

Abstract
Estrogen receptor-alpha (ER) antagonists have been widely used for breast cancer therapy. Despite initial responsiveness, eventually hormone-sensitive ER-positive cancer cells develop resistance to ER antagonists. It has been shown that, in most of these resistant tumor cells, the ER is expressed and continues to regulate tumor growth. Recent studies propose that tamoxifen initially acts as an antagonist but later functions as an ER agonist, promoting tumor growth. This suggests that targeted ER degradation may provide an effective therapeutic approach for breast cancers, even those which are resistant to conventional therapies. With this in mind, we previously demonstrated that PROTACs effectively induce degradation of the ER as a proof of concept experiment. Herein, we further refined the PROTAC approach to target the ER for degradation. The ER-targeting PROTACs are composed of an estradiol on one end and Hypoxia Inducing Factor 1α (HIF-1α)-derived synthetic pentapeptide on the other. The pentapeptide is recognized by an E3 ubiquitin ligase called the von Hippel Lindau tumor suppressor protein (pVHL), thereby recruiting the ER to this E3 ligase for ubiquitination and degradation. Specifically, the pentapeptide is attached at three different locations on estradiol to generate three different types of PROTACs. When the pentapeptide is linked through the C-7α position of estradiol, the resulting PROTAC showed the most effective ER degradation and best affinity for the estrogen receptor. This result provides an opportunity to develop a novel type of ER antagonist that may overcome the resistance of breast tumor to conventional drugs, such as tamoxifen and fulvestrant (Faslodex™).
Keywords: PROTACs, Proteasome, E3 ubiquitin ligase, Targeted degradation
Introduction
Breast cancer is the most common form of malignant disease in women worldwide. The majority of breast tumors, about two thirds of those initially diagnosed, are estrogen receptor-α(ER)-positive. In most cases the ER plays a significant role in the stimulation and growth of these breast tumors.[1, 2] For example, the protein level of the ER is elevated in premalignant and malignant breast lesions as opposed to normal tissue.[1] In addition, the clear correlation between ER positive breast tumors and their response to antiestrogen therapy has been demonstrated.[3]
Most ER-positive tumors initially respond well to antiestrogens, which block the site to which estrogen can bind thereby halting the growth of cancer cells. Tamoxifen, a non-steroid selective ER modulator (SERM), is an antiestrogen therapeutic which has been widely used for more than 20 years in all stages of ER-positive breast cancers.[4, 5] Specifically, tamoxifen induces a conformational change of the ER that blocks the ER’s function.[6] Another example is fulvestrant (Faslodex™),[7, 8] which is a pure steroidal antiestrogen approved for the treatment of postmenopausal women with hormone-sensitive, advanced or metastatic breast cancers. Like tamoxifen, fulvestrant competitively binds the ER and blocks ER-promoted cell proliferation. While tamoxifen causes the accumulation of the ER, fulvestrant, like ER-targeting PROTAC molecules, induces selective degradation of the ER mediated by the proteasome and therefore does not exhibit the agonistic effects commonly associated with SERMs.[9] Unfortunately, despite the relative safety and significant antineoplastic activities of these antiestrogens, most initially responsive breast tumors acquire resistance.[10–14] As such, the major challenge is to develop novel therapeutic agents that not only inhibit the growth of hormone-sensitive breast tumors but also prevent the development of drug-resistance.
While the mechanism by which ER-positive cancer cells acquire drug-resistance is not clearly understood, it is unlikely that any single mechanism or gene confers antiestrogen resistance. Given that less than 25% of tumors that recur following treatment with fulvestrant or tamoxifen lack the ER,[15, 16] ER loss does not seem to be the major mechanism driving acquired resistance. It has also been shown that a loss of antiestrogen responsiveness by initially sensitive tumors is unlikely due to mutations or deletion of the ER gene.[1, 14] In fact, most tumors at recurrence retain levels of ER expression that would define them as ER-positive.[17–21] In addition, the ER appears to maintain the ability to continuously regulate tumor growth in most antiestrogen-resistant tumors.[20] It has been suggested that one likely cause of tamoxifen-resistance is the ability of tumors to switch from recognizing an antiestrogen as an antagonist to recognizing it as an agonist, perhaps through differential coregulator recruitment to the ER.[22–26] For fulvestrant, a conformational change in the ER is suggested to affect the binding of coregulator(s) and influence the stability of the ER, thereby causing its rapid degradation. Additionally, fulvestrant causes a loss of the ER protein without affecting ER mRNA levels.[27, 28] Consistent with this observation, it has been reported that resistance of human breast cancer cells to fulvestrant is not associated with a general loss of ER expression or lack of estrogen responsiveness. [29] Taken together, it seems that tamoxifen and fulvestrant maintain their ER-binding affinity despite the loss of their antiproliferative or ER-antagonistic effects, and that the ER continues to regulate the growth of resistant ER-positive breast cancer cells. Consequently, a small molecule that targets the ER for degradation, regardless of changes that induce drug resistance, may prove useful as an alternative therapeutic option for ER-positive breast cancer therapy.
