% Bonferroni correction (0.01/7850 = 1.27 × 10–6 ≈ 1 × 10–6) was used to determine if the match was significant while accounting for multiple testing.
Results and Discussion
The overall significance and the correctness of the matched sequence of the simulated query-to-target matches were used to assess the capability of each search algorithm to detect neuropeptides and other prohormone peptides. This evaluation step allowed discrimination between obvious and dubious, yet correct, peptide identifications. A peptide match was deemed to be significant if the detection signal (e.g., E- or p-value) was lower (more significant) than a 1 × 10–6 threshold. This stringent threshold aimed to minimize the number of false peptide identifications because the percentage of matches that could be considered by chance (false positives) is less than a 1% Bonferroni corrected significance threshold. There were three outcomes for each simulated spectra: the neuropeptide correctly matched the simulated peptide (true positive), incorrectly matched (false positive) or failed to match (false negative). A lower threshold E- or p-value < 1 × 10–2 was also investigated because neuropeptides tend to be short and thus true positives from the database matching process are unlikely to generate extreme E- or p-values.
Impact of Program on Peptide Identification in Baseline Conditions
Table 2 summarizes the results from the three search methods across three peptide charge states. Most peptides (approximately 7764 out of 7850 peptides across charges when all ions are available) were identified by these three programs regardless of E- or p-value level. Among the 7764 peptides correctly matched by all three programs, 7022 were detected by all three programs followed by OMSSA and X!Tandem (385 peptides), OMSSA alone (302 peptides), OMSSA and Crux (7 peptides) at E- or p-value < 1 × 10–6 meanwhile 48 peptides were detected at E- or p-value > 1 × 10–6 by all three programs. Thus, among the peptides detected by all programs regardless of statistical significance, OMSSA detected these peptides with higher statistical significance, followed by X!Tandem and last Crux (Table 2).
Table 2. Number of Peptides Correctly Matched by X!Tandem, OMSSA and Crux for Precursor Charge States +1, +2, and +3 Across Various Scenarios.
| correctly matcheda | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |
OMSSA+X!Tandem+Crux |
OMSSA+Crux |
X!Tandem+Crux |
Crux | |||||||||||
| |
significantb |
||||||||||||||
| scenarioc | charge | All | OC | OX | O | C | N | OC | O | C | N | XC | C | N | N |
| b + y ions | +1 | 7028 | 8 | 378 | 327 | 0 | 23 | 0 | 0 | 1 | 84 | 1 | 0 | 0 | 0 |
| + neutral | +2 | 7012 | 7 | 397 | 313 | 0 | 35 | 0 | 0 | 0 | 85 | 1 | 0 | 0 | 0 |
| mass loss | +3 | 7027 | 5 | 379 | 265 | 0 | 87 | 0 | 0 | 3 | 82 | 2 | 0 | 0 | 0 |
| b + y ions | +1 | 6874 | 5 | 503 | 339 | 0 | 3 | 41 | 0 | 1 | 84 | 0 | 0 | 0 | 0 |
| – neutral | +2 | 6888 | 5 | 485 | 340 | 0 | 3 | 44 | 0 | 0 | 85 | 0 | 0 | 0 | 0 |
| mass loss | +3 | 6978 | 3 | 389 | 337 | 0 | 8 | 50 | 0 | 1 | 84 | 0 | 0 | 0 | 0 |
| b ions | +1 | 6837 | 109 | 105 | 184 | 46 | 484 | 0 | 0 | 1 | 84 | 0 | 0 | 0 | 0 |
| + neutral | +2 | 6831 | 99 | 109 | 175 | 60 | 491 | 0 | 0 | 0 | 85 | 0 | 0 | 0 | 0 |
| mass loss | +3 | 6887 | 99 | 57 | 105 | 57 | 560 | 0 | 0 | 1 | 84 | 0 | 0 | 0 | 0 |
| y ions | +1 | 6911 | 126 | 118 | 221 | 17 | 370 | 2 | 0 | 1 | 84 | 0 | 0 | 0 | 0 |
| + neutral | +2 | 6905 | 116 | 133 | 202 | 11 | 397 | 1 | 0 | 1 | 84 | 0 | 0 | 0 | 0 |
| mass loss | +3 | 6897 | 99 | 133 | 157 | 24 | 454 | 1 | 0 | 2 | 83 | 0 | 0 | 0 | 0 |
| 50% ions | +1 | 6646 | 230 | 69 | 394 | 14 | 410 | 1 | 0 | 0 | 85 | 1 | 0 | 0 | 0 |
| + neutral | +2 | 6638 | 244 | 69 | 382 | 10 | 421 | 1 | 0 | 2 | 83 | 0 | 0 | 0 | 0 |
| mass loss | +3 | 6668 | 254 | 36 | 278 | 27 | 502 | 0 | 0 | 1 | 84 | 0 | 0 | 0 | 0 |
| 25% ions | +1 | 5370 | 661 | 21 | 156 | 318 | 989 | 198 | 4 | 8 | 52 | 0 | 1 | 29 | 43 |
| + neutral | +2 | 5358 | 673 | 25 | 160 | 295 | 987 | 200 | 4 | 6 | 82 | 0 | 0 | 30 | 30 |
| mass loss | +3 | 5378 | 675 | 4 | 170 | 267 | 1010 | 210 | 5 | 5 | 63 | 0 | 0 | 22 | 41 |
Correctly matched peptide regardless of E- or p-value level on all three, two or one of the database search programs. Missing columns (OMSSA+Crux, X!Tandem, OMSSA) are columns with “0” in all rows.
Detection at E- or p-value < 1 × 10–6. All: OMSSA, X!Tandem and Crux; OC: only OMSSA and Crux E- or p-value < 1 × 10–6; OX: only OMSSA and X!Tandem E-value < 1 × 10–6; XC: only X!Tandem and Crux E- or p-value < 1 × 10–6; O: only OMSSA E-value < 1 × 10–6; C: only Crux p-value < 1 × 10–6; N: No program E- or p-value < 1 × 10–6. Missing columns (XC and X within OMSSA+X!Tandem+Crux, X in X!Tandem+Crux) are columns with “0” in all rows.
