

Abstract
Ischemic stroke represents a leading cause of morbidity and mortality in the developed world. This disabling and sometimes fatal event puts an ever increasing burden on the family members and medical professionals who care for stroke victims. Preclinical ischemic stroke research has predominantly utilized young adult, healthy animals, a clear discrepancy when considering the clinical population affected by stroke. A broad spectrum of risk factors such as age, obesity, diabetes, and hypertension has been associated with an increased stroke risk. The effect of these comorbidities on both stroke pathophysiology and outcome has not been emphasized and has been recognized as a shortcoming of preclinical studies. By addressing these conditions in experimental models of ischemic stroke, it may be possible to more accurately represent the clinical scenario and improve therapeutic translation from bench-to-bedside. In this work, we review many of the risk factors associated with increased stroke risk, particularly as each risk factor relates to inflammation. Additionally, we explore potential animal models that could be utilized in identifying the contribution of these risk factors to stroke outcome. By investigating the risk factors for stroke and how these may alter stroke pathophysiology, the present discrepancies between preclinical studies and the clinical reality can be reconciled in an effort to improve therapeutic development and translation from bench-to-bedside.
Keywords: age, animal models, diabetes, hypertension, inflammation, ischemic stroke, metabolic syndrome, obesity
Introduction
Stroke is the second largest contributor to mortality worldwide and the primary cause of disability among the elderly in Western Europe and the United States [1,2]. Among the various types of stroke, ischemic stroke is the most prominent and accounts for the most long-term disability [3]. This review will deal only with preclinical models of ischemic stroke. It is widely accepted that age is the greatest risk factor for stroke. The process of aging results in a large number of inflammatory changes. It is predicted that the number of people in the United States past the age of 65 will double within the next 30 years [4]. Thus, the need for new treatments and therapeutic options for stroke is a pressing concern as the population ages. Current available therapeutics are limited to thrombolytics such as recombinant tissue plasminogen activator (r-tPA) and mechanical means of thrombolysis [5]. Although r-tPA drastically improves patient outcome when used within the suggested time period, only a small percentage of presenting patients are candidates to receive r-tPA due to an extensive list of contraindications. As such, a large medical need remains unmet for the vast majority of patients afflicted with ischemic stroke.
As more than 100 potential therapeutic agents have progressed from pre-clinical studies with young animals to unsuccessful clinical trials, the need for improved preclinical models is clear [7][7]. These failures call into question the validity of the models being used to represent ischemic stroke. An area of specific concern is how the age and health of the animals used in these experiments replicates disease pathogenesis and pathophysiology observed in the clinic. Previously, stroke work was done principally with young adult, healthy animals despite the fact that the people most susceptible to stroke are older and are often overweight, diabetic, and hypertensive [8]. Recent recommendations from the Stroke Therapy Academic and Industry Roundtable (STAIR) encourage research to be performed with healthy, young adult, male animals first and then proceed to aged, diabetic, hypertensive, and female animals [9]. Currently, these recommendations have not been broadly implemented. In the following sections, physiologic changes associated with aging, obesity, diabetes, and hypertension will be examined for their roles in predisposition to stroke. It will be argued that more accurate models are necessary to improve the applicability of future research endeavors and to adhere to the suggestions made by STAIR. We identify potential models allowing for investigation of ischemic stroke in relation to a given comorbid condition and, in particular, identify pathophysiologic changes associated with each respective disease state that may represent therapeutic targets. By better understanding pathologic alterations induced by common comorbid conditions as related to ischemic stroke, it is our hope that therapeutics may more successfully move from preclinical studies to accepted treatments clinically.
We specifically focus on molecules associated with inflammatory processes and cascades, as not only does inflammation represent a potentially promising therapeutic target but is also heightened or altered in aging and various comorbid conditions associated with ischemic stroke. Pharmacologic agents targeting inflammation have been developed and, in the case of minocycline, appear promising for the treatment of ischemic stroke. Inflammation involves interplay between cellular and molecular components that results in brain tissue loss. Leukocytes, microglia, astrocytes, and neurons are but a few cell types involved in inflammation related to stroke. On the molecular side, a broad range of chemokines, cytokines, and adhesion molecules play a role in infarct development and subsequent deficits. The activity of these components can be altered by the presence of various disease states, aging, and lifestyle factors [6]. These factors have not been emphasized in most preclinical models of ischemic stroke and may represent a potential reason for the lack of successful therapeutic translation from bench-to-bedside. This review addresses the importance of considering other comorbid processes and how they relate to stroke.
Aging and Stroke
Age: The Greatest Risk Factor
Age remains the greatest risk factor for stroke, yet aging is rarely considered in preclinical models of ischemic stroke. Despite evidence demonstrating that ischemic changes in young adult animals do not replicate the pathophysiology of the aged brain, young animals are still commonly utilized [10,11]. Previous work has shown that aging leads to worsened outcome following neurologic injuries such as ischemic stroke. Identifying changes in the normal aged brain compared to the young adult brain may lead to improved therapeutics for ischemic stroke [12,13]. Changes associated with the aged brain have been ascribed to the following theories: genetic-based aging, cellular aging, neuroendocrine aging, immunologic aging, free-radical aging, and the network theory. Much of the work leading to the development of these theories has been completed in both humans and animal models alike, indicating the potential suitability of aged preclinical models [14-17]. In the following sections, we attempt to briefly discuss implications of aging, how aging changes the response to an injurious event, and the benefits and limitations of aged animal models.
The Aged Brain: Fundamentally Different
Aging has been well documented to be associated with an overall dysregulation of the immune system hallmarked by a shift toward a pro-inflammatory condition [17-20]. Age causes an increased secretion of tissue necrosis factor-alpha (TNF-α), a pro-inflammatory cytokine, and decreased interleukin-10 (IL-10), an anti-inflammatory cytokine [19]. These cytokines act on specific cells in the central nervous system. Numerous cell types throughout the body can release cytokines, and these cells can change with age. The concept of “inflamm-aging” (inflammation increasing with age) has been attributed to chronic macrophage stimulation [18]. Despite the mechanistic details being unclear, aging clearly results in inflammatory variations that are especially obvious in the brain. Some of these variations are due to genetics, lifestyle, and the environment as seen in Figure 1. These alterations in inflammation are common to age-related diseases and serve to prime the central nervous system. This often leads to an exaggerated response and worsened outcome following a systemic challenge [17,19]. Consistent with the link between age-related diseases and inflammatory changes are recent findings in humans and animals alike in which inflammation has been linked to cognitive impairment and altered brain structure and metabolism [21]. In fact, high levels of interleukin-6 (IL-6), a pro-inflammatory cytokine, have been described as exhibiting a strong correlation with morbidity and mortality in the aged population. Some discrepancy does exist as even “healthy” centenarians possess a highly elevated IL-6 level [18]. Cell-specific changes in the CNS mediate how the brain responds to and alters the immune system following injury. These cell-specific changes and the brain’s global response to injury will be discussed below.
Figure 1.
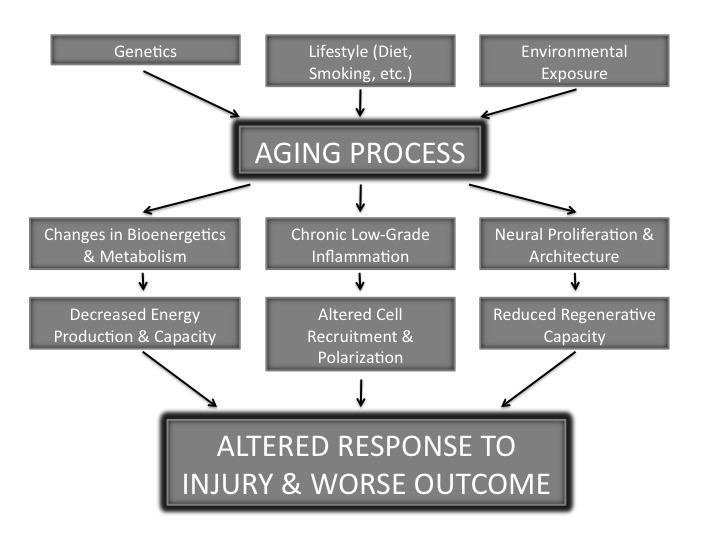
Aging and Stroke. A schematic diagram of the factors contributing to the aging process and how aging alters both the function and structure of the brain. These changes contribute to an altered and generally more deleterious response to injury when compared to the young adult brain.