Towards this end, we have utilized a novel small molecule-based technology termed “PROteolysis TArgeting ChimeraS (PROTACs)” to target the ER for degradation at the post-translational level. PROTAC molecules are composed of three major components (Figure 1A); a small-molecule ligand for a targeted protein, an E3 ubiquitin ligase recognition motif, and a linker. For the E3 ligase recognition domain, a 12-mer polypeptide containing two phophoserines (derived from IκBα) was initially used as a proof of concept.[30] However, the poor bioavailability of the 12-mer PROTAC necessitates microinjection for delivery, hence limiting its use as a molecular probe. To circumvent this, an alternative E3 recognition motif was required, so HIF-1α polypeptides were developed towards this end. A pentapeptide was found by our group and others to be sufficient for HIF-1α-based PROTACs to cross cell membranes and effectively degrade target proteins.[31–33]
Figure 1.
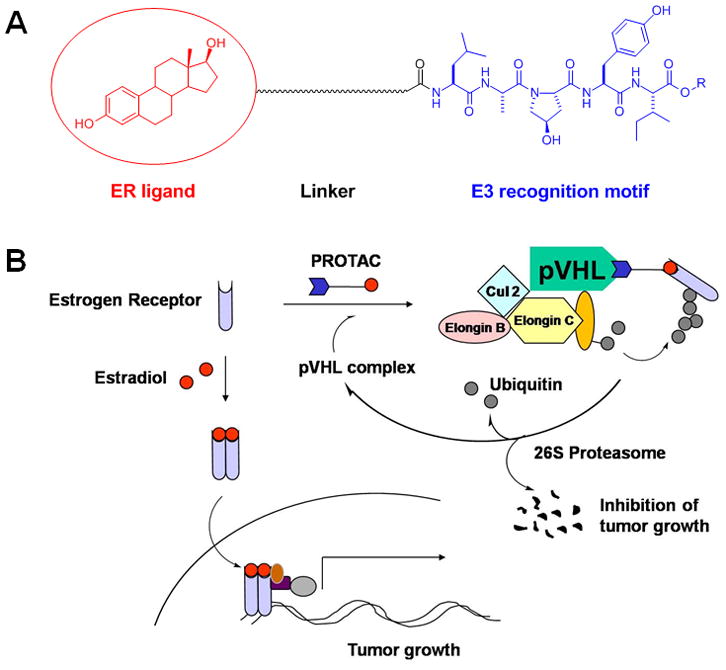
PROTAC-induced targeted degradation of the ER. A. The PROTAC is composed of three components: an E3 ligase recognition motif, linker, and ligand (in this case, targeting the ER). B. The PROTAC recruits the ER to the pVHL E3 ligase complex for ubiquitination and subsequent degradation by the 26S proteasome. This degradation of the ER should decrease the amount of dimerized ER bound to estradiol available to drive gene transcription, a major factor in the growth and survival of breast cancers.
Previously, we developed PROTACs that target the ER by using estradiol as the ligand and the optimized pentapeptide as the E3 recognition motif. These proof of concept PROTACs utilized an ester bond to attach the linker moiety to the O-17 position of estradiol. A significant drawback to this attachment site is its vulnerability to esterases, which would inactivate the PROTAC as well as release estradiol. Another critical issue with the O-17-linked PROTAC was the loss of the 17-OH group, which is reported to be important for ER binding of E2.[34] Therefore, E2-based PROTACs that can spare the 17-hydroxyl group are desirable for the design of improved PROTACs targeting the ER.
Other potential positions for derivation from 17β-estradiol have been reported in the literature. A geldanmycin tagged estradiol compound derivated at the C-16 position on estradiol was reported to maintain its interaction with the ER.[35, 36] Similarly, derivation at the C-7a position of estradiol has been shown to preserve its binding affinity with the ER.[7, 8, 37] For instance, fulvestrant has a high ER binding affinity, and it is an E2 derivative with a long hydrocarbon tail at the C-7α position. Thus, we endeavored to create a more stable linker by attaching the pentapeptide at the C-16 and C-7 positions of estradiol to prepare PROTACs (Scheme 1). In this report, we show a rational design and optimization of PROTACs that target the ER for ubiquitination and degradation. The information contained herein also provides an example for PROTAC design which will produce molecules with superior effects in cell-based systems.
Scheme 1.
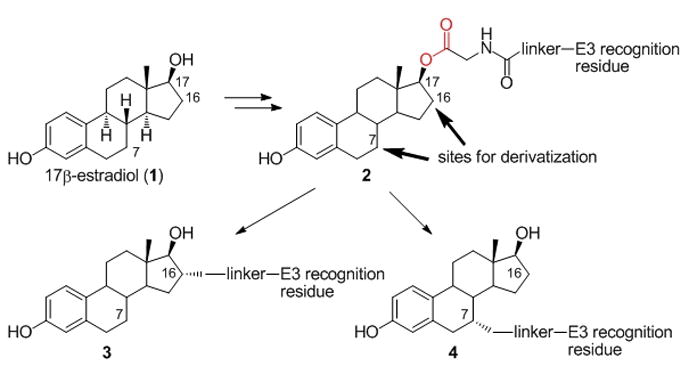
Synthetic strategy for next-generation ER-targeting PROTACs. Attaching the linker via a carbon-carbon bond should produce PROTACs having higher ER binding affinity and stability in living cells than the parent ER-targeting PROTAC 2.