Simulated query scenarios: b + y ions + neutral mass loss: Match using all b- and y-ion series including neutral mass losses; b + y ions - neutral mass loss: Match using all b- and y-ion series excluding neutral mass losses; b ions + neutral mass loss: Match only using the b-ion series including neutral mass losses; y ions + neutral mass loss: Match only using the y-ion series including neutral mass losses; 50% ions + neutral mass loss: Match only using random 50% of all ions including neutral mass losses; 25% ions + neutral mass loss: Match only using random 25% of all ions including neutral mass losses.
Crux was the only program that detected all peptides regardless of significance level (Table 2). Across charges, approximately one peptide was detected by Crux and X!Tandem reaching E- or p-value < 1 × 10–6. Approximately 85 peptides were detected by OMSSA and Crux only but, of these, only approximately two peptides reached E- or p-value < 1 × 10–6 and this was in Crux.
From 23550 simulated spectra (7850 peptides × 3 peptide charge states), OMSSA, X!Tandem and Crux had 23548 (99.9%), 22932 (97.4%) and 23139 (98.3%) correct identifications (true positives) at E- or p-value < 1 × 10–2. At E- or p-value < 1 × 10–6, OMSSA, X!Tandem and Crux had 23281 (98.9%), 22117 (93.9%) and 20890 (88.7%) true positive results, respectively. The similar performance of the programs at the less significant threshold reiterates the findings summarized in Table 2 that the Crux algorithm provides lower significance results. Our results are consistent with previous reports of a higher number of spectra matched by OMSSA than by X!Tandem.17,33 Figure 1 includes a Venn diagram depicting the peptides correctly identified by the three database search programs for charge state 3 using information from all ions. Insights into the features that are differentially accommodated by the assumptions and models used by each database search algorithm were drawn from the investigation of the peptides that were not identified by all three programs.
Figure 1.

Venn diagram depicting the common and distinct true positive peptides identified from the three database search programs, X!Tandem, OMSSA, and Crux with peptide charge state +3 using (A) all ion information; (B) only y-ion series information; (C) only b-ion series information.
Impact of Peptide Length on Peptide Identification
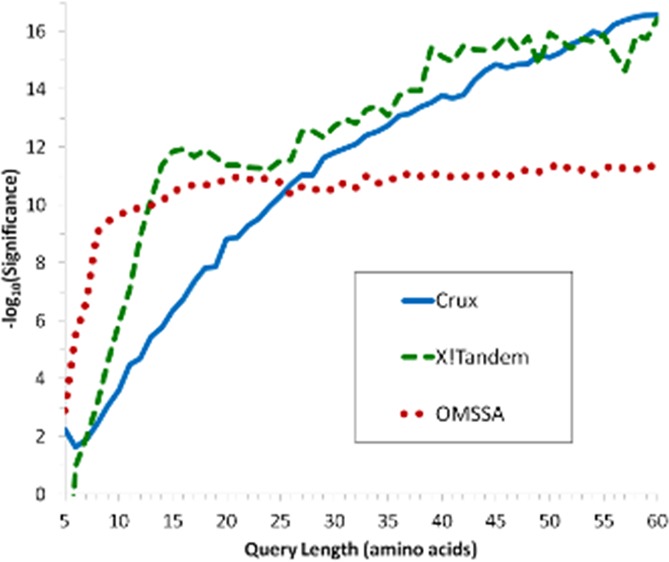
The length of the peptide had an impact on the statistical significance of the match in all the programs. The majority of the 109 (1%) charge +1 peptides that were identified (using b and y ions and including neutral mass loss) by all three programs and reached E-value < 1 × 10–6 in at least one program were five amino acids long peptides. Figure 2 depicts the relationship between the log10 transformed E- or p-values on peptides across peptide length. Overall the correlation between the length of the query sequence and log10 transformation of the E- or p-values for OMSSA, Crux and X!Tandem was 0.1%, 86.8% and 46.7%, respectively although the relationship was nonlinear.
Figure 2.

Comparison of OMSSA, Crux, and X!Tandem log10 (E- or p-values) averaged across peptide length and precursor charge states for all peptides and (inset) magnified for peptides up to 60 amino acids in length.
There was a gradual increase in the number of peptide matches that have p-value < 1 × 10–6 with increased peptide length in Crux although only peptides 46 amino acids long and higher surpassed this threshold. Examination of the relation between query length and log-transformed E-values showed rapid increases up to 11 and 15 amino acids long peptides in OMSSA and X!Tandem, respectively, before the log-transformed E-values stabilized. In contrast, the Crux log-transformed p-values showed a gradual increase up to approximately 50 amino acids before the log-transformed p-values started to stabilize. Consistent with our findings, small peptides between 600 and 700 Da tend to be missed.14 A similar effect of peptide length on the distribution of the MaxQuant program p-scores between target and decoy database was observed.34 In that study, peptides with less than 15 amino acids had a higher likelihood of being incorrectly matched than peptides with 15 or more amino acids.
The increase in E-values with decreasing peptide length is due to the corresponding increase in the number of expected matches by chance. The mean of the underlying Poisson distribution used by OMSSA decreases with smaller peptides resulting in larger E-values due to the increased probability of a random match. In particular, the detection of short peptides by OMSSA is negatively influenced by the tendency of small peptides to exhibit neutral mass losses. For peptides less than 12 amino acids, the correlations of log-transformed E- or p-values between OMSSA and X!Tandem, OMSSA and Crux, and X!Tandem and Crux were 78%, 63% and 52%, respectively. This result also indicates that the selected threshold was more stringent in Crux and X!Tandem than for OMSSA.
No program correctly detected five amino acids long peptides at E- or p-value < 1 × 10–6. OMSSA and X!Tandem correctly detected all peptides longer than 7 and 11 amino acids, respectively at E-value < 1 × 10–6, when all ions were available excluding neutral mass loss. Crux was the only program able to correctly match all peptides although only 12 peptides with less than ten amino acids had p-values < 1 × 10–6. At p-value < 1 × 10–2, 33%, 64%, 75%, 87%, 98%, 99% and 100% of the 5, 6, 7, 8, 9, 10, and 11 amino acids long peptides were detected with Crux. This result suggests that the significance calculation in Crux is more stringent than in the other two programs. The positive association between the fraction of peptides detected and peptide size is partly due to the number of Weibull samples because with 100 permutations only 61 of the peptides with charge state +1 had p-value < 1 × 10–5 threshold (results not shown). Consequently, adding further Weibull samples especially for small peptides may increase the significance levels by providing more accurate density estimation.