Age and Cell-Specific Changes
Astrocytes
With increased awareness of the role of glia in neurologic injury and the neurovascular unit, modulation of astrocytes and their associated functions have been identified as a potential target for ischemic stroke. Astrocytes are implicated in many functions associated with injury pathophysiology such as buffering of potassium ions and maintaining the integrity of the blood-brain barrier (BBB) [22]. Despite increased understanding of astrocyte functions pre- and post-injury, the effect of age on astrocyte function is not entirely clear. Inflammatory cytokines do, however, alter astrocyte ability to respond to injury. Recent studies have shown that astrocytes do express markers consistent with senescence in both aged humans and rodents. These markers include increases in glial fibrillary acidic protein (GFAP), cytokine release, and protein aggregates thought to induce cellular death [23]. Therefore, it is evident that astrocyte function changes with aging and may influence both normal homeostatic mechanisms as well as the response to injury. It will be important for future work to elucidate how age-related inflammatory cytokines modify astrocytes.
Endothelial Cells: A Source of Inflammatory Regulators
Since stroke is a vascular disease, it is important to understand the effect of aging on the vasculature system. One of the primary components of the vasculature system is endothelial cells. Studies in aged rats that were designed to correspond to a 70- to 75-year-old human have revealed a marked increase in pro-inflammatory cytokine expression. This increase in cytokines from endothelial cells may be responsible for altering vascular function and increasing permeability in the CNS. Key cytokines identified include TNF-α, interleukin-1β (IL-1β), IL-6, interleukin-17 (IL-17), and interleukin-6Rα (IL-6Rα). Aging was also associated with decreased expression of endothelial nitric oxide synthase (eNOS) and increased production of inducible nitric oxide synthase (iNOS), factors that clearly alter vascular function [24].
Microglial Cells
Microglia, the resident immune cells of the central nervous system (CNS), undergo significant functional changes with aging ranging from altered iron storage to cytokine production and accumulation of lipofuscin [25]. These age-related altercations are exemplified by morphologic changes and are likely involved in the transition of microglia from a neuroprotective phenotype in the young brain to a neurotoxic and destructive form in the aged brain. The destructive form of microglia is known to secrete increased amounts of IL-6 and TNF-α in the aged brain [26,27]. Interestingly, microglial senescence occurring during the aging process may mediate the transition from protective to deleterious effects. This transition is consistent with findings of senescent microglia in close proximity to degenerating neural cells [20,26]. Furthermore, recent work by Baker and colleagues demonstrated that increasing removal of senescent cells results in delayed acquisition of age-related disorders. Microglial senescence may perhaps be a promising therapeutic target for many neurodegenerative conditions [12]. Microglia in aged subjects possess altered surface markers such as major histocompatibility complex II (MHCII) and ED1. How exactly age-associated changes are induced in microglia remains to be elucidated, but it is clear that microglia in the aged brain are basally activated and respond differently to stimuli such as lipopolysaccharide (LPS) and Nogo B [20,28-30].
Neuron-Microglia Interaction
One method of regulating microglial activation in the healthy, young adult brain is through neuron-microglia communication. Disruptions in this signaling mechanism may explain the age-associated shift of microglia from a protective to a destructive phenotype [35]. Neurons can communicate and regulate microglia in multiple ways with ligand-receptor binding via the CD200-CD200R and CX3CL1-CX3CR1 pathways. CD 200, when bound to its receptor (CD200R), is commonly expressed on cells of the myeloid lineage such as microglia. This binding results in microglia being maintained in the quiescent state. As previous works have demonstrated, targeting CD200-CD200R interaction represents a potential therapeutic target to modulate microglial activation with age. In fact, Cox and colleagues have demonstrated that administration of CD200 fusion protein (CD200fc) restores microglia from a quiescent state and results in improved long-term potentiation (LTP) in aged animals [36]. Fractalkine (CX3CL1), a protein expressed by neurons, has been identified as playing a role in neuroimmune modulation by binding to the corresponding receptor, CX3CR1, on microglia. Consistent with the shift to a pro-inflammatory state seen in the aged brain, fractalkine levels are reduced in the aged rat hippocampus as early as 12 months of age. By correcting this fractalkine deficiency exogenously, hippocampal progenitor cell proliferation and neurogenesis is largely restored. This further illustrates the ability to modulate inflammation for neurologic benefit [37].
Global Changes in Neural Proliferation and Architecture
The aged brain exhibits decreased neurogenesis in the subventricular zone (SVZ) and subgranular layer of the hippocampus in comparison to young adult animals. Proteins such as ubiquitin and GFAP are altered in the aged brain and may play a role in age-associated effects [31,32]. It is currently unknown whether diminished regenerative capabilities are the product of stem cell impairment or, rather, changes in the surrounding environment due to age [15]. Besides changes in neurogenesis, the structure of neuron spines is altered as well. The aged brain is characterized by a loss up to nearly 50 percent of thin spines. No change in mushroom or stubby spine quantity has been noted [33]. Experiments assessing changes in the hypothalamus have gone as far as implicating the G protein-coupled receptor and cytoskeletal-associated protein, GIT2, as a critical regulator of the aging process [34].
Responding to Injury: Role of Brain Aging
Age not only affects risk, but also has a profound impact on recovery [15]. Following injury, aged animals exhibit a rapid development of the glial scar and an increased release of associated signaling molecules. The rapid progression demonstrates a dysregulated cellular and inflammatory response [13]. This altered response is likely a product of physiologic differences pre-stroke and also an associated increase in oxidative stress. The inflammation predisposes to a more deleterious response after injury [37-39]. The contribution of each of these factors and the role aging plays in the pathophysiologic differences is expanded upon below.
Disruption of the Blood-Brain Barrier
Aging has been associated with diminished BBB integrity following ischemic stroke [40,41]. The precise mechanism leading to this altered permeability is unclear, but an increase in matrix metalloproteinase-9 (MMP-9) has been associated with increased permeability. Similarly, claudin-5, a protein integral in BBB structure, is decreased in the aged brain following injury [41]. A disrupted BBB allows an influx of inflammatory cells that are responsible for most of the damage seen following stroke. Modulating BBB integrity therefore represents a promising therapeutic target because BBB disruption precedes neuronal damage and often correlates with the extent of injury [40].
Cell Survival and Degradation
The altered response to injury exhibited by the aged brain results in diminished cell survival and the potential for changes in other key cellular processes such as autophagy. Work in various neural injury models utilizing aged rodents has demonstrated an increase in apoptotic cell death attributed to inflammatory processes and heightened oxidative stress [42,43]. Besides apoptosis, cellular autophagy represents another pathway for degradation. The pathway remains under investigation with respect to aging and deleterious injuries. It might represent yet another potential therapeutic target [44].
Inflammation
Following injury, inflammatory responses differ between young adult and aged subjects. In ischemic stroke, suppressor of cytokine signaling 3 (SOCS3) is elevated in aged animals in the subacute period. It mitigates the effects of an elevated phosphorylated signal transducer and activator of transcription 3 (pSTAT3) [45]. This is consistent with other reports in aged animals. On the other hand, microglia are activated more quickly following ischemic stroke and contribute to the release of deleterious cytokines and reactive oxygen species [13]. Other studies of inflammatory diseases utilized young and aged animals to investigate microglial phenotype and demonstrated age-dependent differences. Specifically, microglia expressed a pro-inflammatory phenotype characterized by ED1 and IL-1β in older rats but not young adult rats in an adjuvant arthritis model [46]. IL-1β is involved in cell proliferation, differentiation, and apoptosis.
