

t-Butyl cation[1] is an iconic intermediate of organic chemistry and hyperconjugation is the textbook explanation for its stability. Positive charge is delocalized via donation of electron density from aligned C-H bonds into the formally empty 2pz orbital on cationic C, giving partial double bond character to the C-C bonds, increasing the δ+ charge on the H atoms and lengthening their C-H bonds (Scheme 1).
Scheme 1.
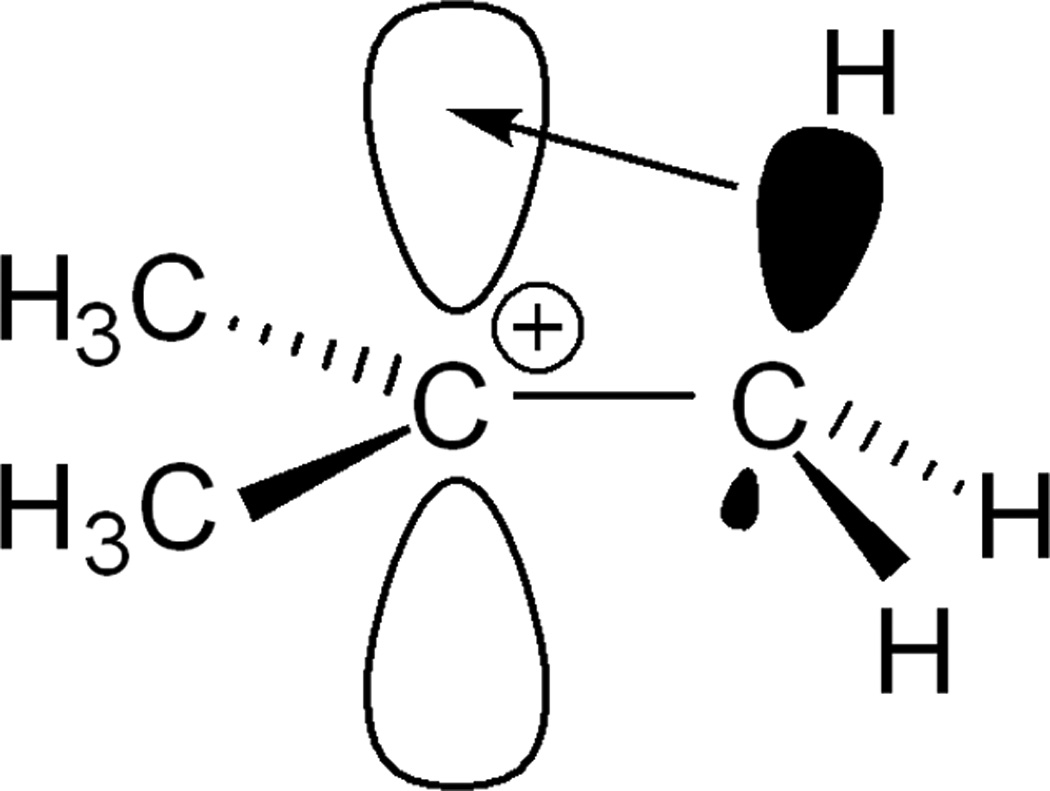
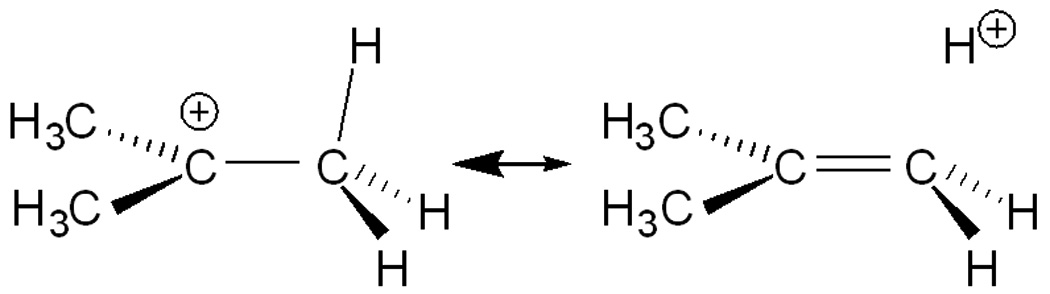
Two representations of hyperconjugative delocalization of positive charge in t-butyl cation. In (a) black represents a filled orbital.
