

Abstract
We show that mushroom tyrosinase catalyzes formation of reactive o-quinones on unstructured, tyrosine-rich sequences such as hemagglutinin (HA)-tags (YPYDVPDYA). In the absence of exogenous nucleophiles and at low protein concentrations, the o-quinone decomposes with fragmentation of the HA-tag. At higher protein concentrations (>5 mg/ml), cross-linking is observed. Besthorn’s reagent intercepts the o-quinone to give a characteristic pink complex, which can be observed directly on a denaturing SDS-PAGE gel. Similar labeled species can be formed using other nucleophiles such as Cy5-hydrazide. These reactions are selective for proteins bearing HA- and other unstructured poly-tyrosine-containing tags and can be performed in lysates to create specifically tagged proteins.
Keywords: labeling, o-quinone, cross-link, mechanism, protein derivatization)
Introduction
Many protocols have been established to introduce unnatural chemical motifs into proteins, including genetic manipulation[1] and the hijacking of protein modification pathways.[2] Some of the earliest protein derivatizations include oxidation/reduction of cysteines and methionines, [3] alkylations/disulfide exchange of cysteines (especially those within cysteine-proteases),[4] sulfonylation of active site serine in serine proteases followed by nucleophilic displacement,[5] and reaction of lysines with acid anhydrides/NHS esters.[6] In recent years more complex transformations have been explored, such as Mukaiyama-aldol of protein aldehydes[7] and O-alkylation of tyrosines with palladium π-allyls. [8]
Most chemical protein derivatization methods suffer drawbacks including side reactions and low conversion, which limits use in protein mixtures. Thus, modern methodologies focus on specific derivatization of protein domains, such as avidin,[9] Halo,[10] or SNAP/CLIP[11] to direct an unnatural chemical species to a protein of interest. These differ from chemical modifiers in that the reaction is choreographed by a fusion protein.
Enzymatic modification of proteins has also been exploited in derivatization reactions. Such processes ideally benefit from the traditional pluses of enzyme-mediated reactions: they are selective, efficient, require catalytic amounts of the derivatizing agent, and are biocompatible. Furthermore, the recognition sequences are usually a few residues rather than whole domains. One such process is oxidation of a protein’s N-terminus to generate carbonyl-containing species.[12] Other recent examples include the Q-, PRIME and YbbR tags, which attach dyes to specific peptide sequences [13, 14]
Relatively few reagents specifically derivatize tyrosines, and these are far from optimal. Iodine is employed to form iodotyrosine. This technique is regularly used to radiolabel enzymes with I131. However, this process targets other residues (methionine, cysteine, histidine[15] and tryptophan).[16] Tetranitromethane usually affords nitrotyrosine, but tyrosine dimers can form in a side reaction.[17] Tetranitromethane can also modify glycine.[18] Newer methodologies to functionalize tyrosine involving diazonium ions and Mannich reactions are now being developed, although the reaction conditions may not be biocompatible.[19]
o-Quinones are chemically malleable species which undergo a range of chemical transformations including conjugate addition,[20] Diels-Alder,[21] and Wittig reactions.[22] However, oxidation of tyrosine to its o-quinone-congener is not commonly employed to derivatize proteins, presumably because o-quinones are hard to control, and difficult to form from tyrosine. The simplest chemical approach to o-quinones is derivatization of catechols, such as L-dihydroxyphenylalanine (DOPA) using periodate,[23] but DOPA is not a naturally encoded amino acid, and periodate is not biocompatible.[24]
o-Quinones are naturally occurring intermediates in the production of melanin, the protective pigmentation in skin and damaged fruit, formed by the action of tyrosinase. This enzyme catalyzes two related processes: the first and slower step, is oxidation of tyrosine to DOPA (Scheme 1); the second is conversion of DOPA to the o-quinone, which spontaneously cross-links to form melanin. This process is often referred to as the Raper-Mason Scheme.[25] Cysteine can intercept o-quinone intermediates,[26] and 5-cysteinyl DOPA[27] hails from this source.[28]
Scheme 1.
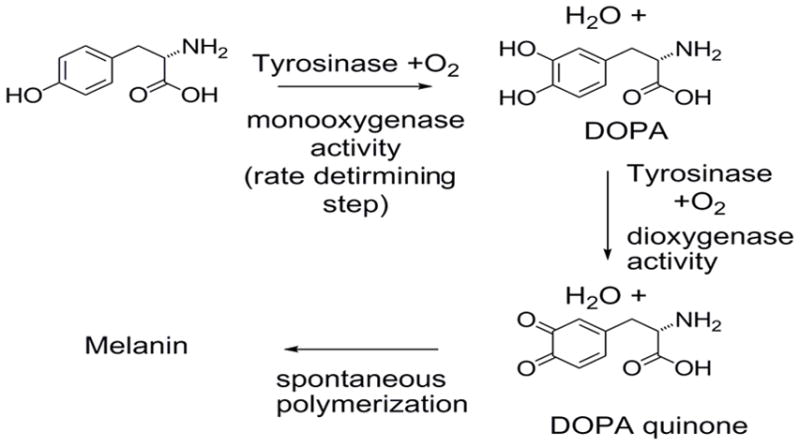
Reactions of tyrosinase.
Many tyrosinases will oxidize N-acyl tyrosine [29] and tyrosine containing oligopeptides in vitro.[30] However, folded proteins are typically not good substrates for tyrosinase, presumably due to steric protection of the tyrosine.[31] Cross-linking is the typical result of tyrosinase catalyzed protein oxidations.[32] This has been recorded for numerous tyrosinases on numerous substrate proteins, although prolonged reaction times and addition of caffeic acid is often required.[33]
The above observations suggested that the reactivity of o-quinones could be harnessed to create a method of selectively derivatizing tyrosines under mild conditions. This could open a range of possibilities for selective chemical functionalization, given the diverse chemistry associated with o-quinones. Importantly, the rate of oxidation of tyrosines on proteins is dependent upon structure, which implies that selectivity may be achievable.[34] We report that mushroom tyrosinase (m-tyrosinase) selectively converts tyrosine residues in hemagglutinin (HA) tags to o-quinones, which can be further derivatized.