Oxidative Stress
Oxidative stress has long been recognized as a potential contribution to the aging process and associated pathologies, but the precise origin of oxidative stress has not been entirely clear. In addition to the changes in metabolism and energy production discussed previously, recent studies demonstrate an association between aging and NADPH oxidase (NOX). NOX is a key enzyme in producing reactive oxygen species. NOXs and their associated subunits appear to vary with the aging process. Similarly, lifestyle choices such as diet may influence the production of reactive oxygen species in the aged population [47]. A proposed neuroprotective agent apocynin, a NOX2 inhibitor, demonstrates contrasting effects in young adult and aged animals. While protective in young adult animals, apocynin worsened outcome from ischemic stroke in aged animals. This further proves the importance of utilizing aged animals in preclinical studies [48].
Preclinical Models of Aging
Assessing the role of aging in preclinical models of ischemic stroke has been accomplished using both in vitro and in vivo models. While neither model system has resulted in the successful translation of therapeutics from bench-to-bedside, preclinical studies using aged rats have demonstrated findings in opposition to similar studies in young adult animals. Aging is one of many components that may lead to improved models.
The primary in vivo models for studying the effects of the aging process on ischemic stroke employ aged rats. These studies generally report the use of animals as disparate as 12 to 28 months old. However, female rats typically undergo reproductive senescence around 9 months of age. Therefore, 9 months of age can been equated to a period similar to the late 40s or early 50s in humans. Studying animals around 18 months of age may equate to a similar period in humans of 75+ years old. In humans, this is the period of greatest stroke risk. Utilizing aged animals, 18 to 20 months of age, typically requires the establishment of an aging colony either by the investigator or the animal supplier.
An alternative to aging animals in the traditional fashion described above is made possible by the development and selection of Senescence-Accelerated Mouse (SAM) strains. These mice originate from inbred strains suffering from early onset of age-related diseases and demonstrate other pathologic features consistent with aging such as dysregulation of the immune system. The SAMP10 strain in particular experiences both histologic (loss of spines, synapses, and neurons) and functional (impaired learning and memory, depressive-like behavior) changes. Notably, this strain exhibits many of the pro-inflammatory characteristics observed in the aged brain of both humans and rodents.
Limitations
Despite the potential for clear benefit not only in investigation of disease pathophysiology but also therapeutic development, models of aging have some drawbacks that must be considered. For example, due to per diem costs for housing and caring for animals, aging can be prohibitively expensive for many groups. Despite the expense, research with this model has been conducted successfully by groups in both North America and Europe. Conversely, SAMs may represent a quicker and more cost-efficient method of studying the aged brain, but the question of how well SAMs correlate with normal aging remains to be answered. It will be important for future work to investigate how closely SAMs replicate the process of aging.
The Metabolic Syndrome and Stroke
Obesity: Inflammatory Mechanisms Driving the Epidemic
The number of overweight and obese people in the United States has doubled within the past 30 years [49]. Obesity dramatically increases the risk for stroke, and several groups have proposed mechanisms to explain this phenomenon. The first mechanism is a decrease of the cytokine called adiponectin. Low adiponectin levels can cause an increase in inflammation, insulin resistance, and vascular degradation [50]. Savopoulos and colleagues showed that the cytokine resistin is altered with obesity. Resistin causes endothelial dysfunction by augmenting the release of endothelin-1 [51]. Endothelin-1 constricts blood vessels and links obesity to hypertension. Still further, the Hishinuma group has shown that visceral fat increases TNF-α as shown in Figure 2. TNF-α acts on pathways that initiate appropriate responses to inflammation and some others that causes apoptosis [52]. TNF-α in association with lymphotoxin induces macrophages to adhere to endothelial cells and exit the blood vessel by diapedesis. These macrophages engulf pathogens and release inflammatory cytokines following an ischemic injury. Furthermore, a phenomenon known as the obesity paradox has been reported during vascular surgery. Obesity decreases the risk of post-operative stroke compared to the risk seen in non-obese individuals [53]. Further work has shown that even though obesity elevates the risk for stroke in the general population, it increases survival rates following stroke. Two prevailing theories about this paradox are an excess nutrient reserve available in obese and overweight individuals following stroke as well as an upregulation of TNF-α receptors in adipose tissue following infarct [54]. The activation of TNF-α receptors might increase the likelihood of stroke in obese individuals but it also provides greater neuroprotection following stroke. This occurs because TNF-α is known to activate apoptotic pathways through tissue necrosis factor receptor 1 (TNFR1) and neuroprotective pathways through tissue necrosis factor receptor 2 (TNFR2) [55].
Figure 2.

Obesity and Stroke. Obesity causes a decrease in adiponectin and an increase in resistin and TNF-alpha. These molecular changes predispose the body to increased inflammation. The inflammation leads to increased stroke risk.
Animal Models of Obesity
In order to elucidate the role of obesity in stroke infarct damage, researchers have sought to develop appropriate models of obesity. Two primary forms of obesity exist in the general public. The first is uncontrollable genetic disorders such as leptin insensitivity and the second is obesity caused by poor diets. A model of the genetic form of obesity is the obese Zucker rat. Zucker rats are insensitive to satiety signals relayed to the hypothalamus ventromedial nucleus and also have deficient leptin receptors [56]. The model has been used to gather important data regarding the release of vasodilator and vasoconstrictor hormones associated with stroke development [57]. A model for the diet-induced obesity uses high fat diets in rats. Rats are fed these high-fat diets from 3 weeks of development onward. After these animals become obese, transient middle cerebral artery occlusion (tMCAO) is induced through a fibrin clot. Infarct damage is increased in these animals compared with non-obese controls [58]. This elevated level of damage is due to neurovascular matrix degradation and BBB disruption mediated by increases in MMP-9 [59]. Furthermore, matrix metalloproteinase 2 (MMP-2) is up-regulated and causes degradation of type IV collagen, which is then replaced by inappropriate collagen type I deposits [60]. Since obesity is associated with many vascular disorders, it will be important in the future to investigate how other stroke risk factors such as age, diabetes, and hypertension interact with obesity.
Diabetes: A Growing International Problem
Diabetes mellitus type 2 has become the most common serious metabolic disorder not only in the United States but across the world [61]. One research group estimates that 171 million people across the globe were inflicted with diabetes mellitus in the year 2000 and that number is projected to double by 2030 [62].The most recognizable aspect of type 2 diabetes mellitus is the uncontrolled levels of hyperglycemia. Hyperglycemia can stimulate the formation of advanced glycosylated end-products (AGEs). In the clinical setting, diabetes mellitus is diagnosed as a fasting plasma-glucose of 110 mg/dL or more, or a random plasma-glucose of 200 mg/dL or more [63]. The pathophysiology of diabetes mellitus, however, is more complex than a simple elevation in serum glucose levels. Clinical manifestations also include glycosuria, polydipsia, polyuria, and renal failure from AGEs. Perhaps the most interesting aspect of the disease is the starvation of body tissues despite the fact that glucose remains abundant in the blood stream. This phenomenon is due to peripheral cells being either completely or partially resistant to the effects of insulin [63]. The body shifts from primarily receiving energy from glucose to receiving energy from lipids. Consequentially, this leads to ketone buildup in the body and a lowered pH level [61].
Causes of Diabetes: An Unknown Conglomeration of Many Factors
A few theories have been proposed regarding the causes of diabetes. In reality, little is known about the causes, and like many other diseases, it is likely multifactorial. Many clinicians point to certain habits, such as sedentary lifestyle and high calorie and fat intake, as the main culprit of diabetes [61]. Interventions focusing on weight loss, diet modifications, and regular physical exercise of at least 150 minutes per week reduce the incidence of diabetes in at-risk patients. These activities also reduce hemoglobin A1C levels in type 2 diabetics [64]. Genetic links have likewise been recognized. Researchers discovered at least 36 genes associated with diabetes [65]. A high concordance level among identical twins shows that type II diabetes is heritable [66].