Results and Discussion
Synthesis of PROTACs
The parent ER-targeting PROTAC 2 was synthesized as described.[38] In addition, PROTAC 2 was also deprotected at the C-terminal of the pentapeptide to give PROTAC 2*, using the same procedure as below (13 to 14). To synthesize PROTAC 3, a ‘free amine handle’ was introduced at the C-16α position of E2 (17β-estradiol) via a carbon-carbon linkage, following a similar procedure to the one previously reported (Scheme 2).[36, 39, 40] First, estrone (5) was treated with tert-butyldimethylsilyl chloride (TBDMSCl) to yield compound 6. Alkylation at the C-16 position of 6 was carried out using lithium diisopropylamide (LDA) and 1,4-dibromo-2-butene. The resulting alkyl bromo residue was converted to the azido compound, 8, by treatment with sodium azide. Sequential reduction of the keto and azide groups with sodium borohydride (NaBH4) and lithium aluminium hydride (LiAlH4) yielded the free amine “handle”, 9. The “handle” was further extended with glycine to give 11, which was activated to the succinimidyl ester 12 using disuccinimidyl suberate (DSS). 12 was finally coupled with the HIF-1α pentapeptide, and the TBDMS group was deprotected with tetrabutylammonium fluoride (TBAF) to yield PROTAC 13. The benzyl group of PROTAC 13 was removed by hydrogenolysis to obtain PROTAC 14.
Scheme 2.
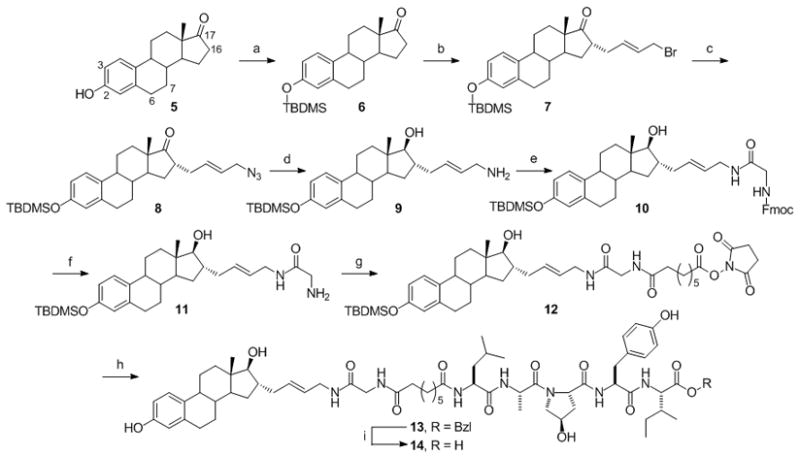
Synthesis of the C-16 derivatized estradiol-based PROTACs. (a) TBDMSCl, imidazole, 99% (b) LDA, C4H6Br2, THF, 45% (c) NaN3, THF, DMSO, H2O, 96% (d) 1. NaBH4, MeOH; 2. LiAlH4, THF, 35% (e) Fmoc-Gly-OH, HBTU, DIPEA, CH2Cl2, 47% (f) 20% piperidine in DMF, 67% (g) DSS, DMF, 38% (h) 1. HIF-1α pentapeptide, DMAP, DMF; 2. TBAF, THF, 47% (i) H2/Pd-C, EtOAc, MeOH, 89%.
As was the case for the PROTAC 13, the most critical step for the synthesis of PROTAC 24 is the introduction of a primary amine “handle” at the C-7α position of estradiol (Scheme 3). A procedure similar to that reported by Skaddan and Katzenellenbogen[41, 42] was followed to introduce this “handle”. As a first step, THP-protected 15 was synthesized following a procedure similar to that previously described.[43–45]
Scheme 3.
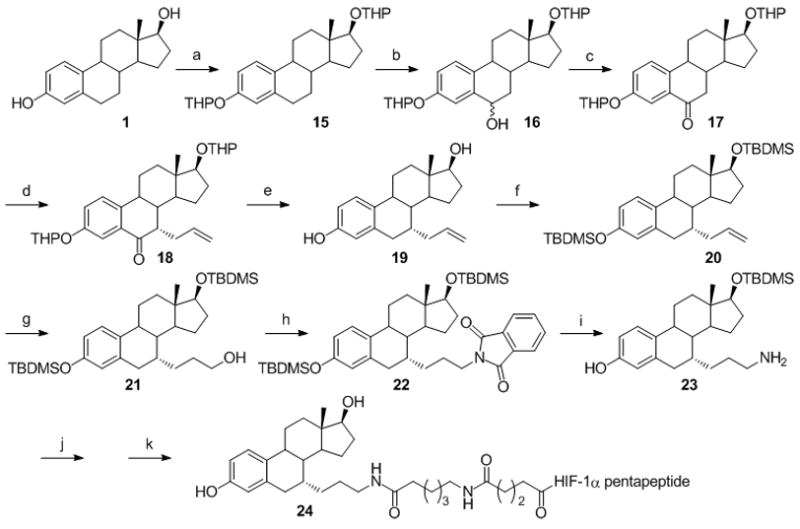
Synthesis of an estradiol containing the amine functional group at the C-7 position. (a) DHP/TsOH, 94% (b) 1. LiDAKOR; 2. B(OMe)3; 3. H2O2, 66% (c) PCC, 80% (d) 1. KOtBu, C3H5I, 2. NaOMe, 36% (e) Et3SiH, BF3.Et2O, 90% (f) TBDMSCl/imidazole, 85% (g) 1. 9-BBN; 2. H2O2, KOH, 66% (h) PPh3, DIAD, Phthalimide, 83% (i) NH2NH2, 76% (j) Disuccinimidyl glutarate, 37% (k) 1. HIF-1α pentapeptide; 2. TFA, 25% over two steps. HIF-1α pentapeptide = -Leu-Ala-ProOHTyr-Ile-Bzl.