The importance of hyperconjugation in isolated (i.e. gas phase) t-butyl cation has been thoroughly established by theory[2] and is substantiated by the outstanding agreement of the experimental gas phase IR spectrum of argon-tagged t-Bu+ obtained by photodissociation with that calculated for the Cs symmetry structure.[3]
Hyperconjugation was invoked by Olah in 1964 to explain the unusually low IR frequency of the C-H stretch (νmax 2830 cm−1) of t-Bu+ in an SbF5 superacid matrix.[4] Near coincidence with νmax in the gas phase (2834 cm−1)[3] has left the impression that the same explanation solely rationalizes the stability of t-Bu+ in condensed phases. We now show that this convergence of gas and condensed phase spectral data is fortuitous. The IR spectrum of t-Bu+ in the solid state requires explanation not only in terms of hyperconjugation but also of H-bonding, an idea first put forward by Laube[5] in describing close contacts between methyl groups and the Sb2F11− counterion in the X-ray structure of a t-butyl cation salt.
t-Butyl cation is remarkably stable as the salt of an inert carborane anion.[6] Such salts are readily isolated either by abstraction of hydride from butane with methyl carborane reagents[7] or by heating diethylchloronium ion salts to 150 °C.[8] Figure 1 shows a representative solid state IR spectrum of t-Bu+ as the CHB11Cl11− salt with νCHmax at 2792 cm−1. Comparison to the spectrum of the d9-deuterated cation (dashed line) shows an H/D ratio of 1.366, confirming the essentially pure C-H stretching mode of this band.
Figure 1.

Solid state IR spectrum of [t-Bu+][CHB11Cl11−] (solid line) and its cation-deuterated analog (dashed).
Sensitivity of t-Bu+ to its environment distinguishes intermolecular H-bonding stabilization from intramolecular hyperconjugative stabilization. The νCHmax frequency of the CHB11Cl11− carborane salt is already 42 cm−1 lower than that in SbF5 superacid media and it moves even lower (as much as 88 cm−1) as the carborane anion becomes more basic. In Figure 2, νCHmax for t-Bu+ salts is plotted versus anion basicity on the νNH scale.[9] A linear correlation is observed. Because the νNH scale is based on an IR measure of H-bonding ability, so too must νCH.
Figure 2.
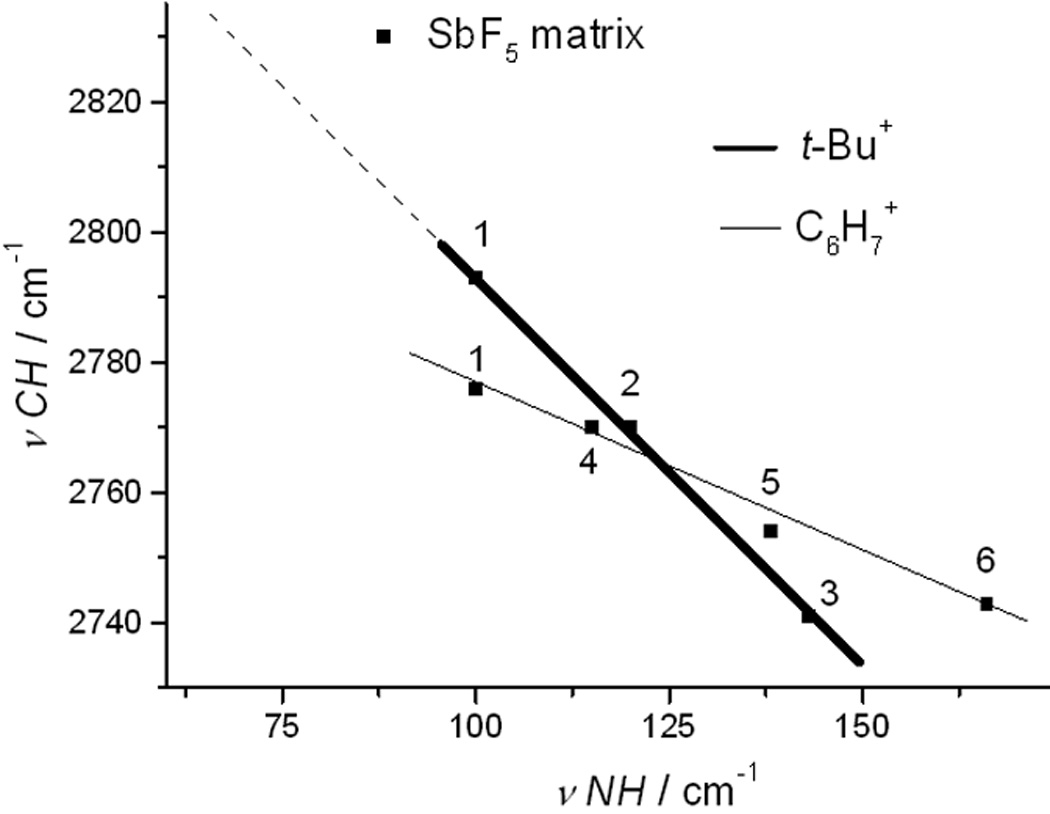
Correlation of νCH with anion basicity on the νNH scale for t-Bu+ (bold line) and C6H7+ (thin line) cations with CHB11Cl11− (1), CHB11Me5Cl6− (2), CHB11Me5Br6− (3), CHB11H5Cl6− (4), CHB11H5Br6− (5) and CHB11H5I6− (6) counterions. Point refers to SbF6−.