Diabetes: Role in Chronic Inflammation and Ischemic Stroke Risk/Outcome
Additionally, many groups began to focus on the influence of inflammation and innate immunity in diabetes. It is uncertain at this point if inflammation occurs prior to or after the onset of diabetes. Specific cytokines such as TNF-α, IL-6, and C-reactive protein (CRP) are elevated in type 2 diabetic patients in comparison to non-diabetic controls [67]. TNF-α and IL-6 cause generalized systemic responses such as fever and increased vascular permeability while CRP is a key player in complement mediated immunity. CRP is the foundational block of the classical pathway. CQ binds to the pathogen, and CR and CS cleave C4 and C2 to make the convertase C4bC2a. The convertase then marks the pathogen for phagocytosis by cleaving C4 to C4b and C4a, which allows C4b to become membrane bound. Another innate immune response is driven by toll-like receptors (TLR). TLRs are activated by LPS or lipoteichoic acid on cell membranes or by DNA or RNA in endosomes. TLRs trigger nucleus specific changes that often lead to increased release of acute phase reactants [68]. These acute phase reactants are beneficial in situations in which the body’s immune system is compromised with an infection or after an acute trauma. Over a longer period of time, these reactants become destructive to the systemic vascular system and cause damage to tissues throughout the body [67]. This destruction of the vascular system contributes to other diseases and injuries such as hypertension, cerebrovascular diseases, renal disease, and ischemic stroke [69,70]. The damage is mediated through numerous pro-inflammatory pathways. The signaling produces enhanced oxidative stress and the toxic build-up of degraded cells, proteins, and molecules [69].
Although the role of inflammation in diabetes is still under investigation, it is important to consider which pathways may be targeted therapeutically. For example, TNF-α induces pathways that lead to increased insulin resistance throughout the body [71]. Insulin resistance is a strong risk factor for ischemic stroke [69]. Targeting TNF-α pathways may therefore be useful for preventing strokes in diabetic patients. Ultimately, any inflammatory trauma to the vascular system will have a large effect on the vasculature of the brain. If the inflammatory changes of diabetes can be controlled, it might greatly reduce the incidence of stroke in this population.
Diabetes in Animal Models
Many well-established rat models are used in type 2 diabetes research. This section will address three of the most prominent models as mentioned in Figure 3. The Israeli sand rat model focuses on the dietary factors of diabetes mellitus. As mentioned previously, an increased caloric intake can intensify the occurrence of diabetes mellitus type 2. When the normal vegetarian diet is changed to a specific high-calorie laboratory diet, the rats eventually develop obesity, glucose intolerance, and hyperinsulinemia [72]. With time, the rat’s intact pancreatic islet cells begin to degrade and die, thus leading to overt diabetes [73,74]. Essentially, this model relies on nutritional means to develop diabetes mellitus type 2 in the animals. The model is beneficial because it mimics the excessive calorie and glucose levels found in the human population. One limitation is that it does not address the genetic factors of diabetes. Many genetic factors have been identified in the human population and lead to activation of different pathways. These pathways present different therapeutic intervention opportunities. Therefore, genetic factors of diabetes are essential in the development of a proper diabetic model.
Figure 3.

Rat Models of Diabetes Type II. Three well-known models of diabetes include the Israeli Sand Rat, Otsuka Long-Evans Tokushima Fatty Rat, and Ventromedial Hypothalamus Lesion (VMH) Dietary Obese Rat. Each of these models represents different characteristics of diabetes such as obesity, glucose intolerance, hyperinsulinemia, polyphagia, and hepatic lipogenesis.
The Otsuka Long-Evans Tokushima fatty (OLETF) rat is one example of a diabetic model that incorporates genetic factors. This rat model is developed by selective breeding from an outbred rat colony of Long-Evan rats. Although selective breeding is used, it is important to note that this is not a transgenic population. At 16 weeks of age, the OLETF rats’ differentiating characteristics are thoroughly developed. This rat model demonstrates the following physical manifestations: glucose intolerance, hyperinsulinemia, mild obesity, and polyphagia [74]. The combination of polyphagia, obesity, and impaired glucose intolerance is a more accurate syndrome of the Western diabetic population. The polyphagia is assumed to be partially explained by a null allele coding for the cholecystokinin A receptor [72]. A problem with this model is that the diabetic characteristics are induced by breeding instead of the actual pathophysiology of diabetes mellitus. Using such models explains why current treatments of diabetes mellitus focus only on eliminating physical manifestations of diabetes instead of treating the underlying cause of the disease.
Another interesting model combines the use of a lesion in the ventromedial hypothalamus region of the brain and a high-fat/high-sucrose diet. This is known as the ventromedial hypothalamus lesion (VMH) dietary obese rat model [75]. The lesion itself is thought to cause the development of hyperphagia, obesity, decreased glucose metabolism, defective regulation of insulin, and increased hepatic lipogenesis [74]. As already discussed, the high-fat and high-sucrose diet is a key factor in exacerbating the lesion’s effects. Within 3 weeks, these rats are found to have fasting hyperinsulinemia, hypertriglyceridemia, and impaired glucose tolerance [75]. Although this model is beneficial in studying the effects of the lesion and the diet on diabetes, it again does not accurately portray the entire pathophysiology of diabetes. Some diabetic pathways are highlighted while others are completely ignored. It is clear that these models do not accurately represent the pathophysiology of diabetes. A more comprehensive model will be necessary to elucidate the mechanisms of diabetes and how these altered pathways increase risk for stroke. Fan and colleagues studied stroke in a streptozotocin diabetic rat model. The model they used is limited because it represents type I diabetes mellitus and hemorrhagic stroke instead of the more common type II diabetes and ischemic stroke. Furthermore, this study focused only on tPA activity instead of investigating other mechanisms and pathways [76].
Hypertension and Stroke: Increasing Prevalence in the United States
Two broad classifications of hypertension include primary hypertension and secondary hypertension. Primary hypertension is not caused by an independent disease such as athersclerosis, but it can lead to higher risk for vascular and heart problems and predispose individuals to disease. The prevalence of primary hypertension is increasing in the United States due to the rise of obesity rates [77]. Three subcategories for primary hypertension are systolic and diastolic hypertension (SDH), isolated systolic hypertension (ISH), and isolated diastolic hypertension (IDH). SDH and ISH are risk factors for ischemic stroke as shown in Figure 4 [78].
Figure 4.

Hypertension and Stroke. Primary hypertension, specifically systolic and diastolic (SDH) and isolated systsolic (ISH), increases inflammation and the risk for stroke. Improved treatments for all classifications of hypertension are necessary to manage the increasing rate in the population.
Hypertension as a Contributing Factor in Stroke Outcome: An Inflammatory Process
Hypertension causes an increase in cell adhesion molecules, vascular adhesion molecules, and selectins. Subsequently, these molecules induce inflammatory responses by increasing the affinity of binding between immune cells and damaged tissue. The molecules also increase the build-up of atherosclerotic plaques in blood vessels through similar binding mechanisms. If a cerebral vessel becomes completely occluded, an ischemic event ensues leading to a cascade of inflammatory responses and still further immune cell binding [79]. Many of these responses are time-specific and depend on precise pathway activation. For example, Interleukin-6 (IL-6) and MMP-9 can paradoxically lead to either neuroprotection or neurotoxicity depending on the specific period of observation [80]. IL-6 also causes a systemic fever useful for disrupting pathogen viability. It is therefore necessary to use models that relate well with human pathology and to observe changes at multiple time points post-ischemia.