Results and Discussion
m-Tyrosinase cleaved C-terminal HA-tags on E. coli DHFR
We chose to investigate the reaction of m-tyrosinase with HA-tagged proteins because: (1) the HA-tag is tyrosine-rich (YPYDVPDYA); (2) HA-tags are usually solvent exposed; and (3) HA-tags are widely used in epitope tagging and pulldown experiments. Escherichia coli dihydrofolate reductase (eDHFR) bearing a C-terminal HA-tag and an N-terminal His6-tag (eDHFR-HA) was treated with m-tyrosinase (Figure 1A). A progressive decrease in the 21 kDa full length protein and corresponding appearance of both a 19 kDa protein and a 45 kDa protein was observed. The latter approximates to twice the weight of the starting protein, and is the expected product of cross-linking. Similar cross-linked products have been observed in tyrosinase-mediated oxidation of unstructured proteins such as β-casein. Structured, folded proteins, such as bovine serum albumin (BSA), do not undergo this process as readily.[35]
Figure 1. Cleavage is unaffected by eDHFR ligands and occurs at the HA-tag.
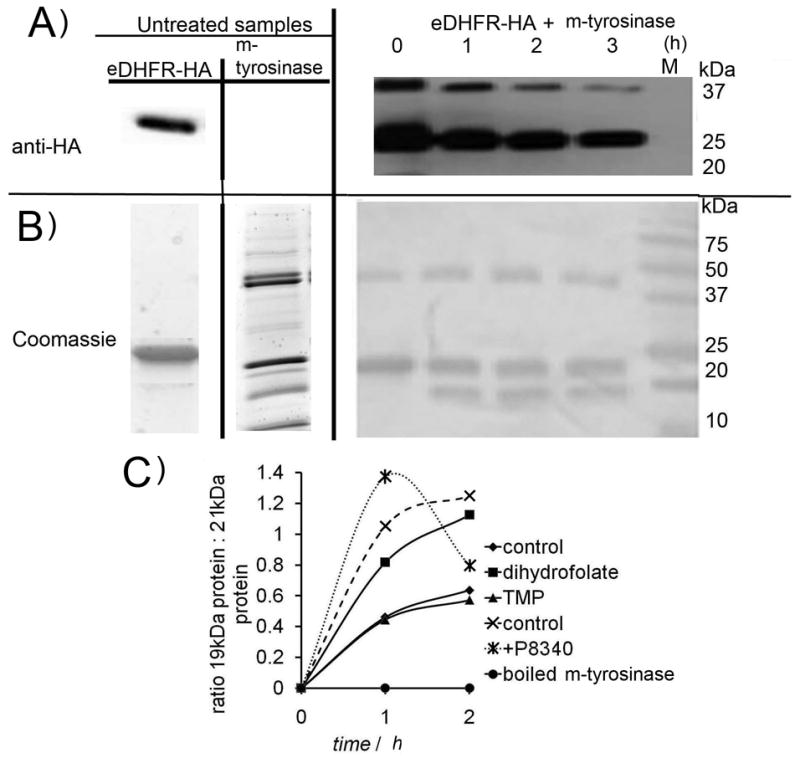
A Cleavage of eDHFR-HA (1.5 mg/ml) as function of time, measured by anti-HA blot (upper) and B Coomassie. [m-tyrosinase] = 0.15 mg/ml (based upon mass of lyophilized product). M = molecular weight marker. C Quantitation eDHFR-HA cleavage under various conditions (200 μM dihydrofolate; 200 μM trimethoprim (TMP); 2x Sigma protease inhibitor cocktail P8340 [containing: 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), pepstatin A, E-64, bestatin hydrochloride, leupeptin hemisulfate, EDTA and aprotinin)]. For the untreated m-tyrosinase sample ~ 7 x the final amount loaded for each time point is shown.
Addition of eDHFR ligands [dihydrofolate or trimethoprim (TMP)] had no significant effect on the cleavage rate (Figure 1B). In addition, eDHFR-HA did not lose activity when treated with m-tyrosinase (Figure S1). These observations suggest that cleavage does not alter the structure of eDHFR and instead occurs in the tags. Mass spectrometry sequencing of the full length and 19 kDa proteins showed two cleavage sites, one at the N-terminus (GSSH_HHH) and a second which could only be localized to after the second Y within the HA tag. Consistent with these data, the 19 kDa protein bound TALON(R) resin, indicating that the cleaved protein contained enough histidines to bind to a metal resin (Figure S2). Furthermore, anti-HA antibodies did not recognize the 19 kDa protein (Figure 1A) indicating that the HA-tag underwent a significant change. Importantly, m-tyrosinase (0.15 mg/ml) also cleaved His6-Halo-HA at the HA-tag (Figure S3). The C-terminus of Halo has no homology to eDHFR’s C-terminus (sequences of both enzymes are shown in Figure S4).
No cleavage was observed when the HA-tag was moved to the N-terminus immediately downstream of the N-terminal His6-tag (NT-HA) in eDHFR (Figures S4 and S9). Thus, this process was position-selective in the eDHFR manifold.
Copper-generated reactive oxygen species (ROS) were not involved in the cleavage process
To our knowledge, no other examples of tyrosinase-mediated protein cleavage have been reported and we thus investigated this process further. Tyrosinases are dinuclear copper enzymes, suggesting that the m-tyrosinase sample may contain free metal ions. Cu+1 can catalyze hydroxyl radical (ROS) formation by a process similar to Fenton chemistry.[36] ROS can oxidize amino acids to form protein radicals leading to fragmentation of the primary structure, although this process is not specific. The involvement of unbound copper was ruled out as follows. Desalted m-tyrosinase was still able to cause cleavage, implying that only enzyme bound copper was required for cleavage. Addition of copper in either the +1 or +2 oxidation state also did not induce cleavage (Figure S5). Finally, catalase had no effect on the rate of cleavage (Figure S5).
HA-tag cleavage is catalyzed by m-tyrosinase
We performed several experiments to determine if cleavage was caused by adventitious proteases. Importantly, boiling m-tyrosinase prior to addition to eDHFR-HA stopped both cleavage events (Figure 1B), which suggests that a protein in the m-tyrosinase sample is responsible for cleavage. SDS-PAGE analysis of the reaction as a function of time showed that there was an initiation phase prior to formation of the 19 kDa product (Figure 2C). Such initial slow phases are a general feature of tyrosinase-mediated oxidation of monophenol substrates,[37] which has been ascribed to redox activation of the dicopper center.[38] Importantly, the lag period is similar to lag times previously reported (5–10 minutes).[39]
Figure 2. 19 kDa formation is stopped by an m-tyrosinase inhibitor.