Deprotonation at the C-6 position under “superbase” conditions followed by treatment with trimethyl borate yielded an intermediate borate ester which was oxidized using hydrogen peroxide to provide 16 as epimers. Further oxidation of 16 with PCC afforded the keto functionality at the C-6 position in 17. Treatment of 17 with potassium tert-butoxide (KOtBu) and allyl iodide afforded 18. Simultaneous deprotection of the THP groups and the C-6 keto functionality was accomplished using triethylsilane (Et3SiH) and boron trifluoride diethyl etherate (BF3.Et2O) to give 19. The free OH groups were reprotected with tert-butyldimethylsilyl (TBDMS) to give 20, which was then treated under hydroboration-oxidation conditions to produce the alcohol, 21. Treatment of 21 with phthalimide under Mitsunobu conditions followed by hydrazinolysis provided the primary amine “handle”, 23. The “handle”, 23, was further extended and activated using disuccinimidyl suberate (DSS). Sequential coupling of HIF-1α pentapeptide and deprotection of TBDMS groups yielded PROTAC 24.
Biological Characterization of PROTACs
These PROTACs (2, 2*, 13, 14, 24) were tested for their ability to degrade the endogenous ER in MCF7 breast cancer cells. After 48 hour incubation with PROTACs, their effects on ER protein levels were evaluated using western blotting analysis and immunofluorescence (IF).
Previous reports indicated that the O-17 linked PROTAC 2 would degrade the ER in a proteasome-dependent manner.[31, 32, 38] Our initial data confirmed this finding (Fig 2A) while demonstrating that the C-terminal protected PROTAC 2 was superior to the deprotected PROTAC 2*. A major drawback to these compounds is their susceptibility to endogenous esterases, so we moved forward with the testing of carbon-based linkages to confirm the results from these proof of concept PROTACs. A similar trend in ER degradation was found when the C-16α-based PROTACs (13 & 14) were tested, with the protected pentapeptide providing superior degradation (Fig 2B). The C-7α-based PROTAC (24) showed a comparable ability to degrade the ER when compared the C-16α-based PROTAC 13 (Fig 2B).
Figure 2.
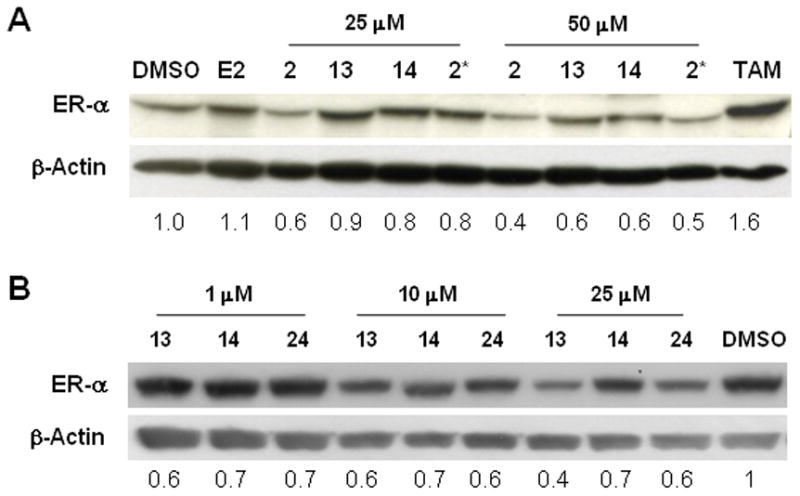
C-terminal protection of the HIF-1α pentapeptide provides improved degradation of the ER by PROTACs linked at any position on estradiol. A. O-17 (2 and 2*) and C-16 (13 and 14) derivatives of E2-based PROTACs with the benzyl protected HIF-1α pentapeptide (2 and 13) are more effective in degrading the endogenous ER than benzyl deprotected PROTACs (2* and 14). B. A similar pattern was observed for the C-7 derivative of E2-based PROTAC, as the benzyl protected compounds (13 and 24) showed greater ER degradation than the deprotected compound (14).
Next, we further confirmed the PROTAC-mediated degradation of the ER in MCF7 cells using immunofluorescence studies. Estradiol (E2) was used as a positive control. It should be noted that E2 is shown to induce the proteasome-mediated degradation of the ER as a result of transcriptional activation, resulting in cell proliferation. The ER degradation by E2 was blocked by co-treatment with epoxomicin, a natural product proteasome-specific inhibitor. Compared to DMSO control, the attenuation of ER signal (green) was observed with the treatment of 13 and 24 (Fig 3A, top row). These compounds showed an accumulation of the green signal when co-treated with epoxomicin, which is also seen in control/epoxomicin treated cells (Fig 3A, bottom row). This result was confirmed via western blotting using 24 and E2 (Fig 3B). To validate the superior position for PROTAC linkage, a competitive ligand binding affinity assay was performed. This in vitro data indicated that the C-7α-based PROTAC (24) had the highest affinity of the E2-based PROTAC compounds tested, superior even to tamoxifen. The binding data for the C-16α-based PROTACs 13 and 14 also support the data from Figure 2 which indicates that benzyl protection of the C-terminal of the pentapeptide gives a superior compound. Since a PROTAC with a higher binding affinity for the target protein should induce optimal ubiquitination and subsequent degradation by the proteasome, further optimization of the PROTAC approach for the ER should utilize the C-7α linkage.
Figure 3.