Hypertension in Animal Models
Since hypertension is the largest factor in predicting stroke severity, it is essential that models of stroke incorporate the issue of hypertension [81]. It was proposed that 9 percent to 16 percent of ischemic strokes could be avoided if hypertension were kept under control in the general population [82]. A model that addresses the issue of hypertension and stroke is known as the spontaneous hypertensive rats-stroke prone (SHR-SP). These rats have spontaneous increases in blood pressure, and upward of 80 percent develop stroke when placed on normal diets [83]. Not surprisingly, some groups have found that diets high in sodium chloride accelerate the time to first infarct in these animals [84]. A primary reason why this model works well is that it allows researchers to investigate how inflammation changes from a baseline state of hypertension to a post-ischemic state. Inflammation triggered by the sheer on an atherosclerotic vessel facilitates an increase in oxidative stress and the activation of the ubiquitin-proteasome system. Initially this leads to small vessel remodeling, but as long as the pressure remains elevated, these changes eventually result in occlusion and infarct [85]. Since this process is progressive, it also allows for measurement of time-specific changes. Unfortunately this model also has some limitations since the animals die before 1 year of age [86]. Since age is the biggest risk factor for stroke, it is important to consider how hypertension changes with age. The majority of people past the age of 65 will eventually develop hypertension due to age-related arterial changes [87]. It will therefore be beneficial for future models to use animals that develop hypertension later in their lives similar to when most humans develop it.
Conclusion and Future Directions
Although it has been challenging to find therapeutics that translate successfully from bench-to-bedside, animal models are still a valuable and promising tool for therapeutic development. Animals experience several of the same physiological changes that humans encounter. With the use of transgenic animals and careful breeding, it is possible to simulate the majority of risk factors seen in complex human diseases [88]. Age, obesity, diabetes, and hypertension were examined in this review as they relate to stroke risk and inflammation. The models that represent these individual risk factors have been used successfully in relation to other disease states and furthermore have provided helpful insights into important components of stroke. The question that remains is how all of these risk factors fit together. In order to answer this question, it will be necessary to design a more comprehensive model that incorporates most of these risk factors.
In human diagnosis, the factors of obesity, dyslipidemia, glucose intolerance, and hypertension have all been linked into a single metabolic syndrome. Until recently, these factors were viewed as separate components and risk elements. Since they often occur together in individual patients, researchers and clinicians were encouraged to propose the idea of the metabolic syndrome [89]. Some cardiovascular researchers have adopted the idea of the metabolic syndrome and now use a transgenic rat model SHR/NDcp, which spontaneously develops obesity, hypertension, hypderlipidemia, and insulin resistance [90]. If such a model could be adapted to stroke research, it may offer a more comprehensive approach necessary to isolate the important pathways. Another possibility is to use the high-fat diet mentioned in the obesity section but initiate it at a later time point in development. This high-fat diet has been deemed the western diet in animal research and has consistently resulted in the development of the metabolic syndrome [58]. The results from this comprehensive model can then be compared with previous work in the other models. Other models have been used to isolate key inflammatory pathways important in stroke morbidity and mortality. A good starting point for using the more comprehensive model would be to look at common pathways consistently showing changes in most of the previous less-sophisticated models.
When looking through the literature, a few inflammatory pathways appear to be activated consistently post-ischemia and would be worth investigating with this comprehensive model. For example, some of the cytokines released post-ischemia such as IL-6, oncostatin M, and ciliary neurotrophic factor bind to the gp130 receptor [91]. These cytokines cause increased inflammation and the recruitment of immune cells. Not surprisingly, these same cytokines are altered by age and the factors of the metabolic syndrome [45,92]. Once the gp130 receptor is activated, it induces downstream Janus kinase 2 (JAK2) phosphorylation of the tyrosine residue on STAT3. STAT3 is unique in its capacity to activate different genes in different cell types [93]. Another pathway that looks promising is TNF-α’s activation of secretory phospholipase A2 IIA (sPLA2 IIA). sPLA2 IIA activation leads to a destruction of phosphatidylcholine in membranes and also to an increase in inflammation-dependent infarct size [94]. The cytokine TNF- α specifically causes endothelial widening and diapedesis of macrophages. As mentioned previously, TNF-α levels are altered by obesity, age, and other factors of the metabolic syndrome. Finally, protein kinase C (PKC) is a well-known modulator of inflammatory pathways. PKC has 12 different isoforms, many of which are altered by the metabolic syndrome and age [95,96]. These responses can sometimes work against each other depending on which isoform of PKC is active. PKCδ is associated with neurotoxicity where PKCε initiates neuroprotection [97].
Although much remains to be discovered about stroke, the use of a comprehensive animal model has been a useful tool for translational research in other disease states and may prove useful for ischemic stroke research as well. It will allow investigators to highlight neurotoxic and neuroprotective pathways and discover how they function with consideration to age and the metabolic syndrome. With increased therapeutic options, it may be possible to increase survival and recovery following an ischemic event.
Abbreviations
- AGEs
advanced glycosylated end-products
- BBB
blood brain barrier
- CD200fc
CD200 fusion protein
- CNS
central nervous system
- CRP
C-reactive protein
- eNOS
endothelial nitric oxide synthase
- GFAP
glial fibrillary acidic protein
- iNOS
inducible nitric oxide synthase
- IL-1β
interleukin-1β
- IL-6
interleukin-6
- IL-6Rα
interleukin-6Rα
- IL-10
interleukin-10
- IL-17
interleukin-17
- IDH
isolated diastolic hypertension
- ISH
isolated systolic hypertension
- JAK2
Janus kinase 2
- LPS
lipopolysaccharide
- LTP
long-term potentiation
- MHCII
major histocompatibility complex II
- MMP-2
matrix metalloproteinase 2
- MMP-9
matrix metalloproteinase 9
- NOX
NADPH oxidase
- PKC
protein kinase C
- r-tPA
recombinant tissue plasminogen activator
- sPLA2IIA
secretory phospholipase A2IIA
- SAM
Senescence-Accelerated Mouse
- STAT3
signal transducers and activators of transcription 3
- SHR-SP
spontaneous hypertensive rats-stroke prone
- STAIR
Stroke Therapy Academic and Industry Roundtable
- SVZ
subventricular zone
- SDH
systolic and diastolic hypertension
- tMCAO
transient middle cerebral artery occlusion
- TNF-α
tissue necrosis factor-alpha
- TNFR1
tissue necrosis factor receptor 1
- TNFR2
tissue necrosis factor receptor 2
- TLR
toll-like receptor
- VMH
ventromedial hypothalamus lesion
References
- Donnan GA, Baron J, Ma H, Davis SM. Penumbral selection of patients for trials of acute stroke therapy. Lancet Neurol. 2009;8(3):261–269. doi: 10.1016/S1474-4422(09)70041-9. [DOI] [PubMed] [Google Scholar]