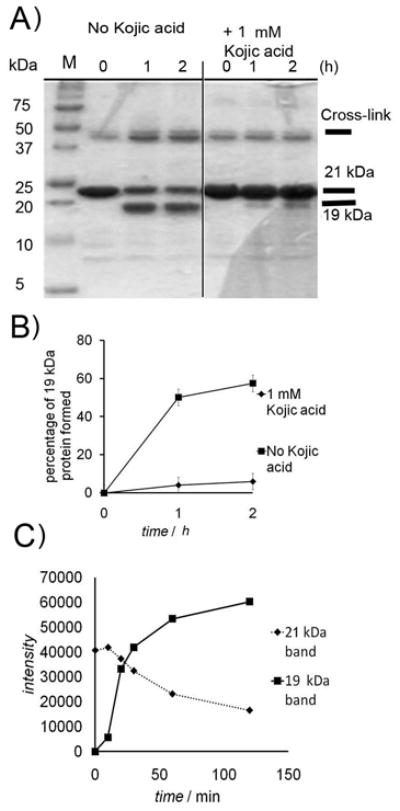
A) eDHFR-HA (1.5 mg/ml) was treated with m-tyrosinase (0.15 mg/ml) (left) and (right) experiment was repeated in presence of 1 mM kojic acid. B) Quantitation of Coomassie-stained SDS-PAGE gels showing inhibition of cleavage by kojic acid (1 mM) N=5 (conditions as in A) errors are standard deviations. C) Formation of 19 kDa and loss of 21 kDa band with time, showing lag (conditions as in A). Gel shown in Figure S7.
Addition of either MG132 or the Sigma Aldrich protease inhibitor cocktails P-8340 or P-214 did not affect formation of the 19 kDa band (Figures 1B and S5 C). These observations suggest cleavage is not due to contaminating proteases. Addition of a tyrosinase inhibitor, kojic acid (1 mM), stopped formation of the 19 kDa fragment (Figure 2A), though a new band appears just below eDHFR (Figure S6 A). This new band contained the HA tag (Figure S6 B). Thus the N-terminal cleavage can be ascribed to an adventitous protease, whereas the HA cleavage is mediated by m-tyrosinase.
HA-tag cleavage proceeds via an o-quinone
We next sought to trap the putative o-quinone. Besthorn’s reagent is a monofunctionalized hydrazine species that reacts with o-quinones to give a stable pink complex.[40] The pink complex is considered to be diagnostic for o-quinones. Treatment of eDHFR-HA with Besthorn’s reagent in the presence of m-tyrosinase produced a pink complex resolvable as a single 25 kDa band on a denaturing SDS-PAGE gel (Figure 3A). Both HA-cleavage and protein cross-linking were inhibited by this reagent (Figure 3B), consistent with the o-quinone being required for both reactions. Numerous dyes/chemical tags are available as hydrazine derivatives, so this experiment also validates the o-quinone as a route to functionalized proteins. Importantly, this process is catalytic in m-tyrosinase and proceeds under mild conditions.
Figure 3. The o-quinone can be intercepted using Besthorn’s reagent.
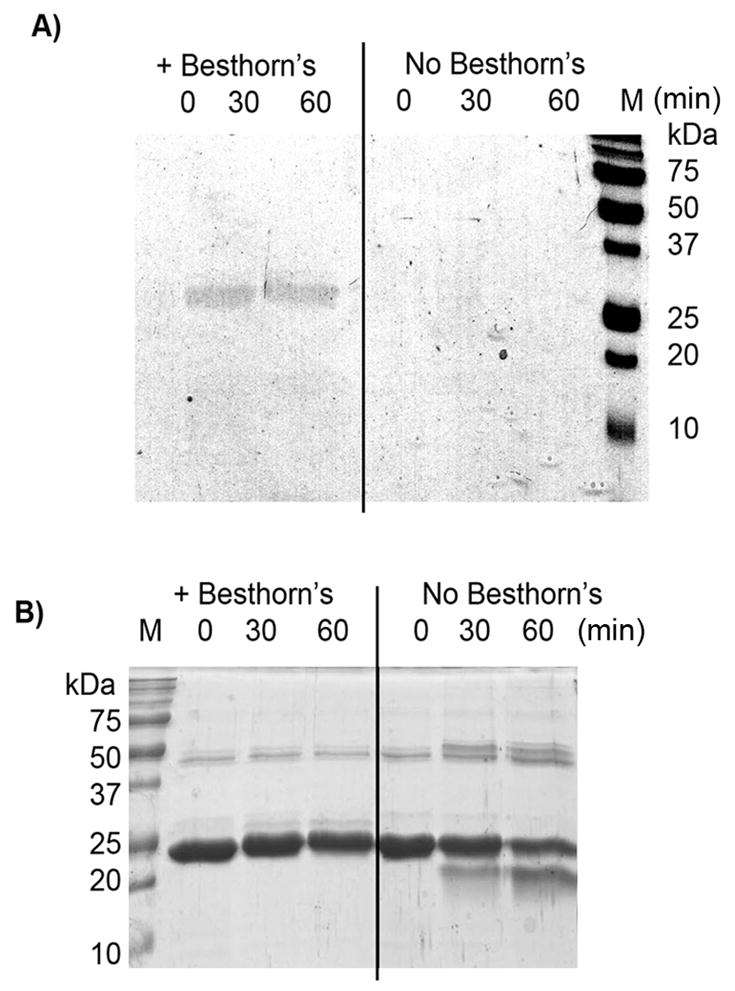
A) eDHFR-HA (1.5 mg/ml) was treated with m-tyrosinase (0.15 mg/ml) and Besthorn’s reagent (30μM) (left). In the control eDHFR-HA (1.5 mg/ml) was treated with m-tyrosinase (0.15 mg/ml) only (right). Aliquots were analyzed by SDS-PAGE without staining. B) The same gel was stained with Comassie. Apparent gain of mass in lanes 2–3 is probably due to Besthorn’s addition (2 x Besthorn’s ~ 400 Da; 3 x Besthorn’s ~ 600 Da). Note the change in mass is small relative to 0 time point in lane 4. A) is shown in color in Figure S14 G).
m-Tyrosinase specifically oxidizes the first and third tyrosines within the HA-tag
Six mutants were made in which tyrosines within the HA-tag (YPYDVPDYA) were mutagenized to phenylalanine (Figure 4A). The nomenclature for these mutants is defined relative to tyrosines within the HA-tag: eDHFR-HA is denoted YYY; whereas eDHFR-FPFDVPDFA is called FFF. As expected FFF did not form the 19 kDa fragment, react with Besthorn’s reagent or cross-link in the presence of m-tyrosinase (Figure S9). Surprisingly, only YFY underwent all three reactions (Figure 4B and C). FYY was the only other variant to react with m-tyrosinase, as evidenced by the formation of the pink complex with Besthorn’s reagent. However, this variant did not form the 19 kDa band. Thus, the first and third tyrosines within the HA-tag are required for the cleavage reaction, but only the third is required to form an o-quinone.