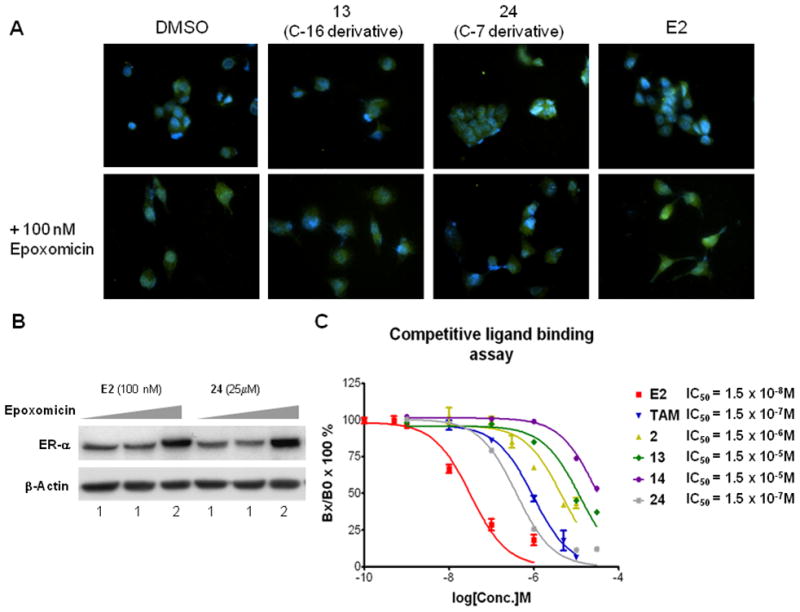
PROTACs bind to the ER and cause its proteasome-mediated degradation. A. Immunofluorescent images of MCF7 cells after 48h treatment with 25 μM PROTACs, 10 nM E2, or DMSO vehicle. The ER is visualized by conjugation with FITC (green) and DAPI staining (blue) indicates the nucleus. Co-treatment with epoxomicin causes accumulation of the ER. B. Western blot data demonstrating the proteasome-dependence of PROTAC-mediated ER degradation. C. Competitive binding assay indicating that the C-7 PROTAC is a superior position for derivation due to its enhanced binding to the ER when compared to the O-17 and C-16α-based PROTACs.
Conclusion
The ubiquitin-proteasome degradation pathway is a destructive, irreversible process and controls many important biological processes such as cell cycle progression, differentiation, and inflammation. We have shown previously that the ubiquitination machinery in cells can be hijacked by a PROTAC containing a pentapeptide that is recognized by the E3 ligase pVHL, and that a PROTAC containing a ligand of a target protein induces ubiquitination and degradation of that protein in living cells.[31, 38, 46] This ‘chemical knockout’ approach provides flexible spatial and temporal control, which is critical to dissect complex signaling pathways in cells.
As many diseases are driven by the expression of a few proteins, a potential therapeutic strategy is to remove these essential proteins. Small interfering RNAs (siRNAs), which knock down the expression of a gene of interest by destroying its mRNAs, have drawn considerable attention as a potential therapeutic approach.[47, 48] However, this siRNA knock-down strategy has some inherent problems,[47–49] such as off-target effects, sequence-independent effects, activation of unrelated signaling pathways, and drug delivery. The PROTAC approach does not seem to suffer these same limitations and thus may provide an additional strategy to treat diseases by destroying disease-promoting proteins.
As ER-positive breast cancers require ER-promoted cell proliferation, the current therapeutic paradigm relies on the blockage of this hormone-mediated cell growth. Thus, a strategy which allows for the selective degradation of the ER could provide a valuable and novel therapeutic option for many breast cancer patients. Additionally, most breast cancers that develop resistance to current treatments retain functional ERs. Optimized PROTACs, which induce the degradation of the ER, may thus provide an additional therapeutic option for the treatment of tamoxifen or fulvestrant-resistant breast tumors.
In summary, we have shown that a chimeric small molecule induces the proteasome-dependent degradation of the ER in living cells. Whether the targeted degradation of ER is useful in treating hormone-sensitive or antiestrogen-resistant breast tumors remains to be determined. Generally, PROTACs should be optimized by determining a position for derivation which retains a maximal binding affinity. Additionally, pentapeptide-based PROTACs benefit from C-terminal protection of the peptide regardless of the linker position. Finally, the small molecule strategy reported here presents a generic approach to target any cancer-promoting protein for degradation.
Experimental Section
Chemical Reagents
All chemical reagents were purchased from Aldrich Chemical Co. PROTAC 2 was synthesized following a procedure similar to that previously reported.[50] Epoxomicin was synthesized as previously reported.[51] For more detailed information on the chemical synthesis, see the supporting information.