The νCHmax value for t-butyl cation in SbF5 superacid media[4] cannot be added to this graph with certainty because the exact nature of the counterion (SbF6−, Sb2F11−, Sb3F16− etc.) is unknown. Only the νNH value for SbF6− is known (88 cm−1)[9] and the corresponding point for this counterion is placed on Figure 2 (•). Since higher oligomeric [SbF6·nSbF5]− anions will have νNH values somewhat lower than SbF6−, we estimate that the correct datum point will lie quite close to the linear correlation line (arrow in Figure 2).
Also plotted in Figure 2 are νCH data for another iconic intermediate of organic chemistry, the C6H7+ arenium ion. Stable salts of the benzenium ion are readily prepared by protonation of benzene with carborane acids[10] and [C6H7][CHB11Me5Br6] has been characterized by X-ray crystallography.[11] Low frequency νCH bands were noted in its IR spectrum and ascribed to ion pair C–H∙∙∙∙Br interactions at the protonated C atom. The linearity of the plot of νCH versus anion basicity on the νNH scale further validates the importance of H-bonding in carbocations. The lesser slope observed for the benzenium ion relative to t-butyl cation indicates weaker H-bonding by C6H7+. This is readily understood in terms of efficient π-delocalization of positive charge in the larger ion.
Not only is the frequency of νCHmax for a t-Bu+ salt unusually low but the band is unusually broad compared to related cations such as the dimethylchloronium ion[8] or protonated methanol.[12] This is an additional indicator of H-bonding and can be understood in terms of the variety of specific C–H∙∙∙∙Cl interactions seen in the X-ray crystal structure of [t-C4H9][CHB11Cl11].[13] Previous X-ray structures of t-butyl cation[5,7] had lower resolution whereas in the present structure the H atoms were located with reasonable certainty. As illustrated for a representative cation/anion interaction in Figure 3, most of the interactions of the cation C–H groups with the anion Cl atoms involve asymmetric bifurcated H-bonding. All nine C–H groups have at least one H∙∙∙∙Cl distance in the range 2.91(2)−3.29(2) Å, consistent with some cation/anion interaction. Two involve normal (monofurcated) H-bonds with C–H∙∙∙∙Cl angles near 163° and relatively short H∙∙∙∙Cl distances in the range 2.91–2.95 Å. All nine C–H bonds in the cation are crystallographically independent so each adds to the broad composition of the νCH band.