Figure 4. Mutagenesis experiments show that cleavage requires 1st and 3rd tyrosine in HA-tag.
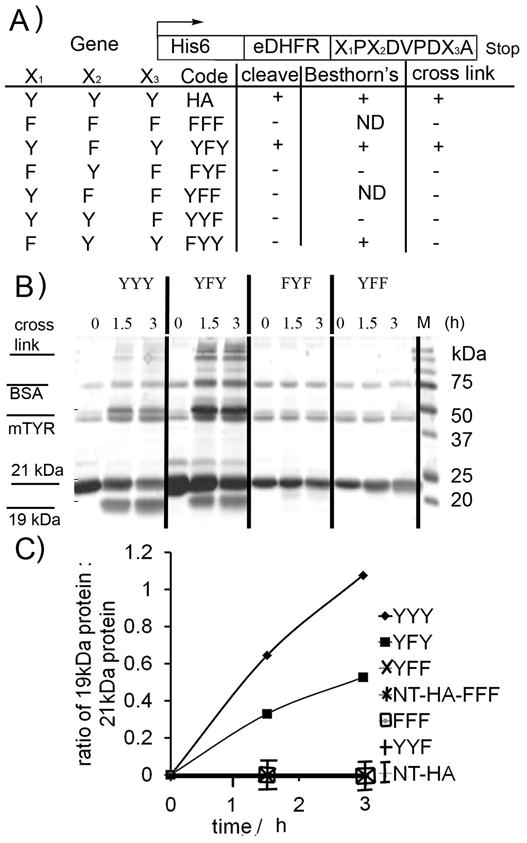
A) eDHFR proteins bearing C-terminal mutant HA-tags used in this study (ND = not determined). B) Representative Coomassie stained SDS-PAGE gel for action of m-tyrosinase (0.15 mg/ml) on mutants (1.5 mg/ml). C) Quantitation of cleavage of all mutants (from Coomassie stained SDS-PAGE gel).
NOTE only YYY and YFY show cleavage: all other mutants lie on X-axis (no cleavage). Conditions as per B). NOTE: In these samples trace BSA (residual from desalting column) was present.
HA-tag cleavage occurs in fusion proteins
Attention turned to assessing the chemical nature of the products of the cleavage reaction. Passing the reaction mixture of eDHFR-HA and m-tyrosinase through a YM-10 membrane and analyzing the flow through by LCMS showed no peaks (see suplementary methods). This is probably because the small fragmentation product undergoes further reactions with m-tyrosinase. We accordingly investigated a system in which the HA-tag was used as a linker, GFP-TEV-HA-GST-α1 (an enhanced green fluorescent protein human glutathione-S-transferase-α1 fusion). Cleavage of the fusion protein with TEV protease yielded fragments of ~25 kDa, one of which was recognized by anti-HA. m-Tyrosinase also cleaved the fusion protein to yield 25 kDa products (Figure 5A). However, these cleavage bands were not recognized by anti-HA antibodies.
Figure 5. Fusion proteins in which the HA-tag acts as a linker are substrates for m-tyrosinase and the product is oxidized.
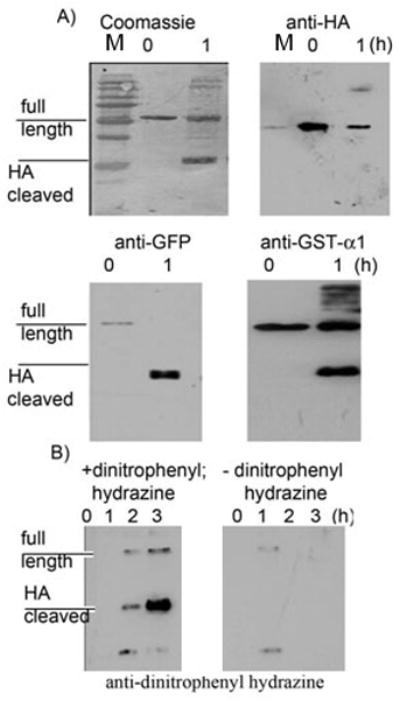
A) GFP-TEV-HA-GST-α1 (1.5 mg/ml) was treated with m-tyrosinase (0.15 mg/ml) and aliquots were extracted at the stated time points. These time points were analyzed using antibodies for HA, GFP and GST-α1 and also stained using Coomassie. B) As in A) but the progress was analyzed by Oxyblot(R), where sample is treated with dinitrophenylhydrazine to produce the hydrazone derivative that is subsesquently recognized by anti-dintrophenyl antibody.
HA-tag cleavage produces a product that is detected by oxyblot
α-Keto amides are known fragmentation products of peptides under oxidative conditions.[41] Ketones can be detected by an OxyblotR. Analysis of the reaction of GFP-TEV-HA-GST-α1 and m-tyrosinase revealed that cleavage bands had a high level of ketone content (Figure 5B). No ketones were detected in untagged eDHFR even after 2 hours exposure to m-tyrosinase. o-Quinones have a short half life thus the cleaved band recognized by the Oxyblot must contain a more stable ketone containing chemotype, such as an α-keto amide.
Although more complex mechanisms of hydrolysis cannot be ruled out, these results are consistent with the following mechanism (Scheme 2): m-tyrosinase converts tyrosine to DOPA, which is converted to the o-quinone. This species rearranges to give an acylenamine species with concomitant rearomatization of the arene ring. Similar intermediates have been observed in model systems by Rzepecki et al.[42] Importantly, Rzepecki’s findings indicate that dehydroDOPA formation is favored under conditions similar to ours (phosphate buffer pH 6–6.5). The acyl enamine is labile and is hydrolyzed [either via an acyl imminium ion (1), or by attack at the acyl group itself] to give an α-keto amide and a primary amide.