- Floel A, Warnecke T, Duning T, Lating Y, Uhlenbrock J, Schneider A. et al. Granulocyte-colony stimulating factor (G-CSF) in stroke patients with concomitant vascular Disease—A randomized controlled trial. PLoS ONE. 2011;6(5):e19767. doi: 10.1371/journal.pone.0019767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montaner J, Mendioroz M, Delgado P, García-Berrocoso T, Giralt D, Merino C. et al. Differentiating ischemic from hemorrhagic stroke using plasma biomarkers: The S100B/RAGE pathway. J Proteomics. 2012;75(15):4758–4765. doi: 10.1016/j.jprot.2012.01.033. [DOI] [PubMed] [Google Scholar]
- Odden MC, Coxson PG, Moran A, Lightwood JM, Goldman L, Bibbins-Domingo K. The impact of the aging population on coronary heart disease in the United States. Am J Med. 2011;124(9):827–833. doi: 10.1016/j.amjmed.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taussky P, Tawk RG, Daugherty WP, Hanel RA. Medical therapy for ischemic stroke: Review of intravenous and intra-arterial treatment options. World Neurosurg. 2011;76(6 Suppl):S9–S15. doi: 10.1016/j.wneu.2011.05.048. [DOI] [PubMed] [Google Scholar]
- Shah IM, Ghosh SK, Collier A. Stroke presentation in type 2 diabetes and the metabolic syndrome. Diabetes Res Clin Pract. 2008;79(1):e1–e4. doi: 10.1016/j.diabres.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Dronne M, Grenier E, Chapuisat G, Hommel M, Boissel J. A modelling approach to explore some hypotheses of the failure of neuroprotective trials in ischemic stroke patients. Prog Biophys Mol Biol. 2008;971:60–78. doi: 10.1016/j.pbiomolbio.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Schmidt S, Bruehl C, Frahm C, Redecker C, Witte OW. Age dependence of excitatory-inhibitory balance following stroke. Neurobiol Aging. 2012;33(7):1356–1363. doi: 10.1016/j.neurobiolaging.2010.11.019. [DOI] [PubMed] [Google Scholar]
- Fisher M, Feuerstein G, Howells DW, Hurn PD, Kent TA, Savitz SI. et al. Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke. 2009;40(6):2244–2250. doi: 10.1161/STROKEAHA.108.541128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Napoli M, Shah IM. Neuroinflammation and cerebrovascular disease in old age: A translational medicine perspective. J Aging Res. 2011;2011:857484. doi: 10.4061/2011/857484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won SJ, Xie L, Kim SH, Tang H, Wang Y, Mao X. et al. Influence of age on the response to fibroblast growth factor-2 treatment in a rat model of stroke. Brain Res. 2006;1123(1):237–244. doi: 10.1016/j.brainres.2006.09.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, de Sluis BV. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;4797372:232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa-Wagner A, Buga A, Kokaia Z. Perturbed cellular response to brain injury during aging. Ageing Res Rev. 2011;10(1):71–79. doi: 10.1016/j.arr.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Bilbo SD, Smith SH, Schwarz JM. A lifespan approach to neuroinflammatory and cognitive disorders: A critical role for glia. J Neuroimmune Pharmacol. 2012;7(1):24–41. doi: 10.1007/s11481-011-9299-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buga AM, Vintilescu R, Pop OT, Popa-Wagner A. Brain aging and regeneration after injuries: An organismal approach. Aging Dis. 2011;2(1):64–79. [PMC free article] [PubMed] [Google Scholar]
- Corona AW, Fenn AM, Godbout JP. Cognitive and behavioral consequences of impaired immunoregulation in aging. J Neuroimmune Pharmacol. 2011;7(1):7–23. doi: 10.1007/s11481-011-9313-4. [DOI] [PubMed] [Google Scholar]
- Pizza V, Agresta A, D’Acunto CW, Festa M, Capasso A. Neuroinflammation and ageing: Current theories and an overview of the data. Rev Recent Clin Trials. 2011;6(3):189–203. doi: 10.2174/157488711796575577. [DOI] [PubMed] [Google Scholar]
- Franceschi C, Bonafe M, Valensin S, Olivieri F, De Luca M, Ottaviani E. et al. Inflamm-aging. an evolutionary perspective on immunosenescence. Ann NY Acad Sci. 2000;908:244–254. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- Hein AM, O’Banion MK. Neuroinflammation and cognitive dysfunction in chronic disease and aging. J Neuroimmune Pharmacol. 2012;7(1):3–6. doi: 10.1007/s11481-011-9340-1. [DOI] [PubMed] [Google Scholar]
- von Bernhardi R, Tichauer JE, Eugenin J. Aging-dependent changes of microglial cells and their relevance for neurodegenerative disorders. J Neurochem. 2010;112(5):1099–1114. doi: 10.1111/j.1471-4159.2009.06537.x. [DOI] [PubMed] [Google Scholar]
- Rosano C, Marsland AL, Gianaros PJ. Maintaining brain health by monitoring inflammatory processes: A mechanism to promote successful aging. Aging Dis. 2012;3(1):16–33. [PMC free article] [PubMed] [Google Scholar]
- Bitto A, Sell C, Crowe E, Lorenzini A, Malaguti M, Hrelia S. et al. Stress-induced senescence in human and rodent astrocytes. Exp Cell Res. 2010;316(17):2961–2968. doi: 10.1016/j.yexcr.2010.06.021. [DOI] [PubMed] [Google Scholar]
- Salminen A, Ojala J, Kaarniranta K, Haapasalo A, Hiltunen M, Soininen H. Astrocytes in the aging brain express characteristics of senescence-associated secretory phenotype. Eur J Neurosci. 2011;34(1):3–11. doi: 10.1111/j.1460-9568.2011.07738.x. [DOI] [PubMed] [Google Scholar]
- Csiszar A, Ungvari Z, Koller A, Edwards JG, Kaley G. Aging-induced proinflammatory shift in cytokine expression profile in coronary arteries. FASEB J. 2003;17(9):1183–1185. doi: 10.1096/fj.02-1049fje. [DOI] [PubMed] [Google Scholar]
- Brown DR. Role of microglia in age-related changes to the nervous system. ScientificWorldJournal. 2009;9:1061–1071. doi: 10.1100/tsw.2009.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XG, Ding JQ, Chen SD. Microglia in the aging brain: Relevance to neurodegeneration. Mol Neurodegener. 2010;5:12. doi: 10.1186/1750-1326-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njie EG, Boelen E, Stassen FR, Steinbusch HW, Borchelt DR, Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging. 2012;33(1):195.e1–195.e12. doi: 10.1016/j.neurobiolaging.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry GJ, Kraft AD. Microglia in the developing brain: A potential target with lifetime effects. Neurotoxicology. 2012;33(2):191–206. doi: 10.1016/j.neuro.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart AD, Wyttenbach A, Hugh Perry V, Teeling JL. Age related changes in microglial phenotype vary between CNS regions: Grey versus white matter differences. Brain Behav Immun. 2012;26(5):754–765. doi: 10.1016/j.bbi.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KJ, Miller A, Thelma R, Cowley F, Fionnuala Cox F, Lynch MA. The age- and amyloid-β-related increases in nogo B contribute to microglial activation. Neurochem Int. 2011;58(2):161–168. doi: 10.1016/j.neuint.2010.11.009. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Kim JY, Sun W. Age-dependent changes in the subcallosal zone neurogenesis of mice. Neurochem Int. 2012 Mar 6; doi: 10.1016/j.neuint.2012.02.027. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- McGinn MJ, Colello RJ, Sun D. Age-related proteomic changes in the subventricular zone and their association with neural stem/progenitor cell proliferation. J Neurosci Res. 2012;90(6):1159–1168. doi: 10.1002/jnr.23012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Baxter MG. The ageing cortical synapse: Hallmarks and implications for cognitive decline. Nat Rev Neurosci. 2012;13(4):240–250. doi: 10.1038/nrn3200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadwick W, Martin B, Chapter MC, Park SS, Wang L, Daimon CM. et al. GIT2 acts as a potential keystone protein in functional hypothalamic networks associated with age-related phenotypic changes in rats. PLoS One. 2012;7(5):e36975. doi: 10.1371/journal.pone.0036975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurgens HA, Johnson RW. Dysregulated neuronal–microglial cross-talk during aging, stress and inflammation. Exp Neurol. 2012;233(1):40–48. doi: 10.1016/j.expneurol.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox FF, Carney D, Miller A, Lynch MA. CD200 fusion protein decreases microglial activation in the hippocampus of aged rats. Brain Behav Immun. 2012;26(5):789–796. doi: 10.1016/j.bbi.2011.10.004. [DOI] [PubMed] [Google Scholar]