X!Tandem was not able to correctly detect the 85 peptides that were five amino acids long which accounted for 1% of all peptides. Across the different scenarios evaluated (ion availability, etc.), most of these peptides were not detected (80%) and the rest were incorrectly identified (mismatched). X!Tandem was able to correctly match at E-value < 1 × 10–6 peptides at least ten amino acids long, and 94% of the peptides that were seven amino acids long and all peptides that were at least eight amino acids long were detected at E-value < 1 × 10–2 threshold, when all ions were available including neutral mass loss. Similarly, the performance of OMSSA was also influenced by peptide size because most peptides at least ten amino acids long were detected at E-value < 1 × 10–6. However, 100%, 27%, 6%, and 1% of the peptides five, six, seven and eight amino acids long, respectively, did not reach significance in OMSSA.
The statistical significance of the X!Tandem matches was inferred using the lowest scores from the matches. Consequently, the significance values assigned by X!Tandem is negatively influenced when there are insufficient matches to provide an accurate estimate of the X!Tandem score. At the other computational extreme, Crux uses resampling to provide a statistical significance value for each peptide identified. Resampling consists of random permutations of the peptide sequence that are subsequently matched to the database and scored. The implementation of resampling in Crux is with replacement, which potentially allows the same sequence to be repeatedly sampled. This event is more likely with shorter peptides and high resampling specifications. Unlike Crux and X!Tandem, the OMSSA E-value is derived from the assumption that the number of matches can be represented by Poisson distribution and does not depend on the matches or generated sequences although it relies on the database size. The OMSSA formulation is also dependent on peptide size so that small peptides tend to be on the lower bound of significance due to the smaller proportion of ion matches than larger peptides. For example, if the Poisson mean is equal to one, then the probability of zero ion matches is 0.37%.
Understanding Neuropeptide Misidentification
The few detection failures in this study were investigated using OMSSA because the E-value calculation offers more sensitivity. Five neuropeptide sequences were not first ranked peptides in OMSSA across all simulated conditions. Four of the peptides are processed from the highly homologous Oxytocin-neurophysin 1 and Vasopressin-neurophysin 2-copeptin prohormones. These peptides were further reduced to two sets of peptides after consideration of ambiguous cleavage sites that lead to two possible peptides within the homologue. The simulated spectra of the b- and y-ions without neutral mass loss were similar between these peptides with the maximum difference being 19.9 m/z and occurring at the b6 ion. Another mismatch occurred with a PENK (UniProt id P22005) peptide due to the multiple occurrences of the Met-enkephalin in a longer peptide. For simulated charge states +1 and +2 including neutral mass loss, a mismatch occurred between the two Met-enkephalin peptides located at the C- and N-terminal. Due to the similarity in sequence and E-values, these peptides were treated as “homeometric peptides”35,36 and were considered as correct matches.
Unlike X!Tandem and Crux, OMSSA failed to detect the peptide for all three charge states and a neutral mass loss for one peptide, a chromogranin B peptide (positions 592 to 652) predicted by the NeuroPred mouse model. Also, OMSSA had one mismatch; a secretogranin II peptide (positions 475 to 547) from a NeuroPred nonmammalian model matched to neurotensin (positions 87 to 156) with charge state +3 and neutral mass loss at an E-value >60. This peptide was detected by OMSSA in the other two charge states. Both peptides were correctly detected when the simulation excluded neutral mass losses. This suggests a weakness (or lower sensitivity) of the OMSSA algorithm to accommodate neutral mass losses. Examination of both peptides indicated that 54% of the amino acids in each sequence were prone to lose water (28% of the amino acids) and ammonia (25% of the amino acids). As a result approximately 2/3 of the ions can include neutral mass losses and OMSSA was not able to distinguish the series with and without neutral losses.
Combining identifications that were significant in at least two programs improves the average identification rate across all three charge states from 89% to 94% when all ions were available for scoring and including neutral mass loss. Using a consensus approach, as has been advocated in the identification of proteins,13 can improve peptide identification because the probability of all programs incorrectly identifying a peptide is equal to or less than probability of the least accurate program being incorrect. While this consensus approach assists in the correct identification of peptides, it is less suitable to the goals of the present study because the individual programs helps us to understand the particular distributional features of the prohormone peptide population of mass spectra relative to protein database searches and recommends the best tools for particular neuropeptides. For example, a closer inspection of the few peptides (5%) that were not consistently identified across programs revealed that these peptides were correctly matched by at least Crux and OMSSA although exhibited low scores, irrespectively of the programs, due to the small size of these peptides (ranging between 5 and 11 amino acids long). This result suggests that for these few neuropeptides all three programs have comparable disadvantages but have complementary strengths and weaknesses to detect neuropeptides.
Impact of Neutral Mass Losses on Peptide Identification
The performance of the three programs in the identification of peptides when all ions from both series are available including and excluding neutral mass losses in the simulated spectra is summarized in Table 3 for charge state +1 and in Supporting Information Tables 1 and 2 for all charge states. The inclusion of neutral mass losses in the simulated query spectra had minor influence on the overall detection of peptides across the programs. The average percentage of peptides that were detected at E- or p-value < 1 × 10–6 including neutral mass loss over all charge states was 98.3%, 94.4% and 89.5% in OMSSA, X!Tandem, and Crux, respectively. The average percentage of peptides that were detected at E- or p-value < 1 × 10–6 excluding neutral mass loss over all charge states was 98.9%, 93.9% and 88.6% in OMSSA, X!Tandem, and Crux, respectively. The opposite trend observed in OMSSA relative to the other two programs may be due to the extremely high percentage of peptides already identified including or excluding neutral mass loss. The percentage of peptides detected at low or nonsignificant values was five- and 10-fold higher in X!Tandem and Crux relative to OMSSA, respectively. For these peptides, inclusion of neutral mass loss improved the detection of peptides at low or nonsignificant levels in OMSSA and X!Tandem by 1% and have the opposite effect in Crux. Peptide detection by Crux or OMSSA was not largely affected by neutral mass loss. The inclusion of neutral mass loss noticeably influenced the significance levels of X!Tandem matches that were already highly significant (E-value < 1 × 10–10).