Scheme 2.
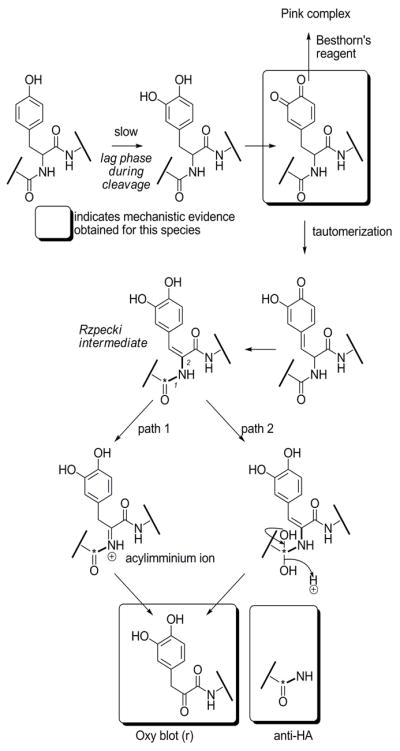
Proposed mechanism of HA-tag cleavage.
The o-quinone intermediate can be functionalized using nucleophilic reagents
To demonstrate selectivity of tyrosine modification, a lysate of E. coli expressing eDHFR-HA was treated with m-tyrosinase and Besthorn’s reagent (Figure 6). A control lysate of E. coli expressing human glutathione-S-transferase-μ (GST; a protein of similar size to eDHFR, bearing 12 tyrosines, none of which are in unstructured regions) was treated similarly. A time-dependent increase in pink color was observed only in the eDHFR-HA containing lysate. No pink color was observed in the control lysate even after 3 hours (Figure S12). Analysis of the lysate expressing eDHFR-HA showed a single 25 kDa pink band which increased in intensity as a function of time. Similar results were obtained with His6-Halo-HA (Figure S13).
Figure 6.

Functionalization of HA-tag using m-tyrosinase and Besthorn’s reagent is selective in E. coli lysates overexpressing a HA-tagged enzyme.
A) BL21 (DE3) E. coli lysate over expressing eDHFR-HA forms a single pink band when treated with m-tyrosinase (0.15 mg/ml) and Besthorn’s reagent (30 μM) and analyzed by SDS PAGE without staining. This gel is shown in color in Figure S13 A. B) Gel in A) stained with Coomassie.
The m-tyrosinase reaction was also used to introduced Cy5 labels into proteins. A single red band was produced when an E. coli lysate expressing Halo-HA was treated with m-tyrosinase and Cy5-hydrazide (Figure S13). Similarly, eDHFR-HA was selectively labeled with Cy5-hydrazide in lysates of HEK 293T cells (Figure S12 D). We also used this method to produce Cy5-labeled YFY and GST-α1-HA (Figure S13). These examples demonstrate the utility of m-tyrosinase-mediated protein modification.
Protein cross-linking catalyzed by m-tyrosinase can be favored by increasing the number of tyrosines within the tag
Recently, enzyme cross-linking has become of interest to identify protein-protein interactions.[43] Many methodologies involve metal-based oxidants which are not optimal.[44] As noted in the Introduction, m-tyrosinase catalyzes the polymerization of tyrosine via the o-quinone. We hypothesized that eDHFR-HA might be incorporated into tyrosine polymers. Adding 1 mM L-tyrosine to eDHFR-HA in the presence of m-tyrosinase led to rapid loss of the 21 kDa eDHFR-HA band (Figure S15 A). No cleavage was detected, indicating that the o-quinone is trapped prior to fragmentation.
In contrast, cleavage was still observed when 1 mM alanine was added to eDHFR-HA and m-tyrosinase. A similar lag period was also observed for this HA-tag cleavage reaction (Figure S15 B). This shows that the amine within α-amino acids is not reactive enough to trap the o-quinone. Thus, polymerization of eDHFR-HA in the presence of tyrosine probably involves o-quinone polymerization, as has previously been recorded.[45]
We decided to investigate whether increasing the number of tyrosines at the C-terminus of eDHFR would favor cross-linking. Thus, the HA-tag was replaced with a (GY)5-tag on eDHFR-HA m-Tyrosinase treatment produced cross-linked species which were of higher effective molecular weight than those observed with eDHFR-HA (Figure 7B). Importantly, enzyme activity was stable over the first 20 minutes of m-tyrosinase exposure, even though significant amounts of protein cross linking were observed (Figure 8 and S16). Enzyme activity was lost after prolonged exposure to m-tyrosinase (>1 hour). Finally, as above, treatment of eDHFR-(GY)5 with Besthorn’s reagent stopped the cross-linking reaction and produced a pink complex band (Figure 8C). Thus, this methodology represents a way to selectively functionalize or polymerize proteins by a simple change in conditions.
Figure 7. Cross-linking can be favored by using high substrate concentrations or by increasing the number of tyrosines within the tag.

A) eDHFR-HA was added to m-tyrosinase (0.15 mg/ml) and analyzed by SDS-PAGE as a function of time (left); B) eDHFR-(GY)5 (1.5 mg/ml) was added to m-tyrosinase (0.15 mg/ml). C) eDHFR-(GY)5 (1.5 mg/ml) was added to m-tyrosinase (0.15 mg/ml) and after 20 seconds Besthorn’s reagent (30 μM) was added (left) or DMSO was added (right). Time points were extracted and analyzed by SDS-PAGE without staining. D) gel from C) stained using Coomassie.
Figure 8. Initially activity of eDHFR-(GY)5 is not affected by action of m-tyrosinase, but prolonged exposure inhibits the enzyme.