- Gemma C, Bachstetter AD, Bickford PC. Neuron-microglia dialogue and hippocampal neurogenesis in the aged brain. Aging Dis. 2010;1(3):232–244. [PMC free article] [PubMed] [Google Scholar]
- Dotson V, Horak K, Alwardt C, Larson DF. Relationship of aging and cardiac IL-10. J Extra Corpor Technol. 2004;36(2):197–201. [PubMed] [Google Scholar]
- Meador BM, Krzyszton CP, Johnson RW, Huey KA. Effects of IL-10 and age on IL-6, IL-1beta, and TNF-alpha responses in mouse skeletal and cardiac muscle to an acute inflammatory insult. J Appl Physiol. 2008;104(4):991–997. doi: 10.1152/japplphysiol.01079.2007. [DOI] [PubMed] [Google Scholar]
- DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL. Early disruptions of the blood–brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. 2008;29(5):753–764. doi: 10.1016/j.neurobiolaging.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee P, Kim J, Williams R, Sandhir R, Gregory E, Brooks WM. et al. Effects of aging on blood brain barrier and matrix metalloproteases following controlled cortical impact in mice. Exp Neurol. 2012;234(1):50–61. doi: 10.1016/j.expneurol.2011.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa-Wagner A, Stocker K, Balseanu AT, Rogalewski A, Diederich K, Minnerup J. et al. Effects of granulocyte-colony stimulating factor after stroke in aged rats. Stroke. 2010;41(5):1027–1031. doi: 10.1161/STROKEAHA.109.575621. [DOI] [PubMed] [Google Scholar]
- Itoh T, Imano M, Nishida S, Tsubaki M, Mizuguchi N, Hashimoto S. et al. Increased apoptotic neuronal cell death and cognitive impairment at early phase after traumatic brain injury in aged rats. Brain Struct Funct. 2012 Feb 29; doi: 10.1007/s00429-012-0394-5. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Caballero B, Coto-Montes A. An insight into the role of autophagy in cell responses in the aging and neurodegenerative brain. Histol Histopathol. 2012;27(3):263–275. doi: 10.14670/HH-27.263. [DOI] [PubMed] [Google Scholar]
- DiNapoli VA, Benkovic SA, Li X, Kelly KA, Miller DB, Rosen CL. et al. Age exaggerates proinflammatory cytokine signaling and truncates signal transducers and activators of transcription 3 signaling following ischemic stroke in the rat. Neuroscience. 2010;170(2):633–644. doi: 10.1016/j.neuroscience.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu Z, Hayashi Y, Nakanishi H. Age-dependent neuroinflammatory responses and deficits in long-term potentiation in the hippocampus during systemic inflammation. Neuroscience. 2012;216:133–142. doi: 10.1016/j.neuroscience.2012.04.050. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, White CL, Gupta S, Knight AG, Pistell PJ, Ingram DK. et al. NOX activity in brain aging: Exacerbation by high fat diet. Free Radical Biology and Medicine. 2010;49(1):22–30. doi: 10.1016/j.freeradbiomed.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly KA, Li X, Tan Z, VanGilder RL, Rosen CL, Huber JD. NOX2 inhibition with apocynin worsens stroke outcome in aged rats. Brain Res. 2009;1292:165–172. doi: 10.1016/j.brainres.2009.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knecht S, Ellger T, Levine JA. Obesity in neurobiology. Prog Neurobiol. 2008;84(1):85–103. doi: 10.1016/j.pneurobio.2007.09.003. [DOI] [PubMed] [Google Scholar]
- Katsiki N, Ntaios G, Vemmos K. Stroke, obesity and gender: A review of the literature. Maturitas. 2011;69(3):239–243. doi: 10.1016/j.maturitas.2011.04.010. [DOI] [PubMed] [Google Scholar]
- Savopoulos C, Michalakis K, Apostolopoulou M, Miras A, Hatzitolios A. Adipokines and stroke: A review of the literature. Maturitas. 2011;70(4):322–327. doi: 10.1016/j.maturitas.2011.09.002. [DOI] [PubMed] [Google Scholar]
- Hishinuma A, Majima M, Kurabayashi H. Insulin resistance in patients with stroke is related to visceral fat obesity and adipocytokines. J Stroke Cerebrovasc Dis. 2008;17(4):175–180. doi: 10.1016/j.jstrokecerebrovasdis.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Jackson RS, Black JH III, Lum YW, Schneider EB, Freischlag JA, Perler BA. et al. Class I obesity is paradoxically associated with decreased risk of postoperative stroke after carotid endarterectomy. J Vasc Surg. 2012;55(5):1306–1312. doi: 10.1016/j.jvs.2011.11.135. [DOI] [PubMed] [Google Scholar]
- Kastorini C, Panagiotakos DB. The obesity paradox: Methodological considerations based on epidemiological and clinical evidence—New insights. Maturitas. 2012;72(3):220–224. doi: 10.1016/j.maturitas.2012.04.012. [DOI] [PubMed] [Google Scholar]
- Pan W, Kastin AJ. Tumor necrosis factor and stroke: Role of the blood–brain barrier. Prog Neurobiol. 2007;83(6):363–374. doi: 10.1016/j.pneurobio.2007.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orosco M, Rough C, Meile MJ, Nicolaidis S. Spontaneous feeding-related monoamine changes in rostromedial hypothalamus of the obese zucker rat: A microdialysis study. Physiol Behav. 1995;57(6):1103–1106. doi: 10.1016/0031-9384(94)00383-g. [DOI] [PubMed] [Google Scholar]
- Subramanian R, MacLeod KM. Age-dependent changes in blood pressure and arterial reactivity in obese zucker rats. Eur J Pharmacol. 2003;477(2):143–152. doi: 10.1016/j.ejphar.2003.08.003. [DOI] [PubMed] [Google Scholar]
- Langdon KD, Clarke J, Corbett D. Long-term exposure to high fat diet is bad for your brain: Exacerbation of focal ischemic brain injury. Neuroscience. 2011;182:82–87. doi: 10.1016/j.neuroscience.2011.03.028. [DOI] [PubMed] [Google Scholar]
- Amantea D, Corasaniti MT, Mercuri NB, Bernardi G, Bagetta G. Brain regional and cellular localization of gelatinase activity in rat that have undergone transient middle cerebral artery occlusion. Neuroscience. 2008;152(1):8–17. doi: 10.1016/j.neuroscience.2007.12.030. [DOI] [PubMed] [Google Scholar]
- Deutsch C, Portik-Dobos V, Smith AD, Ergul A, Dorrance AM. Diet-induced obesity causes cerebral vessel remodeling and increases the damage caused by ischemic stroke. Microvasc Res. 2009;78(1):100–106. doi: 10.1016/j.mvr.2009.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg GD, Wadhwa S. Diabetes disease management in a community-based setting. Manag Care. 2002;11(6):42, 45-50. [PubMed] [Google Scholar]
- Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- Mahler RJ, Adler ML. Clinical review 102: Type 2 diabetes mellitus: Update on diagnosis, pathophysiology, and treatment. J Clin Endocrinol Metab. 1999;84(4):1165–1171. doi: 10.1210/jcem.84.4.5612. [DOI] [PubMed] [Google Scholar]
- Ripsin CM, Kang H, Urban RJ. Management of blood glucose in type 2 diabetes mellitus. Am Fam Physician. 2009;79(1):29–36. [PubMed] [Google Scholar]
- Herder C, Roden M. Genetics of type 2 diabetes: Pathophysiologic and clinical relevance. Eur J Clin Invest. 2011;41(6):679–692. doi: 10.1111/j.1365-2362.2010.02454.x. [DOI] [PubMed] [Google Scholar]
- Medici F, Hawa M, Ianari A, Pyke DA, Leslie RD. Concordance rate for type II diabetes mellitus in monozygotic twins: Actuarial analysis. Diabetologia. 1999;42(2):146–150. doi: 10.1007/s001250051132. [DOI] [PubMed] [Google Scholar]
- Pickup JC. Inflammation and activated innate immunity in the pathogenesis of type 2 diabetes. Diabetes Care. 2004;27(3):813–823. doi: 10.2337/diacare.27.3.813. [DOI] [PubMed] [Google Scholar]