Table 3. Number of Peptides Unmatched, Mismatched and Correctly Matched at Various Significance Levels by X!Tandem, OMSSA and Crux Including or Excluding Neutral Mass Losses When for All Ions from Both Series in the Query Are Available and for Precursor Charge State +1.
| OMSSA |
X!Tandem |
Crux |
||||
|---|---|---|---|---|---|---|
| significancea | includingb | excluding | including | excluding | including | excluding |
| Unmatchedc | 1 | 0 | 69 | 115 | 0 | 0 |
| Mismatchd | 0 | 0 | 16 | 11 | 0 | 0 |
| 0 | 0 | 0 | 4 | 2 | 1 | 1 |
| 1 | 1 | 0 | 73 | 73 | 118 | 129 |
| 2 | 11 | 1 | 91 | 93 | 214 | 236 |
| 3 | 48 | 2 | 82 | 80 | 171 | 226 |
| 4 | 24 | 10 | 33 | 30 | 160 | 178 |
| 5 | 24 | 75 | 75 | 69 | 151 | 170 |
| 6 | 49 | 4 | 91 | 95 | 172 | 172 |
| 7 | 73 | 5 | 83 | 74 | 171 | 213 |
| 8 | 28 | 63 | 47 | 13 | 194 | 200 |
| ≥9 | 7591 | 7690 | 7186 | 7195 | 6498 | 6325 |
| Prop > 6e | 98.6% | 98.9% | 94.4% | 94.0% | 89.6% | 88.0% |
Significance threshold (t) for matched to be considered significant at E- or p-value < 1 × 10–t.
Including or excluding neutral mass losses.
Unmatched: the program does not provide a match with the program setting.
Mismatched: the program provided an incorrect match.
Percentage of the matches that have E- or p-value < 1 × 10–6.
The impact of neutral mass loss on peptide detection depended on the charge state. At the stringent threshold E-value < 1 × 10–10, peptides that have precursor charge state +1 had more significant matches (93%) than precursor charge state +3 (81%) in OMSSA. This difference decreased with less stringent thresholds and at the E-value < 1 × 10–6 threshold the difference in detection was only 1% between charge states +1 and +3. This may be partially explained by the assumption that +2 product ions are present in precursor charge state +3 and higher spectra but not present in charge state +1 spectra.32 This assumption results in a higher number of possible ions and a consequently a lower significant E-value even if the spectra lacks these highly charged product ions.
Impact of Missing Ions on Peptide Identification
A summary of the performance of the three programs in the identification of peptides when only b-ion series, y-ion series, random 50% of all ions, and random 25% of all ions are available including neutral mass losses, respectively for charge state +1 is presented in Table 4. Likewise, Supporting Information Tables 3, 4, 5, and 6 report the performance of the three programs for all three charge states. The percentage of correct identifications across all programs was 87%, 88%, 85% and 68% when b-ion series, y-ion series, random 50% and random 25% of the ions were available, respectively. The proportion of unidentified peptides in all programs was 8%, 6%, 7% and 14%, when b-ion series, y-ion series, random 50% and random 25% of the ions were available, respectively.
Table 4. Number of Peptides Unmatched, Mismatched and Correctly Matched at Various Significance Levels by X!Tandem, OMSSA and Crux when Either the b-, y-Ion Series, 50%, or 25% of the Ions in the Query Available for Precursor Charge State +1 and Including Neutral Mass Losses.
| OMSSA |
X!Tandem |
Crux |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| significancea | bb | y | 50 | 25 | b | y | 50 | 25 | b | y | 50 | 25 |
| Unmtchc | 0 | 0 | 1 | 73 | 79 | 72 | 73 | 295 | 0 | 0 | 0 | 0 |
| Mismtchd | 0 | 0 | 0 | 4 | 6 | 15 | 13 | 10 | 0 | 0 | 0 | 0 |
| 0 | 160 | 48 | 71 | 492 | 237 | 138 | 316 | 1133 | 0 | 2 | 9 | 60 |
| 1 | 84 | 62 | 72 | 182 | 109 | 113 | 170 | 284 | 93 | 131 | 151 | 322 |
| 2 | 87 | 86 | 85 | 184 | 149 | 156 | 133 | 228 | 229 | 196 | 243 | 302 |
| 3 | 100 | 99 | 87 | 178 | 122 | 113 | 166 | 229 | 215 | 155 | 188 | 218 |
| 4 | 94 | 88 | 106 | 160 | 96 | 98 | 104 | 140 | 154 | 169 | 180 | 183 |
| 5 | 90 | 89 | 88 | 167 | 105 | 109 | 157 | 136 | 167 | 140 | 188 | 209 |
| 6 | 64 | 77 | 81 | 133 | 137 | 139 | 109 | 120 | 188 | 173 | 190 | 273 |
| 7 | 94 | 73 | 86 | 113 | 104 | 105 | 122 | 146 | 167 | 196 | 209 | 326 |
| 8 | 93 | 90 | 74 | 106 | 89 | 103 | 127 | 131 | 168 | 226 | 244 | 365 |
| ≥9 | 6984 | 7138 | 7099 | 6058 | 6617 | 6689 | 6360 | 4998 | 6469 | 6462 | 6248 | 5592 |
| Prop >6e | 92.2 | 94.0 | 93.5 | 81.7 | 88.5 | 89.6 | 85.6 | 68.7 | 89.1 | 89.9 | 87.8 | 83.5 |
Significance threshold (t) for matched to be considered significant at E- or p-value < 1 × 10–t.
b-, y-ion series, 50%, or 25% of the ions in the query are available.
Unmatched: the program does not provide a match with the program setting.
Mismatched: the program provided an incorrect match.
Percentage of the matches that have E- or p-value < 1 × 10–6.