Biological Reagents
Fetal bovine serum (FBS), RPMI 1640, phenol-red free RPMI, conjugated secondary antibody Alexa Fluor 488 (FITC), antibiotics, Hank’s Balanced Salt Solution (HBSS), goat serum, Prolong Gold antifade w/ DAPI, recombinant human estrogen receptors (ER-α and trypsin-EDTA were purchased from Invitrogen (Carlsbad, CA). 17β-estradiol, tamoxifen, Kodak XAR film, Sodium Chloride, Nonidet-P40 (NP40), Protease inhibitors cocktail, TWEEN® 20, Ethanol, Bovine Serum Albumin (BSA), and 2X Laemmi sample buffer were obtained from Sigma Aldrich (St. Louis, MO). Charcoal-dextran treated FBS was purchased from Hyclone Co. (Logan, UT). Anti-ER antibodies were acquired from Santa Cruz (Santa Cruz, CA) for western blotting and Abcam (Cambridge, MA) for immunofluorescence while the anti-beta actin antibody was purchased from Novus Biologicals Inc. (Littleton, CO). Anti-mouse IgG horseradish peroxidase was obtained from Zymed Laboratories (South San Francisco, CA). The anti-rabbit IgG horseradish peroxidase and enhanced chemiluminescence detection reagents (ECL) were acquired from GE Healthcare (Piscataway, NJ). Protein Assay Dye Reagent Concentrate, Tris-Chloride, Triton X-100, Sodium Dodecyl Sulfate (SDS), and PVDF membranes were purchased from Bio-Rad (Hercules, CA). Methylsulfoxide (DMSO) and potassium chloride were obtained from EMD (Darmstadt, Germany) while the CellTiter 96 Aqueous One Solution Cell Proliferation Assay was purchased from Promega (Madison, WI). Para-formaldehyde (PFA) was purchased from Fisher Scientific while potassium phosphate monobasic and potassium phosphate dibasic were acquired from Mallinckrodt Baker (Phillipsburg NJ). Antibody Dilutant w/ Background Reducing Components was purchased from DAKO (Glostrup, Denmark) while [6, 7-3H 17] estradiol was purchased from Amersham Biosciences (Buckinghamshire, United Kingdom).
Cell Culture
The MCF7 human breast cancer cell line was purchased from the American Type Culture Collection (Manassas, VA). MCF7 cells were maintained in RPMI 1640 (Gibco, Carlsbad, CA) medium containing 10 % (v/v) FBS (Gibco, Carlsbad, CA) and 100 U/mL penicillin-100 ug/mL streptomycin (Gibco, Carlsbad, CA). All experiments were performed when the cells were 70% confluent and had been maintained in 5% (v/v) charcoal-dextran treated FBS RPMI with antibiotics for at least 24 hours. Compounds were treated in a DMSO vehicle at the appropriate dilutions for 48 hours unless noted otherwise.
Western Blotting
Whole cell lysates were prepared by incubating cells in nondenaturing lysis buffer (50mM Tris-Cl, 150mM NaCl, 1% NP40, 1% Triton X-100, and 1% protease inhibitor cocktail) on ice for 1 hour. Cells were then centrifuged with supernatants collected and subjected to protein assay via the Bradford method. The sample was mixed with an equal volume of 2X Laemmi sample buffer and heated in boiling water for 10 min. Equal protein concentrations of the samples were subjected to a 10% SDS polyacrylamide gel, electrophoresed, and blotted onto a PVDF membrane. After blocking, the membranes were incubated overnight at 4°C in primary antibody and for 1h at room temperature with secondary antibody. Antibody binding was detected using ECL and film. All membranes were then reprobed with mouse anti-beta-actin to ensure equal protein loading.
Cell Viability Assay by MTS
MCF7 cells were plated at a density of 5 × 103 cells per 96 well plates in RPMI 1640 with 10 % FBS and left overnight. The media was changed to 5% Charcoal RPMI for 24 hours prior to the addition of compounds. The proliferation rate of the cells was determined after 48 hours by using the CellTiter 96 Aqueous One Solution Cell Proliferation Assay according to the supplier’s instruction. Absorbance was measured at 490nm on a microplate reader using the KC4™ program. In Graph Pad Prism™ (San Diego, CA), IC50 values were obtained from at least triplicate results via a sigmoid dose-response curve using a nonlinear regression to a logarithmic function.
ER Binding Affinity Assay
Competitive ligand binding assays were performed according to the manufacturer’s protocol (Invitrogen). 10nM of purified human recombinant estrogen receptor was added to 20nM [3H]-labeled estradiol and the indicated concentrations of estradiol or PROTACs. After an incubation (2 hr at room temperature or overnight at 4°C), a 50% hydroxy apatite slurry was added to bind the receptor/ligand complex. The sample was centrifuged and the pellet resuspended for analysis of tritium activity using scintillation counting. The percent specific binding affinity was calculated and IC50 values were obtained using one site competition curve analyses provided by Graph Pad Prism™. Relative binding affinity (RBA) was calculated using the following equation, RBA = {IC50 (E2)/IC50 (sample)} × 100.
Immunofluorescence
Coverslips were sterilized with ethanol and UV light exposure. MCF7 cells were added directly to the coverslip and allowed to attach for 24 hours. The media was then changed to phenol-red free RPMI with 5% Charcoal/Dextran-treated FBS until treatment with compounds. Compounds were diluted in the phenol-red free media, and treated as before. Cells were then fixed with 4% PFA, washed with Phosphate Buffered Saline (PBS), and permeablized with 0.2% Triton-X in PBS. Between all subsequent steps coverslips were washed in PBST (PBS with 0.05% Tween-20). Coverslips were blocked in 10% goat serum with 0.1% BSA in PBST. Next, primary antibody was added in the antibody dilutant directly to the coverslip, then secondary antibody was added in the same way. Prolong Gold with DAPI was added to clean slides to mount the coverslips and the mounted slides were allowed to dry overnight. After drying, the coverslips were rimmed with clear nail polish and visualized on an inverted fluorescence microscope (Nikon Ti-U microscope) with NIS Element Research image analysis software.