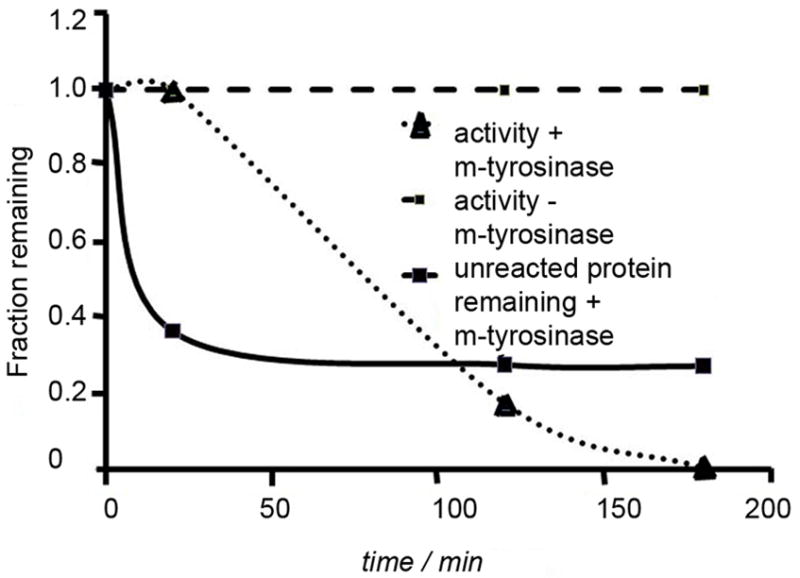
eDHFR-(GY)5 (5.0 mg/ml) was incubated with m-tyrosinase (0.15 mg/ml). At the designated time points aliquots were extracted and (1) assayed for residual activity and (2) added to loading buffer and analyzed by SDS-PAGE. Control represents a sample treated similarly, but without added m-tyrosinase.
Conclusion
This work demonstrates that the HA-tag and other unstructured tyrosine-rich protein tags are amenable to selective oxidation by m-tyrosinase. This mild process, which occurs at a pH of 6.6 at 37°C with catalytic m-tyrosinase, proceeds via an o-quinone intermediate. This o-quinone species can be used either as a cross-linking reagent, or as an alternative means to specifically functionalize proteins. Encouragingly, HA-tag functionalization by m-tyrosinase is selective in E. coli lysates overexpressing the HA-tagged protein and even in lysates of transfected mammalian cells. Since a wide array of proteins have been cloned with HA-tags, this methodology constitutes a general strategy to selectively functionalize proteins, for cross linking studies, immobilization or to investigate transient protein interactions. Importantly, the HA-tag is smaller than the equivalent FlAsH and PRIME tags.[46]
We have also discovered that m-tyrosinase treatment cleaves HA-tags. This process is most efficient in the absence of exogenous nucleophiles at low protein concentrations (< ~ 10 μM). This cleavage reaction is a novel function of m-tyrosinase. We propose that cleavage proceeds via an unusual mechanism involving oxidative fragmentation of the amino acid backbone. The proteolytic function is particularly interesting because phenoloxidases, such as tyrosinase, are implicated in the innate immune system of insects. [47] Whether the proteolysis function we have uncovered has any biological relevance, clearly, remains to be tested, but we hope this new finding will stimulate research in this area.
Experimental Section
Reagents were purchased from Sigma-Aldrich Chemical Company (St Louis, MO) and were of the highest grade. Water was purified on a Milli-Q apparatus (Millipore, Billerica, MA). Kinetic measurements were made on a Carey Bio 100. Protein concentrations were measured using Bradford Assay (Bio-RadR, Hercules, CA) using IgG as a standard. Quantitation of gel bands was made using Image-J (NIH). The following protease inhibitors were used: Sigma Aldrich protease inhibitor cocktails P-8340 and P-214 (both administered at 2x concentration) the former containing: 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF), pepstatin A, E-64, bestatin hydrochloride, leupeptin hemisulfate, and aprotinin; the latter AEBSF, aprotinin, bestatin hydrochloride, E-64, EDTA and Leupeptin hemisulfate). MG132 was from Aldrich and was shown to be active in a proteasome assay.
Cloning
eDHFR-HA was PCR amplified from pHis6-eDHFR-HA using primers that introduced the required mutations at the HA-tag: forward primer was consistent: reverse primer variable. pHis6-eDHFR-HA was linearized using EcoR1. The first PCR product was used to PCR clone the desired HA mutation into the linearized plasmid. All clones were verified by sequencing (Genewiz, Boston, MA).
Enzyme preparation and activity assessment
Bacteria were grown in Luria Broth at 37 °C. Induction was initiated at an OD600 of 1, with 500 μM IPTG at 30 °C (18 h). Bacteria were pelleted (10000 RPM; J-10 rotor) and then resuspended in lysis buffer (100 mM phosphate buffer, pH 7.6, 200 mM KCl, 5 mM imidazole, 1 mM BME) and sonnicated on ice (10 seconds of sonnication followed by 10 s for 3 minutes total). Lysate was clarified by centrifugation (15000 RPM; J-20 rotor). Enzymes were purified in a batch process at 4 °C using Ni-NTA (His-TrapR, Qiagen, Valencia, CA). Final elution: 150 mM imidazole in lysis buffer. Purified enzyme was dialyzed against 100 mM Tris, pH 7.8, 150 mM NaCl, 1 mM BME, 10 % glycerol three times at 4 °C. Tyrosinase activity was assessed using the method of Park et al.[48]
o-Quinone formation
Enzyme was dissolved at 1.5 mg/ml (unless otherwise stated) in 50 mM phosphate buffer pH 6.6 (assay buffer) and incubated at 37 °C for 20 minutes. Enzyme was desalted on Sephadex G10 resin (Amersham, Piscataway, NJ) (approx 1.2 ml resin volume on a bio-spin 6 column, Bio-RadR) that had been washed with 150 μL 0.1 % bovine serum albumin (BSA) in assay buffer and then washed 3 times (150, 100, 100 μL) with assay buffer prior to loading enzyme. Centrifugation was carried out at 800g for 1 minute. Typically no BSA was observed in the eluted sample. Trace BSA could be observed in some runs but it had no effect on cleavage or cross linking. After desalting, mushroom tyrosinase (Aldrich) was added to a final concentration of 0.15 mg/ml (based upon weight measurement of starting lyophilized sample). Aliquots were extracted at different time points and immediately added to SDS-PAGE loading buffer to make a final concentration of 2x. Samples were kept on ice or frozen until required. For o-quinone trapping experiments, the nucleophile (e.g. Besthorn’s) was added after all other ingredients were added.