Figure 3.
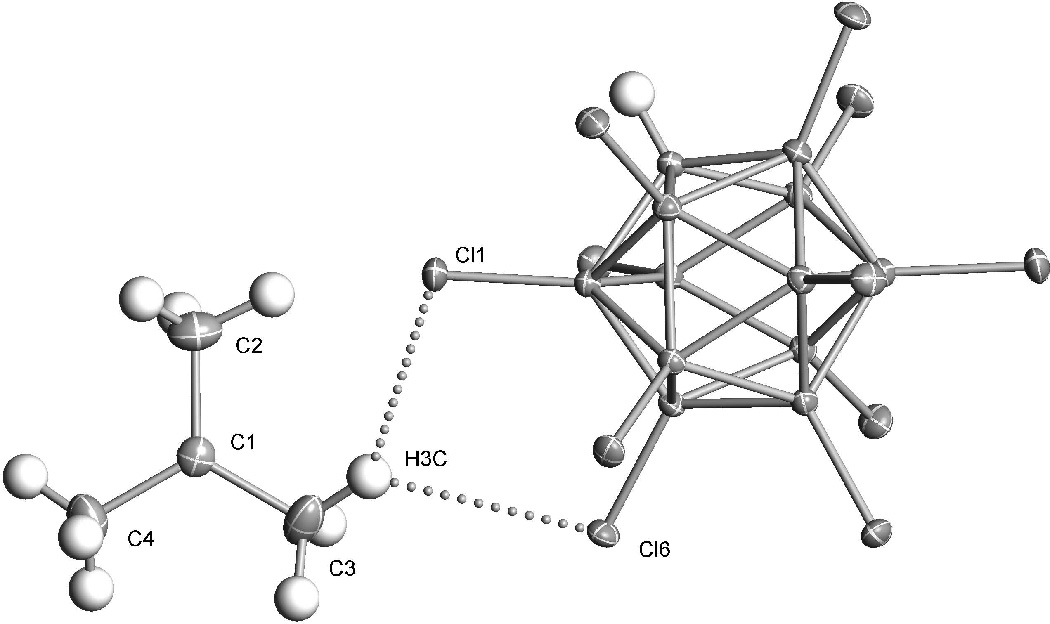
X-ray structure of [t-C4H9][CHB11Cl11] showing arepresentative C-H∙∙∙∙Cl asymmetric bifurcated H-bond. Selected bond distances: C1-C2 = 1.452(1), C1-C3 = 1.443(1), C1-C4 = 1.451(1), C3-H3C = 1.01(2), H3C-Cl1 = 2.98(2), H3C-Cl6 =3.23(2) Å. Angles: C2-C1-C3 = 120.7(1)°, C2-C1-C4 = 119.4(1)°, C3-C1-C4 = 119.9(1)°, C3-H3C-Cl1 = 146(2)°, C3-H3C-Cl6 = 137(2)°.
As discussed for earlier X-ray structures,[5,7] the average 0.06 Å shortening of C–C bond lengths compared to normal Csp2−Csp3 bonds is widely taken as evidence for hyperconjugation. Because of the higher resolution of the present structure, further evidence for hyperconjugation can be found in the angles associated with the H atom locations.[14] Perhaps remarkably, given its flat potential energy surface,[2] the planar view in Figure 3 reveals that the solid state structure of the cation sustains only minor methyl group rotational distortions from idealized gas-phase Cs symmetry. Three C–H bonds are within 5−10° of optimal alignment with the vacant pz orbital on the central C atom, consistent with near optimal hyperconjugative stabilization. As illustrated in Scheme 2 for a representative methyl group (C4), the C-C-H angle of the aligned C–H bond (101°) is ca. 10° more acute than the non-aligned ones and is very close to the calculated gas-phase value (102°).[2a]
Scheme 2.