Supplementary Material
Acknowledgments
This work has been supported by Graduate School Academic Year, Kentucky Opportunity, and American Foundation for Pharmaceutical Education Fellowships (M.W.). K. B. K. gratefully acknowledges the support of the Markey Cancer Foundation, NIH (CA131059), and DoD (W81XWH-08-1-0092). H. S. thanks the NIH for financial support (ES014849). We thank members of the Swanson, Lee, and Kim laboratories for their helpful comments on the manuscript. We are grateful to Dr. Wooin Lee for helping with the collection of fluorescent images.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org or from the author.
Contributor Information
Prof. Dr. Hollie Swanson, Email: hswan@uky.edu.
Prof. Dr. Kyung-Bo Kim, Email: kbkim2@uky.edu.
References
- 1.Sommer S, Fuqua SA. Semin Cancer Biol. 2001;11:339. doi: 10.1006/scbi.2001.0389. [DOI] [PubMed] [Google Scholar]
- 2.Omoto Y, Kobayashi Y, Nishida K, Tsuchiya E, Eguchi H, Nakagawa K, Ishikawa Y, Yamori T, Iwase H, Fujii Y, Warner M, Gustafsson JA, Hayashi SI. Biochem Biophys Res Commun. 2001;285:340. doi: 10.1006/bbrc.2001.5158. [DOI] [PubMed] [Google Scholar]
- 3.Rutqvist LE, Cedermark B, Fornander T, Glas U, Johansson H, Nordenskjold B, Rotstein S, Skoog L, Somell A, Theve T, et al. J Clin Oncol. 1989;7:1474. doi: 10.1200/JCO.1989.7.10.1474. [DOI] [PubMed] [Google Scholar]
- 4.Jordan VC. J Natl Cancer Inst. 2003;95:338. doi: 10.1093/jnci/95.5.338. [DOI] [PubMed] [Google Scholar]
- 5.Jordan VC. Cancer Cell. 2004;5:207. doi: 10.1016/s1535-6108(04)00059-5. [DOI] [PubMed] [Google Scholar]
- 6.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, Greene GL. Cell. 1998;95:927. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 7.Wakeling AE. Cancer Treat Res. 1991;53:239. [PubMed] [Google Scholar]
- 8.Wakeling AE, Bowler J. Biochem Soc Trans. 1991;19:899. doi: 10.1042/bst0190899. [DOI] [PubMed] [Google Scholar]
- 9.Wakeling AE. Endocr Relat Cancer. 2000;7:17. doi: 10.1677/erc.0.0070017. [DOI] [PubMed] [Google Scholar]
- 10.Larsen SS, Heiberg I, Lykkesfeldt AE. Br J Cancer. 2001;84:686. doi: 10.1054/bjoc.2000.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clarke R, Leonessa F, Welch JN, Skaar TC. Pharmacol Rev. 2001;53:25. [PubMed] [Google Scholar]
- 12.Osborne CK, Coronado-Heinsohn EB, Hilsenbeck SG, McCue BL, Wakeling AE, McClelland RA, Manning DL, Nicholson RI. J Natl Cancer Inst. 1995;87:746. doi: 10.1093/jnci/87.10.746. [DOI] [PubMed] [Google Scholar]
- 13.Howell A, DeFriend D, Robertson J, Blamey R, Walton P. Lancet. 1995;345:29. doi: 10.1016/s0140-6736(95)91156-1. [DOI] [PubMed] [Google Scholar]
- 14.Dorssers LC, Van der Flier S, Brinkman A, van Agthoven T, Veldscholte J, Berns EM, Klijn JG, Beex LV, Foekens JA. Drugs. 2001;61:1721. doi: 10.2165/00003495-200161120-00004. [DOI] [PubMed] [Google Scholar]
- 15.Bachleitner-Hofmann T, Pichler-Gebhard B, Rudas M, Gnant M, Taucher S, Kandioler D, Janschek E, Dubsky P, Roka S, Sporn E, Jakesz R. Clin Cancer Res. 2002;8:3427. [PubMed] [Google Scholar]
- 16.Kuukasjarvi T, Kononen J, Helin H, Holli K, Isola J. J Clin Oncol. 1996;14:2584. doi: 10.1200/JCO.1996.14.9.2584. [DOI] [PubMed] [Google Scholar]
- 17.Encarnacion CA, Ciocca DR, McGuire WL, Clark GM, Fuqua SA, Osborne CK. Breast Cancer Res Treat. 1993;26:237. doi: 10.1007/BF00665801. [DOI] [PubMed] [Google Scholar]
- 18.Johnston JA, Johnson ES, Waller PR, Varshavsky A. J Biol Chem. 1995;270:8172. doi: 10.1074/jbc.270.14.8172. [DOI] [PubMed] [Google Scholar]
- 19.Johnston SC, Larsen CN, Cook WJ, Wilkinson KD, Hill CP. Embo J. 1997;16:3787. doi: 10.1093/emboj/16.13.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, Skaar TC, Gomez B, O’Brien K, Wang Y, Hilakivi-Clarke LA. Oncogene. 2003;22:7316. doi: 10.1038/sj.onc.1206937. [DOI] [PubMed] [Google Scholar]
- 21.Ali S, Coombes RC. Nat Rev Cancer. 2002;2:101. doi: 10.1038/nrc721. [DOI] [PubMed] [Google Scholar]