Western Blotting
SDS-PAGE gel was run as per Laemelli. This was transferred onto a PVDF membrane (Millipore, immobilon) in Towbin buffer at 100 V (1 h), followed by 70 V (3 h). Membrane was blocked in 5 % milk in TBS-T (100 mM Tris pH 7.6, 150 mM NaCl-0.05% Tween-20). Membrane was immediately added to primary antibody in 1 % milk in TBS-T (2 h). Membrane was washed 3 times (10 min) in TBS-T, then exposed to the secondary antibody (40 min). Membrane was washed 2 times in TBS-T (10 min) then washed (15 min) in TBS. Membrane was treated with ECL-plus reagent (GE Healthcare, Piscataway, NJ) and visualized using film (Hy-Blot-CL, Denville Scientific, Metuchen, NJ). Oxy blot R kit was from Millipore and was used as per the manufacturer’s instructions.
Supplementary Material
Scheme 3.
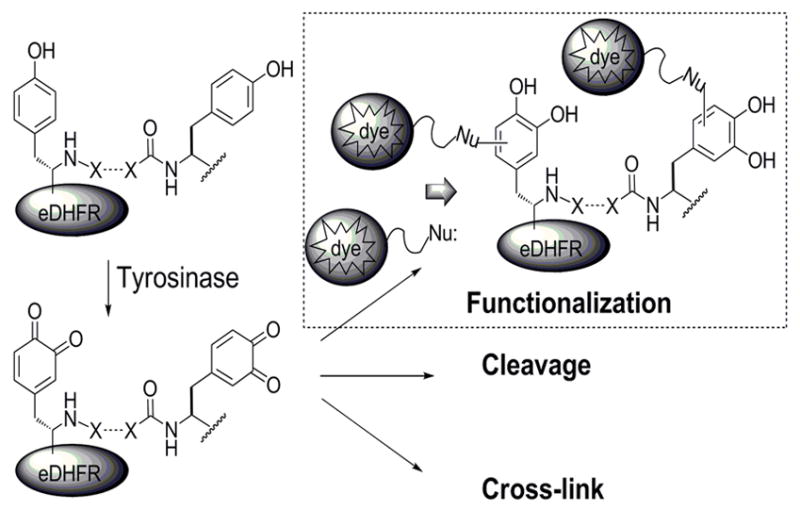
Possible fates of m-tyrosinase treated tyrosine-tagged proteins.
Acknowledgments
This work was supported by NIH grant GM054403 (LH). MJCL acknowledges a Howard Hughes Medical Institute international research fellowship. Dr Yimon Aye is acknowledged for help with graphics and proof reading.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Wang L, Magliery TJ, Liu DR, Schultz PG. J Am Chem Soc. 2000;122:5010–5011. [Google Scholar]
- 2.Muir TW, Sondhi D, Cole PA. PNAS. 1998;95:6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kaiser ET, Lawrence DS, Rokita SE. Ann Rev Biochem. 1985;54:565–595. doi: 10.1146/annurev.bi.54.070185.003025. [DOI] [PubMed] [Google Scholar]
- 4.Levine HJ, Kaiser ET. J Am Chem Soc. 1978;100:7670. [Google Scholar]; Hilbert D, Kaiser ET. Biotechnol Gen Eng Rev. 1987;5:297. doi: 10.1080/02648725.1987.10647841. [DOI] [PubMed] [Google Scholar]
- 5.Neet K, Nanci A, Koshland DE. J Biol Chem. 1968;243:6392. [PubMed] [Google Scholar]
- 6.Schmidt DF, Westheimer FH. Biochemistry. 1971;10:1249. doi: 10.1021/bi00783a023. [DOI] [PubMed] [Google Scholar]
- 7.Alam J, Keller TH, Loh TP. Chem Commun. 2011;47:9066–9068. doi: 10.1039/c1cc12926k. [DOI] [PubMed] [Google Scholar]
- 8.Antos JM, Francis MB. J Am Chem Soc. 2004;126:10256. doi: 10.1021/ja047272c. [DOI] [PubMed] [Google Scholar]
- 9.Zuk PA, Elferink LA. J Biol Chem. 2000;275:26754–26764. doi: 10.1074/jbc.M000344200. [DOI] [PubMed] [Google Scholar]
- 10.Los GV, EnCell LP, McDougall MG, Karassha N, Zimprich C, Wood MG, Learish R, Ohana RF, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 11.Jongsma MA, Litjens RH. Proteomics. 2006;6:2650. doi: 10.1002/pmic.200500654. [DOI] [PubMed] [Google Scholar]
- 12.Gilmore JM, Scheck RA, Esser-Kahn AP, Joshi NS, Francis MB. Angew Chem, Int Ed. 2006;45:5307. doi: 10.1002/anie.200600368. [DOI] [PubMed] [Google Scholar]
- 13.For Q-tag see: Lin C-W, Ting AY. J Am Chem Soc. 2006;128:4542–4543. doi: 10.1021/ja0604111.For PRIME, see: Uttamapinant C, White KA, Baruah H, Thompson S, Fernandez-Suarez M, Puthenveetil S, Ting AY. PNAS. 2010;107:10914–10919. doi: 10.1073/pnas.0914067107.
- 14.Yim J, Straight PD, McLoughlin SD, Zhou Z, Lin A, Golan DE, Kelleher NL, Kolter R, Walsh CT. PNAS. 2005;102:15815–15820. doi: 10.1073/pnas.0507705102.and Yin J, Lin AJ, Golan DE, Walsh CT. Nature Protocols. 2006;1:280–285. doi: 10.1038/nprot.2006.43.
- 15.Boyer PD. The Enzymes XIII. 3. 1976. For iodohistidine, see: Covelli I, Wolff I. J Biol Chem. 1966;241:4444–4451.
- 16.Wolff J, Covelli I. Biochemistry. 1966;5:867–871. doi: 10.1021/bi00867a009. [DOI] [PubMed] [Google Scholar]
- 17.Williams J, Lowe JM. Biochem J. 1971;121:203–209. doi: 10.1042/bj1210203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamada H, Yamashita T, Domoto H, Imoto T. J Biol Chem. 1990;108:432–440. doi: 10.1093/oxfordjournals.jbchem.a123218. [DOI] [PubMed] [Google Scholar]
- 19.Basle E, Joubert N, Pucheault M. Chemistry and Biology. 2010;17:213–227. doi: 10.1016/j.chembiol.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 20.For addition of cysteins to o-quinones, see: Slaughter DE, Hanzlik RP. Chem Res Toxicol. 1991;4:349. doi: 10.1021/tx00021a015.for addition of lipoic acid, see: Tsuji-Nato K, Hatani T, Okada T, Tehara T. Biochem Biophys Res Commun. 2006;343:15–20. doi: 10.1016/j.bbrc.2006.02.118.For radical addition to 1,2-dione moiety, see: Yamaguchi T, Fujita M. Angew, Chem, Int Ed. 2008;47:2067–2069. doi: 10.1002/anie.200705139.