The lower percentage of peptides identified in scenarios that had 50% and 25% of the ions available was mainly due to a poorer performance of X!Tandem regardless of the charge state. The minimum length for a peptide to be detected at significance E-value < 1 × 10–6 were 13, 15, 14, and 81 amino acids for the y-ion series, b-ion series, 50% ions and 25% ions available, respectively, compared to ten amino acids when all ions were available. These trends are likely to be related to the number of ions that can potentially be available rather than the percentage of ions available.
Missing ions also impacted the detection of peptides by OMSSA. On average, 65 peptides across all three charge states were undetected when only 25% of all ions were available. These peptides were between five and eight amino acids long and only peptides with more than 25 amino acids had E-value < 1 × 10–6.
The impact of missing ions on peptide identification depended on charge state. There was a tendency for the number of undetected peptides by OMSSA to increase with increasing charge state when only 50% of ions were available. This result is consistent with the observed trend in the presence of neutral loss simulation and suggests that the presence of neutral loss, rather than the absence of 50% of ions, was the factor driving the lower detection rate. However, when only 25% of the ions were available, charge state +1 peptides were four times more likely to be undetected relative to higher charge states. These results indicate that the absolute number of ions present is potentially more critical to the OMSSA algorithm than the relative percentage of ions available. Overall, these findings highlight a diminishing return on accurate identification for additional ions used by OMSSA, with the detection E-value threshold dependent on the precursor charge state. Longer peptides are expected to generate more ions, suggesting that the OMSSA scoring system based on the actual number of mass spectra peak matches needs to account for the overall peptide length. This adjustment is important for neuropeptides because the goal is identifying each form of these oftentimes short peptides.
Missing ions on the query had minor influence on the identification and significance level of the peptides in Crux, unlike in OMSSA and X!Tandem (Table 4). Venn diagrams depicting the peptides correctly identified at E- or p-value < 1 × 10–6 by all three database search programs, X!Tandem, OMSSA, and Crux using information from all ions or only y- or b-ion series for peptide charge state +3 is depicted in the top, bottom left and bottom right corners of Figure 2, respectively. Figure 2 highlights that overlap among all three programs (particularly between OMSSA and X!Tandem), and the ability of OMSSA to identify peptides scored at E-value < 1 × 10–6. The lesser overlap between programs when either one of the ion series is considered, stresses the relative advantage of Crux when only b-ion series were available, and of OMSSA and X!Tandem when only y-ion series were available for peptide identification. Figure 3 presents the Venn diagrams depicting the peptides identified at E- or p-value < 1 × 10–6 by all three database search programs using only 50% (left Venn diagram) or 25% (right Venn diagram) of all ion information for peptides with charge state +3. The previous Venn diagrams center on peptides detected at high significance levels and ignore the strength of Crux to detect small peptides that have low p-values.
Figure 3.

Venn diagram depicting the common and distinct peptides identified by all three database search programs with peptide charge state +3 using only (A) 50% or (B) 25% of all ion information.
The Venn diagrams highlight the increasing detrimental impact of missing ions on the performance of X!Tandem. The simulation of the random proportion of ions represents one type of incomplete fragmentation that is an important component of the differences between programs in peptide identification.13 Peptides can be identified by the programs when incomplete fragmentation provided sufficient ions are present especially for large peptides. The challenge for these programs centers in assigning an appropriate significance threshold since most of the peptides were correctly matched regardless of program used. The low impact of relative ion availability on Crux is possibly due to the lack of resampled peptides that share similar ion patterns. The OMSSA E-values even increased with fewer ions available because the E-value computation assumes that all possible ions are present. At E-value < 1 × 10–6 threshold, 17.3% and 6.9% of all correct peptide identifications did not reach significance threshold in random 25% and 50% proportion of ions, respectively, relative to 1.7% when all ions were available including neutral mass loss. X!Tandem is clearly negatively influenced by decreased lower number of ions available. A possible explanation is that with fewer ions present, the score of the correct match is insufficiently different from the score of incorrect matches with all ions, both leading to a low score and potentially mismatches.
Impact of Algorithm on the Speed of Search
The search time of the three database search programs was a function of the number of neuropeptides in the search database. This computational comparison is empirical and that the database search can be easily computed in parallel because the experimental spectra can be independently analyzed. The computational time to evaluate all 7850 peptides was measured for all programs separately. X!Tandem returned results the fastest (averaged 23 CPU seconds), followed by Crux with no p-value calculation (3.8× more time than X!Tandem; averaged 89 CPU seconds) followed by OMSSA (5.3× more than X!Tandem; averaged 123 CPU seconds). The Crux p-value calculation adds considerable time due to the permutation-based approach to assess the statistical significance of the database match. This approach requires the generation and scoring of dummy sequences to obtain the Weibull density for each match. The computation of p-values for 100 and 1000 dummy sequences required over 1 and 13 h of CPU time, respectively. The increase in time is linear on the number of sequences evaluated such that each sequence took approximately 47 CPU seconds. This resampling test approach is not limited to Crux and a comparable increase in time would occur when this approach is used with X!Tandem and OMSSA.
Impact of Ion Series-Dependent Scoring on Peptide Identification
An alternative approach to improve the speed of peptide matches offered by some programs is to search only one ion series. Table 5 summarizes the performance of OMSSA and X!Tandem in terms of number of peptides unmatched, mismatched or correctly matched at various significance levels by ion series scored for charge state +1. Supporting Information Tables 7 and 8 summarize the performance of the database search programs across match significance levels when the b- and y-ion series were scored, respectively for charge states +1, +2 and +3. Supporting Information Table 9 reports the number of peptides identified at E-value < 1 × 10–6 by both or either program.