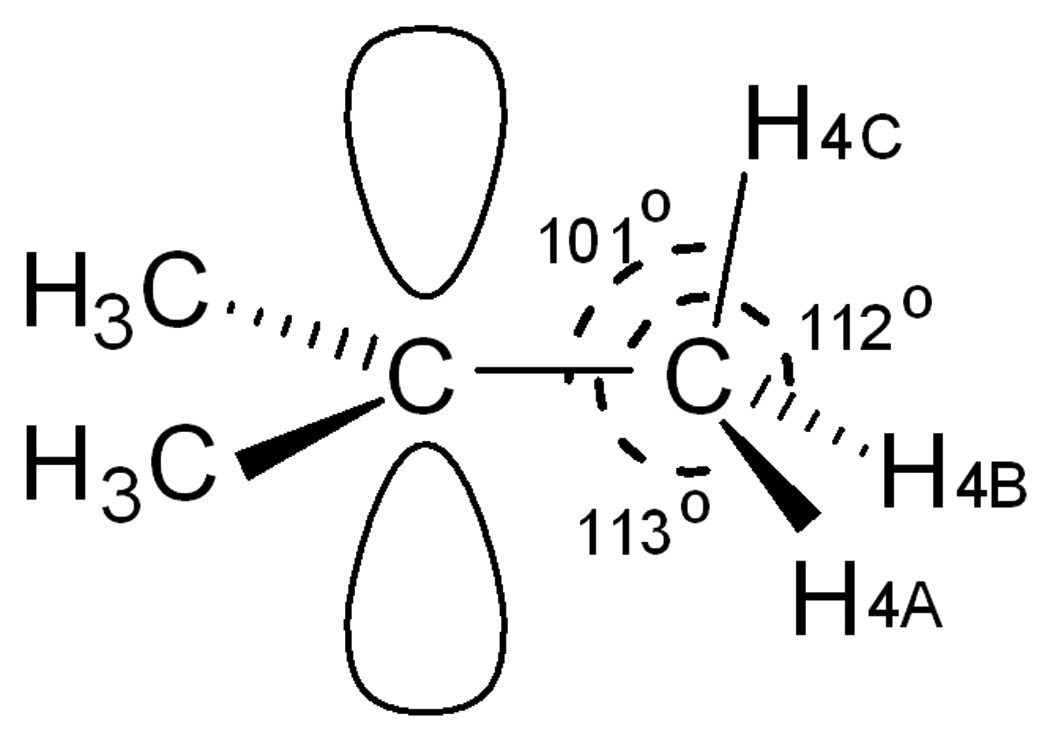
Bond angle evidence in the C4 methyl group for C-H(4C) bond hyperconjugation.
So both hyperconjugation and hydrogen bonding are present in solid state t-Bu+ salts. H-bonding interactions presumably also apply to the liquid phase where anions and solvent molecules can act as H-bond acceptors. Only in the gas phase is hyperconjugation necessarily the sole mechanism for dispersal of positive charge. Since nearly all practical hydrocarbon chemistry is done in condensed phases, the reality of H-bonding possibilities in carbocations deserves wider appreciation. For example, theory indicates that non-classical carbocations are competent C-H bond donors to bases relevant to enzyme-catalyzed terpenoid synthesis.[15]
Traditionally, H-bonding has been the province of hydrogen atoms bonded to elements more electronegative than carbon, typically O and N. However, in the present age of weak interactions, H-bonding involving intrinsically less polar bonds such as C-H and heavier acceptor atoms such as Cl has become more widely discussed.[16] In particular, C–H hydrogen bonding is favoured as C–H bonds become acidic (e.g. in alkynes)[17] and when Cl is negatively charged.[18] It is evident in the present work that the hyperconjugatively delocalized positive charge on t-butyl cation significantly enhances the ability of C-H bonds to engage in hydrogen bonding. In future work we will attempt to partition hyperconjugation from hydrogen bonding effects and explore the extent of H-bonding in other carbocations. Preliminary indications are that in alkyl cations, the phenomenon extends only to CH groups that are α to the site of formal positive charge.