- 21.Agrup G, Falck B, Kennedy BM, Rorsman H, Rosengren AM, Rosengren E. Acta Derm Venereol. 1975;55:1–3. [PubMed] [Google Scholar]
- 22.Ferreira SB, Kaiser CR, Ferreira VF. Synlett. 2008;17:2625–2628. [Google Scholar]
- 23.Weidman SW, Kaiser ET. J Am Chem Soc. 1966;88:5820–5827. [Google Scholar]
- 24.Malaprade L. Bull Chem Soc Fr. 1934;3:833. [Google Scholar]
- 25.Prota G. Melanins and Melanogenesis. Academic Press, Inc; 1992. [Google Scholar]
- 26.Ito S, Kato T, Shinpo K, Fujita K. Biochem J. 1984;222:407–411. doi: 10.1042/bj2220407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Napolitano A, Di Donato P, Prota G. J Org Chem. 2001;66:6958–6966. doi: 10.1021/jo010320g. [DOI] [PubMed] [Google Scholar]
- 28.Agrup G, Falck B, Kennedy BM, Rorsman H, Rosengren AM, Rosengren E. Acta Derm Venereol. 1975;55:1–3. [PubMed] [Google Scholar]
- 29.Kahn V, Ben-Shalom N. Pigment Cell Res. 1998;11:24–33. doi: 10.1111/j.1600-0749.1998.tb00707.x. [DOI] [PubMed] [Google Scholar]
- 30.Marumo K, Waite JH. Biochim et Biophy Acta Protein Structure and Molecular Enzymology. 1986;872:98–103. doi: 10.1016/0167-4838(86)90152-4. [DOI] [PubMed] [Google Scholar]
- 31.Corey JG, Frieden E. Biochemistry. 1967;6:116–120. doi: 10.1021/bi00853a020.and Corey JG, Frieden E. Biochemistry. 1967;6:121–126. doi: 10.1021/bi00853a021.
- 32.Takasaki S, Kawakishi S, Murata M, Homma S. Lebensmittel Wissenschaft und Technologie. 2001;24:507–512. [Google Scholar]
- 33.Fairhead M, Thony-Meyer L. J Biotechnol. 2010;150:546–551. doi: 10.1016/j.jbiotec.2010.10.068.For effects of pH and addition of caffeic acid on cross-linking, see: Thalmann C, Lotzbeyer T. Eur Food Res Tech. 2002;214:276–281.
- 34.Cory JG, Frieden E. Biochemistry. 1967;6:121–126. doi: 10.1021/bi00853a021. [DOI] [PubMed] [Google Scholar]
- 35.Mattinen ML, Lantto R, Selinheimo E, Kruus K, Buchert J. J Biotechnol. 2008;133:395–400. doi: 10.1016/j.jbiotec.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 36.Povirik LF. Mutat Res. 1996;355:71–89. doi: 10.1016/0027-5107(96)00023-1. [DOI] [PubMed] [Google Scholar]
- 37.Lerner AB, Fitzpatrick TB. Physiol Rev. 1950;30:91–126. doi: 10.1152/physrev.1950.30.1.91. [DOI] [PubMed] [Google Scholar]
- 38.Boyer RF, Mascotti PD, Schori BE. Phytochemistry. 1986;25:1281–1283. [Google Scholar]
- 39.Lerner AB, Fitzpatrick TB. Physiol Rev. 1950;30:91–126. doi: 10.1152/physrev.1950.30.1.91. [DOI] [PubMed] [Google Scholar]
- 40.Jacobsohn GM, Iskandar R, Jacobsohn MK. Biochemica et Biophysica Acta (BBA) Protein Structure and Molecular Enzymology. 1993;1202:317–324. doi: 10.1016/0167-4838(93)90022-j. [DOI] [PubMed] [Google Scholar]
- 41.Stadtman ER. Annu Rev Biochem. 1993;62:797–321. doi: 10.1146/annurev.bi.62.070193.004053. [DOI] [PubMed] [Google Scholar]
- 42.Rzpecki LM, Nagafuchi T, Waite JH. Arch Biochim Biophys. 1991;285:17–26. doi: 10.1016/0003-9861(91)90323-b. [DOI] [PubMed] [Google Scholar]
- 43.See: Fancy DA, Denison C, Kim K, Xie Y, Holdeman T, Amini F, Kodadek T. Chem Biol. 2000;7:697–708. doi: 10.1016/s1074-5521(00)00020-x.and Heijnis WH, Wierenga PA, Van Berkel WJH, Gruppen H. J Agric Food Chem. 2011;58:5692–5697. doi: 10.1021/jf100168x.
- 44.Kim K, Fancy DA, Carney D, Kodadek T. J Am Chem Soc. 1999;121:11896–11897. [Google Scholar]
- 45.For polymerization through o-diphenol/o-quinone reaction, see: Solano F, Hearing VJ, Garcia-Borron JC. Neurotoxicity Research. 1999;1:153–169. doi: 10.1007/BF03033287.quinone polymerization is often assumed to be the end point of quinones generated in water, but see Sugumaran M, Semensi V, Dali H, Mitchell W. Bioorganic Chemistry. 1989;17:86–95.for a critical account.
- 46.Kaganman I. Nature Methods. 2010;7:584. doi: 10.1038/nmeth0810-584. [DOI] [PubMed] [Google Scholar]
- 47.Gonzalez-Santoyo I, Cordoba-Aguilar A. Entomologia Experimentalis et Applicata. 2012;142:1–16. [Google Scholar]
- 48.Baek YS, Ryu YB, Curtis-Long MJ, Ha TJ, Rengasamy R, Yang MS, Park KH. Bioorg Med Chem. 2009;17:35–41. doi: 10.1016/j.bmc.2008.11.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.