- Creely SJ, McTernan PG, Kusminski CM, Fisher M, Da Silva NF, Khanolkar M. et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab. 2007;292(3):E740–E747. doi: 10.1152/ajpendo.00302.2006. [DOI] [PubMed] [Google Scholar]
- Mukherjee D. Peripheral and cerebrovascular atherosclerotic disease in diabetes mellitus. Best Pract Res Clin Endocrinol Metab. 2009;23(3):335–345. doi: 10.1016/j.beem.2008.10.015. [DOI] [PubMed] [Google Scholar]
- Lee M, Saver JL, Hong KS, Song S, Chang KH, Ovbiagele B. Effect of pre-diabetes on future risk of stroke: Meta-analysis. BMJ. 2012;344:e3564. doi: 10.1136/bmj.e3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. 1995;95(5):2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DA, Alcolado JC. Animal models of diabetes mellitus. Diabetic Med. 2005;22(4):359–370. doi: 10.1111/j.1464-5491.2005.01499.x. [DOI] [PubMed] [Google Scholar]
- Walder KR, Fahey RP, Morton GJ, Zimmet PZ, Collier GR. Characterization of obesity phenotypes in psammomys obesus (israeli sand rats) Int J Exp Diabetes Res. 2000;1(3):177–184. doi: 10.1155/EDR.2000.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan K, Ramarao P. Animal models in type 2 diabetes research: An overview. Indian J Med Res. 2007;125(3):451–472. [PubMed] [Google Scholar]
- Axen KV, Li X, Fung K, Sclafani A. The VMH-dietary obese rat: A new model of non-insulin-dependent diabetes mellitus. Am J Physiol. 1994;266(3 Pt 2):R921–R928. doi: 10.1152/ajpregu.1994.266.3.R921. [DOI] [PubMed] [Google Scholar]
- Fan X, Qiu J, Yu Z, Dai H, Singhal AB, Lo EH. et al. A rat model of studying tissue-type plasminogen activator thrombolysis in ischemic stroke with diabetes. Stroke. 2012;43(2):567–570. doi: 10.1161/STROKEAHA.111.635250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar I, Kotchen TA. Trends in prevalence, awareness, treatment, and control of hypertension in the united states, 1988-2000. J Am Med Assoc. 2003;290(2):199–206. doi: 10.1001/jama.290.2.199. [DOI] [PubMed] [Google Scholar]
- Inoue R, Ohkubo T, Kikuya M, Metoki H, Asayama K, Obara T. et al. Stroke risk in systolic and combined systolic and diastolic hypertension determined using ambulatory blood pressure: The ohasama study. Am J Hypertens. 2007;20(10):1125–1131. doi: 10.1016/j.amjhyper.2007.04.017. [DOI] [PubMed] [Google Scholar]
- Hjelstuen A, Anderssen SA, Holme I, Seljeflot I, Klemsdal TO. Markers of inflammation are inversely related to physical activity and fitness in sedentary men with treated hypertension. Am J Hypertens. 2006;19(7):669–675. doi: 10.1016/j.amjhyper.2005.11.012. [DOI] [PubMed] [Google Scholar]
- Nilupul Perera M, Ma HK, Arakawa S, Howells DW, Markus R, Rowe CC. et al. Inflammation following stroke. J Clin Neurosci. 2006;13(1):1–8. doi: 10.1016/j.jocn.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Angeli F, Verdecchia P, Reboldi GP, Gattobigio R, Bentivoglio M, Staessen JA. et al. Calcium channel blockade to prevent stroke in hypertension: A meta-analysis of 13 studies with 103,793 subjects. Am J Hypertens. 2004;17(9):817–822. doi: 10.1016/j.amjhyper.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Hisham NF, Bayraktutan U. Epidemiology, pathophysiology, and treatment of hypertension in ischaemic stroke patients. J Stroke Cerebrovasc Dis. 2012 Jun 8; doi: 10.1016/j.jstrokecerebrovasdis.2012.05.001. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Inoue I, Chen L, Zhou L, Zeng X, Wang H. Reproduction of scalp acupuncture therapy on strokes in the model rats, spontaneous hypertensive rats-stroke prone (SHR-SP) Neurosci Lett. 2002;333(3):191–194. doi: 10.1016/s0304-3940(02)01032-7. [DOI] [PubMed] [Google Scholar]
- Masineni SN, Chander PN, Singh GD, Powers CA, Stier CT Jr.. Male gender and not the severity of hypertension is associated with end-organ damage in aged stroke-prone spontaneously hypertensive rats. Am J Hypertens. 2005;18(6):878–884. doi: 10.1016/j.amjhyper.2004.12.014. [DOI] [PubMed] [Google Scholar]
- Shimizu M, Ishikawa J, Yano Y, Hoshide S, Shimada K, Kario K. The relationship between the morning blood pressure surge and low-grade inflammation on silent cerebral infarct and clinical stroke events. Atherosclerosis. 2011;219(1):316–321. doi: 10.1016/j.atherosclerosis.2011.06.030. [DOI] [PubMed] [Google Scholar]
- Sironi L, Maria Calvio A, Bellosta S, Lodetti B, Guerrini U, Monetti M. et al. Endogenous proteolytic activity in a rat model of spontaneous cerebral stroke. Brain Res. 2003;974(1-2):184–192. doi: 10.1016/s0006-8993(03)02578-2. [DOI] [PubMed] [Google Scholar]
- Aronow WS, Fleg JL, Pepine CJ, Artinian NT, Bakris G, Brown AS. et al. ACCF/AHA 2011 expert consensus document on hypertension in the elderly: A report of the american college of cardiology foundation task force on clinical expert consensus documents developed in collaboration with the american academy of neurology, american geriatrics society, american society for preventive cardiology, american society of hypertension, american society of nephrology, association of black cardiologists, and european society of hypertension. J Am Soc Hypertens. 2011;5(4):259–352. doi: 10.1016/j.jash.2011.06.001. [DOI] [PubMed] [Google Scholar]
- Durukan A, Tatlisumak T. Acute ischemic stroke: Overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav. 2007;87(1):179–197. doi: 10.1016/j.pbb.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Goodman E, Li C, Tu Y, Ford E, Sun SS, Huang TT. Stability of the factor structure of the metabolic syndrome across pubertal development: Confirmatory factor analyses of three alternative models. J Pediatr. 2009;155(3):S5.e1–S5.e8. doi: 10.1016/j.jpeds.2009.04.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai K, Sakairi T, Harada S, Shinozuka J, Ide M, Sato H. et al. Diet modification and its influence on metabolic and related pathological alterations in the SHR/NDmcr-cp rat, an animal model of the metabolic syndrome. Exp Toxicol Pathol. 2012;64(4):333–338. doi: 10.1016/j.etp.2010.09.006. [DOI] [PubMed] [Google Scholar]
- Kim SY, Park H, Choi J, Lee JE, Cha J, Choi Y. et al. Ischemic preconditioning-induced expression of gp130 and STAT3 in astrocytes of the rat hippocampus. Mol Brain Res. 2004;129(1-2):96–103. doi: 10.1016/j.molbrainres.2004.06.025. [DOI] [PubMed] [Google Scholar]
- Ahonen TM, Saltevo JT, Kautiainen HJ, Kumpusalo EA, Vanhala MJ. The association of adiponectin and low-grade inflammation with the course of metabolic syndrome. Nutr Metab Cardiovasc Dis. 2012;22(3):285–291. doi: 10.1016/j.numecd.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Maritano D, Sugrue ML, Tininini S, Dewilde S, Strobl B, Fu X. et al. The STAT3 isoforms alpha and beta have unique and specific functions. Nat Immunol. 2004;5(4):401–409. doi: 10.1038/ni1052. [DOI] [PubMed] [Google Scholar]
- Adibhatla RM, Hatcher JF. Secretory phospholipase A2 IIA is up-regulated by TNF-α and IL-1α/β after transient focal cerebral ischemia in rat. Brain Res. 2007;1134:199–205. doi: 10.1016/j.brainres.2006.11.080. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- La Porta CAM, Comolli R. Age-dependent modulation of PKC isoforms and NOS activity and expression in rat cortex, striatum, and hippocampus. Exp Gerontol. 1999;34(7):863–874. doi: 10.1016/s0531-5565(99)00057-1. [DOI] [PubMed] [Google Scholar]
- Sajan MP, Nimal S, Mastorides S, Acevedo-Duncan M, Kahn CR, Fields AP. et al. Correction of metabolic abnormalities in a rodent model of obesity, metabolic syndrome, and type 2 diabetes mellitus by inhibitors of hepatic protein kinase C-ι. Metab Clin Exp. 2012;61(4):459–469. doi: 10.1016/j.metabol.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Alkon DL. Pharmacology of protein kinase C activators: Cognition-enhancing and antidementic therapeutics. Pharmacol Ther. 2010;127(1):66–77. doi: 10.1016/j.pharmthera.2010.03.001. [DOI] [PubMed] [Google Scholar]