Table 5. Number of Peptides Unmatched, Mismatched and Correctly Matched at Various Significance Levels by X!Tandem, OMSSA and Crux When Either the b- or the y-Ion Series Is Used to Score the Match for Precursor Charge State +1 and Including Neutral Mass Losses.
| OMSSA |
X!Tandem |
|||
|---|---|---|---|---|
| significancea | bb | y | b | y |
| Unmatchedc | 415 | 365 | 75 | 74 |
| Mismatchedd | 11 | 11 | 10 | 11 |
| 0 | 122 | 47 | 248 | 151 |
| 1 | 63 | 50 | 113 | 108 |
| 2 | 76 | 69 | 187 | 179 |
| 3 | 76 | 66 | 270 | 214 |
| 4 | 103 | 87 | 746 | 591 |
| 5 | 62 | 81 | 1902 | 1785 |
| 6 | 102 | 90 | 1536 | 1948 |
| 7 | 108 | 80 | 842 | 861 |
| 8 | 116 | 126 | 690 | 648 |
| ≥9 | 6596 | 6778 | 1231 | 1280 |
| Prop >6e | 88.2% | 90.1% | 54.8% | 60.3% |
Significance threshold (t) for matched to be considered significant at E- or p-value < 1 × 10–t.
b- or y-ions used to score the peptide match.
Unmatched: the program does not provide a match with the program setting.
Mismatched: the program provided an incorrect match.
Percentage of the matches that have E- or p-value < 1 × 10–6.
Scoring only one of the ion series was noticeably detrimental to peptide detection for both OMSSA and X!Tandem. The y-ion series provided a higher detection rate than the b-ion series at E-value < 1 × 10–6. The number of mismatched and unmatched peptides when one series was scored increase substantially in OMSSA compared to X!Tandem. OMSSA had more false positive and false negative results when scoring only one ion series. The number of correctly matched peptides at E-value > 1 × 10–8 was higher in X!Tandem than in OMSSA and at E-value < 1 × 10–8 was higher in OMSSA than in X!Tandem when either ion series was used for scoring. Overall, the number of correctly matched peptides regardless of E-value was higher in OMSSA than in X!Tandem when either ion series was used for scoring. The major reason for weaker performance of X!Tandem when scoring one ion series was that the peptides had less significant E-values than when scoring both ion series. Scoring using only y-ion series resulted in fewer unmatched peptides (difference of 34), mismatched peptides (difference of 12) and peptides with less significant E-values (98% of peptides with the E-value < 1 × 10–2) in OMSSA. In both programs, higher charge states were associated with slightly poorer peptide detection with scoring based on the y-ion series being less affected than on the b-ion series.
The length of the peptide was also critical when one ion series was used to score the matches between the query and target database mass spectra. The minimum length among the detected peptides was 10 and 13 amino acids in OMSSA and X!Tandem, respectively, compared to six amino acids when both ion series were scored. The median length of the correctly identified peptides with both programs using the b- and y-ion series was 83 and 76 amino acids, respectively. Also, the median length of the missed (false negative) peptides in both programs was seven and six amino acids when the b- and y-ion series were used, respectively. This result reflects the issues of correctly identifying small peptides at E-value < 1 × 10–6.
Impact of Chimera Spectra on Peptide Identification
Chimera spectra is a likely phenomenon in peptidomics measurements; this occurs when multiple peptides coelute in the same LC fraction and each peptide present contributes to the observed peaks in the tandem MS spectra, usually when the peptides are similar to each other. As neuropeptides span many orders of magnitude in concentration dynamic range, chimera spectra can be caused by peptides at vastly different levels. To evaluate the performance of the programs when spectra from multiple coeluted peptides are present, peptides that have similar mass and are likely to coelute were identified. Of the 7649 peptides identified by all programs at E- or p-value < 1 × 10–2, 2049 peptides had at least one other peptide with theoretical mass ±0.4 Da. These peptides were split into 945 groups each including at least two peptides within a theoretical mass range or tolerance within group. Of these, 804, 126, 12, and 3 groups included 2, 3, 4, and 5 peptides, respectively.
Table 6 summarizes the number of peptides identified from chimera spectra with precursor charge state +1, all ions are available and including neutral mass losses by X!Tandem, OMSSA and Crux. Crux had the best performance and X!Tandem generally failed to identify peptides from chimera spectra regardless of the threshold used. At E- or p-value < 1 × 10–2 threshold, OMSSA, X!Tandem and Crux, correctly identified 81%, 43% and 99% of peptides, respectively. Of these, Crux only had three unmatched peptides at p-value <10–2 meanwhile X!Tandem only reported one peptide in the chimera spectra unless the other matches had the same score. OMSSA had a correct match rate similar to Crux at E-value < 1 × 10–6 and lower at E-value < 1 × 10–2 threshold. A further decrease in the accuracy of peptide identification in chimeras was observed in small peptides for X!Tandem and Crux. At E- or p-value < 1 × 10–2 threshold, the correct identifications by X!Tandem and Crux were 39.1% and 98.5% in peptides less than 20 amino acids in length, and 37.6% and 91.8% in peptides less than 10 amino acids in length, respectively. Consistent with our results, Houel et al.25 reported that Mascot correctly identified peptide A in 87% of chimera spectra containing 50% of peptide A and 50% of peptide B.
Table 6. Number of Peptides Identified from Chimera Spectra of Groups of 2–5 Peptides with Precursor Charge State +1, All Ions Are Available, and Including Neutral Mass Losses by X!Tandem, OMSSA and Crux.
| number of peptides correctly matched in a spectra
with an E- or p-value < 1 × 10–2 |
percentage
of peptides detected |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| program | N pepa | 0 | 1 | 2 | 3 | 4 | 5 | >2b | >6c |
| OMSSA | 2 | 11 | 213 | 580 | 85.4 | 84.1 | |||
| 3 | 3 | 25 | 64 | 34 | 67.5 | 61.9 | |||
| 4 | 1 | 3 | 5 | 2 | 1 | 47.9 | 33.3 | ||
| 5 | 0 | 0 | 1 | 0 | 1 | 1 | 73.3 | 66.7 | |
| Total | 15 | 241 | 650 | 36 | 2 | 1 | 81.1 | 78.7 | |
| X!Tandem | 2 | 0 | 799 | 5 | 50.3 | 12.8 | |||
| 3 | 59 | 67 | 0 | 0 | 17.7 | 0.5 | |||
| 4 | 11 | 1 | 0 | 0 | 0 | 2.1 | 0.0 | ||
| 5 | 3 | 0 | 0 | 0 | 0 | 0 | 0.0 | 0.0 | |
| Total | 73 | 867 | 5 | 0 | 0 | 0 | 42.8 | 10.2 | |
| Crux | 2 | 0 | 10 | 794 | 99.4 | 81.3 | |||
| 3 | 0 | 0 | 3 | 123 | 99.2 | 61.6 | |||
| 4 | 0 | 0 | 0 | 0 | 12 | 100.0 | 20.8 | ||
| 5 | 0 | 0 | 0 | 0 | 0 | 3 | 100.0 | 13.3 | |
| Total | 0 | 10 | 797 | 123 | 12 | 3 | 99.4 | 75.8 | |
Number of peptides simulated in a chimera spectra.