Experimental Section
Synthesis
Reactions and sample handling were carried out in Vacuum Atmospheres dry boxes (O2, H2O < 1 ppm). The t-Bu+ and d9- t-Bu+ salts of CHB11Cl11− were obtained via thermal decomposition of corresponding protio- and deuterio-diethylchloronium salts at 150° C, as previously described.[8] The t-Bu+ salts of CHB11Me5Cl6− and CHB11Me5Br6− anions were prepared as previously described in Supporting Information.[7] Benzenium ion salts with CHB11Cl11−, CHB11H5Cl6−, CHB11H5Br6− and CHB11H5I6− anions were prepared by wetting the corresponding carborane acids with liquid benzene and evaporating the excess benzene.[11]
IR Spectroscopy
IR spectra were recorded on a Perkin Elmer Spectrum-100 spectrometer inside a dry box in transmission or ATR mode (4000-400 cm−1) and the data manipulated using GRAMS/AI (7.00) software from Thermo Scientific.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (CHE-1144838) and the National Institutes of Health (GM 23851).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Olah GA, Tolgyesi WS, Kuhn SJ, Moffatt ME, Bastien IJ, Baker EB. J. Am. Chem. Soc. 1963;85:1328–1334. [Google Scholar]
- 2.a) Feng H, Sun W, Xie Y, Schaefer HF., III Chem. Eur. J. 2010;17:10552–10555. doi: 10.1002/chem.201100797. [DOI] [PubMed] [Google Scholar]; b) Rasul G, Chen JL, Prakash GKS, Olah GA. J. Phys. Chem. A. 2009;113:6795–6799. doi: 10.1021/jp903002z. and references therein. [DOI] [PubMed] [Google Scholar]
- 3.Douberly GE, Ricks AM, Ticknor BW, Schleyer PvR, Duncan MA. J. Am. Chem. Soc. 2007;129:13782–13783. doi: 10.1021/ja0753593. [DOI] [PubMed] [Google Scholar]
- 4.Olah GA, Baker EB, Evans JC, Tolgyesi WS, McIntyre JS, Bastien IJ. J. Am. Chem. Soc. 1964;86:1360–1373. [Google Scholar]
- 5.Hollenstein S, Laube T. J. Am. Chem. Soc. 1993;115:7240–7245. [Google Scholar]
- 6.Reed CA. Acc. Chem. Res. 2010;43:121–128. doi: 10.1021/ar900159e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato T, Reed CA. Angew. Chem. Int. Ed. 2004;43:2908–2911. doi: 10.1002/anie.200453931. [DOI] [PubMed] [Google Scholar]
- 8.Stoyanov ES, Stoyanova IV, Tham FS, Reed CA. J. Am. Chem. Soc. 2010;132:4062–4063. doi: 10.1021/ja100297b. [DOI] [PubMed] [Google Scholar]
- 9.Stoyanov ES, Kim K-C, Reed CA. J. Am. Chem. Soc. 2006;128:8500–8508. doi: 10.1021/ja060714v. [DOI] [PubMed] [Google Scholar]
- 10.Reed CA. Chem. Commun. 2005:1669–1677. doi: 10.1039/b415425h. [DOI] [PubMed] [Google Scholar]
- 11.Reed CA, Kim K-C, Stoyanov ES, Stasko D, Tham FS, Mueller LJ, Boyd PDW. J. Am. Chem. Soc. 2003;125:1796–1804. doi: 10.1021/ja027336o. [DOI] [PubMed] [Google Scholar]
- 12.Stoyanov ES, Stoyanova IV, Reed CA. Chem. A Europ. J. 2008;14:3596–3604. doi: 10.1002/chem.200701746. [DOI] [PubMed] [Google Scholar]
- 13.CCDC 874494. See Supporting Information for details.
- 14.C-C-H bond angles are better defined than C-H bond distances.
- 15.Mojin MD, Tantillo DJ. J. Phys. Chem. A. 2006;110:4810–4816. doi: 10.1021/jp056965s. [DOI] [PubMed] [Google Scholar]
- 16.Steiner T. Angew. Chem. Int. Ed. 2002;41:48–76. [Google Scholar]
- 17.Desiraju GR. J. Chem. Soc. Chem. Commun. 1990:454–455. [Google Scholar]
- 18.Taylor R, Kennard O. J. Am. Chem. Soc. 1982;104:5063–5070. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.