Percentage of correctly matched peptides with an E- or p-value < 1 × 10–2.
Percentage of correctly matched peptides with an E- or p-value < 1 × 10–6.
Conclusions
The present study demonstrated that although most neuropeptides and prohormone peptides with ideal MS/MS spectra can be identified using standard database search methods, a careful assessment of the accuracy of the match is still required. Furthermore, the search must be optimized for the specific biological context, here prohormone cleaved neuropeptides.
The results from the present study indicate that the correct identification of peptides based on a single threshold across all spectra is challenging even when provided with ideal spectra and target database. A major component of this challenge was the scoring and assignment of a single significance threshold for all peptides. This problem is exacerbated when analyzing experimental data because the quality of the data and the specifications of the program have a large impact on the accuracy of peptide identification. Crux was the only program able to correctly match all the peptides regardless of p-value. The E- and p-values rapidly become more significant with increasing peptide length before stabilizing at approximately 50 amino acids in Crux and 13 amino acids in OMSSA and X!Tandem. Of the three programs, Crux is better suited to detect short peptides although the p-value calculation in Crux is more stringent. The typically short prohormone peptides have less significant E- or p-values because of the higher number of expected matches by chance. Overall, results indicated the need to optimize the scoring and associated E- or p-value calculation in database searches for neuropeptides and small peptides. A straightforward approach that does not require program modifications is to accept less significant matches for short peptides meanwhile keeping more significant thresholds for longer peptides. The peptide length that was associated with stabilization of E- and p-values (50 amino acids in Crux and 13 for OMSSA and X!Tandem) offer a good start.
Most small peptides were detected by one or two programs (Crux or OMSSA) and thus the program consensus approach advocated for protein identification is not well suited to identify small peptides. Crux had the best performance in the identification of peptides from chimera spectra and when ions were missing, OMSSA provided the most significant E-values; meanwhile, X!Tandem returned results fastest for peptides detected by all programs.
A comprehensive evaluation of the impact of multiple factors on peptide identification was undertaken. Additional simulations can help to assess the importance of other aspects of MS/MS on peptide identification.37 The evaluations performed in this study assumed that the peptides had ideal uniform spectra to avoid additional confounding factors. Different peptide ion fragmentation methods (e.g., CID, HCD, ETD) have different abilities to fragment that affect the performance of the database search tools.38 Additional studies can consider the impact of the fragmentation method and PTMs on the ability to identify neuropeptides. Also, the identification of peptides using spectrum-to-spectrum search tools has been proposed in recent years.39 A study of the performance of spectrum-to-spectrum searches when applied to small prohormone peptide identification needs to be undertaken.
Acknowledgments
The support of NIH/NIDA (Grant Numbers: R21DA027548 and P30DA018310) and COMSATS Institute of Information Technology are greatly appreciated.
Supporting Information Available
Individual performance of X!Tandem, OMSSA and Crux in each simulated scenario are provided in Tables S1–S9. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hook V.; Funkelstein L.; Lu D.; Bark S.; Wegrzyn J.; Hwang S. R. Proteases for processing proneuropeptides into peptide neurotransmitters and hormones. Annu. Rev. Pharmacol. Toxicol. 2008, (48), 393–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson M.; Skold K.; Svenningsson P.; Andren P. E. Peptidomics-based discovery of novel neuropeptides. J. Proteome Res. 2003, 2(2), 213–9. [DOI] [PubMed] [Google Scholar]
- Nilsson A.; Falth M.; Zhang X.; Kultima K.; Skold K.; Svenningsson P.; Andren P. E. Striatal alterations of secretogranin-1, somatostatin, prodynorphin, and cholecystokinin peptides in an experimental mouse model of Parkinson disease. Mol. Cell. Proteomics 2009, 5(8), 1094–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand F. L.Neuropeptides: regulators of physiological processes; MIT Press: Cambridge, MA, 1999; p 658. [Google Scholar]
- Kastin A. J.Handbook of biologically active peptides; Academic Press: Boston, 2006; p 1595. [Google Scholar]
- Hook V.; Bark S.; Gupta N.; Lortie M.; Lu W. D.; Bandeira N.; Funkelstein L.; Wegrzyn J.; O’Connor D. T.; Pevzner P. Neuropeptidomic components generated by proteomic functions in secretory vesicles for cell-cell communication. AAPS J. 2010, 4(12), 635–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonen K.; Creemers J. W.; Schoofs L. Bioactive peptides, networks and systems biology. Bioessays 2009, 3(31), 300–14. [DOI] [PubMed] [Google Scholar]
- Li L.; Sweedler J. V. Peptides in the brain: mass spectrometry-based measurement approaches and challenges. Annu. Rev. Anal. Chem. 2008, (1), 451–83. [DOI] [PubMed] [Google Scholar]
- Fricker L. D.; Lim J.; Pan H.; Che F. Y. Peptidomics: identification and quantification of endogenous peptides in neuroendocrine tissues. Mass Spectrom. Rev. 2006, 2(25), 327–44. [DOI] [PubMed] [Google Scholar]
- Yates J. R. 3rd; Eng J. K.; McCormack A. L.; Schieltz D. Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 1995, 8(67), 1426–36. [DOI] [PubMed] [Google Scholar]
- Craig R.; Beavis R. C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004, 9(20), 1466–7. [DOI] [PubMed] [Google Scholar]
- Park C. Y.; Klammer A. A.; Kall L.; MacCoss M. J.; Noble W. S. Rapid and accurate peptide identification from tandem mass spectra. J. Proteome Res. 2008, 7(7), 3022–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapp E.; Schutz F. Overview of tandem mass spectrometry (MS/MS) database search algorithms. Curr. Protoc. Protein Sci. 2007, 49, 25.2.1–25.2.19. [DOI] [PubMed] [Google Scholar]
- Kapp E. A.; Schutz F.; Connolly L. M.; Chakel J. A.; Meza J. E.; Miller C. A.; Fenyo D.; Eng J. K.; Adkins J. N.; Omenn G. S.; Simpson R. J. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics 2005, 13(5), 3475–90. [DOI] [PubMed] [Google Scholar]
- Xu H.; Freitas M. A. MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics 2009, 6(9), 1548–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balgley B. M.; Laudeman T.; Yang L.; Song T.; Lee C. S. Comparative evaluation of tandem MS search algorithms using a target-decoy search strategy. Mol. Cell. Proteomics 2007, 9(6), 1599–608. [DOI] [PubMed] [Google Scholar]
- Xu H.; Freitas M. A. A dynamic noise level algorithm for spectral screening of peptide MS/MS spectra. BMC Bioinform. 2010, 11, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P.; Hou X.; Romanova E. V.; Sweedler J. V. Neuropeptidomics: mass spectrometry-based qualitative and quantitative analysis. Methods Mol. Biol. 2011, 789, 223–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.; Bark S.; Hook V.; Bandeira N. NeuroPedia: neuropeptide database and spectral library. Bioinformatics 2011, 19(27), 2772–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rholam M.; Fahy C. Processing of peptide and hormone precursors at the dibasic cleavage sites. Cell. Mol. Life Sci. 2009, 13(66), 2075–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southey B. R.; Sweedler J. V.; Rodriguez-Zas S. L. Prediction of neuropeptide cleavage sites in insects. Bioinformatics 2008, 6(24), 815–25. [DOI] [PubMed] [Google Scholar]
- Arolas J. L.; Vendrell J.; Aviles F. X.; Fricker L. D. Metallocarboxypeptidases: emerging drug targets in biomedicine. Curr. Pharm. Des. 2007, 4(13), 349–66. [DOI] [PubMed] [Google Scholar]
- Lee J. E.; Atkins N. Jr; Hatcher N. G.; Zamdborg L.; Gillette M. U.; Sweedler J. V.; Kelleher N. L. Endogenous peptide discovery of the rat circadian clock: a focused study of the suprachiasmatic nucleus by ultrahigh performance tandem mass spectrometry. Mol. Cell. Proteomics 2010, 2(9), 285–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopmann M. R.; Finney G. L.; MacCoss M. J. High-speed data reduction, feature detection, and MS/MS spectrum quality assessment of shotgun proteomics data sets using high-resolution mass spectrometry. Anal. Chem. 2007, 15(79), 5620–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houel S.; Abernathy R.; Renganathan K.; Meyer-Arendt K.; Ahn N. G.; Old W. M. Quantifying the impact of chimera MS/MS spectra on peptide identification in large-scale proteomics studies. J. Proteome Res. 2010, 8(9), 4152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southey B. R.; Amare A.; Zimmerman T. A.; Rodriguez-Zas S. L.; Sweedler J. V. NeuroPred: a tool to predict cleavage sites in neuropeptide precursors and provide the masses of the resulting peptides. Nucleic Acids Res. 2006, (34), W267–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falth M.; Skold K.; Norrman M.; Svensson M.; Fenyo D.; Andren P. E. SwePep, a database designed for endogenous peptides and mass spectrometry. Mol. Cell. Proteomics 2006, 6(5), 998–1005. [DOI] [PubMed] [Google Scholar]
- Nucleic Acids Res. 2012, 40, D71–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southey B. R.; Rodriguez-Zas S. L.; Sweedler J. V. Characterization of the prohormone complement in cattle using genomic libraries and cleavage prediction approaches. BMC Genomics 2009, 10, 228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southey B. R.; Sweedler J. V.; Rodriguez-Zas S. L. A python analytical pipeline to identify prohormone precursors and predict prohormone cleavage sites. Front. Neuroinform. 2008, 2, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegge A. N.; Southey B. R.; Sweedler J. V.; Rodriguez-Zas S. L. Comparative analysis of neuropeptide cleavage sites in human, mouse, rat, and cattle. Mamm. Genome 2008, 2(19), 106–20. [DOI] [PubMed] [Google Scholar]
- Geer L. Y.; Markey S. P.; Kowalak J. A.; Wagner L.; Xu M.; Maynard D. M.; Yang X.; Shi W.; Bryant S. H. Open mass spectrometry search algorithm. J. Proteome Res. 2004, 5(3), 958–64. [DOI] [PubMed] [Google Scholar]
- Yadav A. K.; Kumar D.; Dash D. MassWiz: a novel scoring algorithm with target-decoy based analysis pipeline for tandem mass spectrometry. J. Proteome Res. 2011, 5(10), 2154–60. [DOI] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 12(26), 1367–72. [DOI] [PubMed] [Google Scholar]
- Hummon A. B.; Amare A.; Sweedler J. V. Discovering new invertebrate neuropeptides using mass spectrometry. Mass Spectrom. Rev. 2006, 1(25), 77–98. [DOI] [PubMed] [Google Scholar]
- Frank A. M.; Savitski M. M.; Nielsen M. L.; Zubarev R. A.; Pevzner P. A. De novo peptide sequencing and identification with precision mass spectrometry. J. Proteome Res. 2007, 1(6), 114–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielow C.; Aiche S.; Andreotti S.; Reinert K. MSSimulator: Simulation of mass spectrometry data. J. Proteome Res. 2011, 7(10), 2922–9. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Tolic N.; Xie F.; Zhao R.; Purvine S. O.; Schepmoes A. A.; Moore R. J.; Anderson G. A.; Smith R. D. Effectiveness of CID, HCD, and ETD with FT MS/MS for degradomic-peptidomic analysis: comparison of peptide identification methods. J. Proteome Res. 2011, 9(10), 3929–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen C. Y.; Houel S.; Ahn N. G.; Old W. M. Spectrum-to-spectrum searching using a proteome-wide spectral library. Mol. Cell. Proteomics 2011, 7(10), M111.007666. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.