- 22.Cui Y, Parra I, Zhang M, Hilsenbeck SG, Tsimelzon A, Furukawa T, Horii A, Zhang ZY, Nicholson RI, Fuqua SA. Cancer Res. 2006;66:5950. doi: 10.1158/0008-5472.CAN-05-3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K, Floore A, Velds A, van’t Veer L, Neefjes J. Cancer Cell. 2004;5:597. doi: 10.1016/j.ccr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- 24.Norris JD, Paige LA, Christensen DJ, Chang CY, Huacani MR, Fan D, Hamilton PT, Fowlkes DM, McDonnell DP. Science. 1999;285:744. doi: 10.1126/science.285.5428.744. [DOI] [PubMed] [Google Scholar]
- 25.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SA, Wong J, Allred DC, Clark GM, Schiff R. J Natl Cancer Inst. 2003;95:353. doi: 10.1093/jnci/95.5.353. [DOI] [PubMed] [Google Scholar]
- 26.Jordan VC. J Natl Cancer Inst. 1998;90:967. doi: 10.1093/jnci/90.13.967. [DOI] [PubMed] [Google Scholar]
- 27.Wijayaratne AL, McDonnell DP. J Biol Chem. 2001;276:35684. doi: 10.1074/jbc.M101097200. [DOI] [PubMed] [Google Scholar]
- 28.Howell A, Osborne CK, Morris C, Wakeling AE. Cancer. 2000;89:817. doi: 10.1002/1097-0142(20000815)89:4<817::aid-cncr14>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 29.Larsen CN, Finley D. Cell. 1997;91:431. doi: 10.1016/s0092-8674(00)80427-4. [DOI] [PubMed] [Google Scholar]
- 30.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Proc Natl Acad Sci U S A. 2001;98:8554. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang D, Baek SH, Ho A, Kim K. Bioorg Med Chem Lett. 2004;14:645. doi: 10.1016/j.bmcl.2003.11.042. [DOI] [PubMed] [Google Scholar]
- 32.Zhang D, Baek SH, Ho A, Lee H, Jeong YS, Kim K. Combinatorial Chemistry & High Throughput Screening. 2004;7:691. doi: 10.2174/1386207043328364. [DOI] [PubMed] [Google Scholar]
- 33.Schneekloth JS, Jr, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM. J Am Chem Soc. 2004;126:3748. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 34.Anstead GM, Carlson KE, Katzenellenbogen JA. Steroids. 1997;62:268. doi: 10.1016/s0039-128x(96)00242-5. [DOI] [PubMed] [Google Scholar]
- 35.Kuduk SD, Harris TC, Zheng FF, Sepp-Lorenzino L, Ouerfelli Q, Rosen N, Danishefsky SJ. Bioorg Med Chem Lett. 2000;10:1303. doi: 10.1016/s0960-894x(00)00208-0. [DOI] [PubMed] [Google Scholar]
- 36.Kuduk SD, Zheng FF, Sepp-Lorenzino L, Rosen N, Danishefsky SJ. Bioorg Med Chem Lett. 1999;9:1233. doi: 10.1016/s0960-894x(99)00185-7. [DOI] [PubMed] [Google Scholar]
- 37.Osborne CK, Wakeling A, Nicholson RI. Br J Cancer. 2004;90(Suppl 1):S2. doi: 10.1038/sj.bjc.6601629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodriguez-Gonzalez A, Cyrus K, Salcius M, Kim K, Crews CM, Deshaies RJ, Sakamoto KM. Oncogene. 2008;27:7201. doi: 10.1038/onc.2008.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fevig TL, Lloyd JE, Zablocki JA, Katzenellenbogen JA. J Med Chem. 1987;30:156. doi: 10.1021/jm00384a026. [DOI] [PubMed] [Google Scholar]
- 40.Fevig TL, Mao MK, Katzenellenbogen JA. Steroids. 1988;51:471. doi: 10.1016/0039-128x(88)90046-3. [DOI] [PubMed] [Google Scholar]
- 41.Skaddan MB, Katzenellenbogen JA. Bioconjug Chem. 1999;10:119. doi: 10.1021/bc980094q. [DOI] [PubMed] [Google Scholar]
- 42.Skaddan MB, Wust FR, Katzenellenbogen JA. J Org Chem. 1999;64:8108. doi: 10.1021/jo990641g. [DOI] [PubMed] [Google Scholar]
- 43.Tedesco R, Thomas JA, Katzenellenbogen BS, Katzenellenbogen JA. Chem Biol. 2001;8:277. doi: 10.1016/s1074-5521(01)00006-0. [DOI] [PubMed] [Google Scholar]
- 44.Tedesco R, Youngman MK, Wilson SR, Katzenellenbogen JA. Bioorg Med Chem Lett. 2001;11:1281. doi: 10.1016/s0960-894x(01)00189-5. [DOI] [PubMed] [Google Scholar]
- 45.Hussey SL, He E, Peterson BR. Org Lett. 2002;4:415. doi: 10.1021/ol0171261. [DOI] [PubMed] [Google Scholar]
- 46.Lee H, Puppala D, Choi EY, Swanson H, Kim KB. Chembiochem. 2007;8:2058. doi: 10.1002/cbic.200700438. [DOI] [PubMed] [Google Scholar]
- 47.Hudson DF, Morrison C, Ruchaud S, Earnshaw WC. Trends Cell Biol. 2002;12:281. doi: 10.1016/s0962-8924(02)02281-x. [DOI] [PubMed] [Google Scholar]
- 48.Henry CM. Chemical & Engineering News. 2003;81:32. [Google Scholar]
- 49.Jackson AL, Bartz SR, Schelter J, Kobayashi SV, Burchard J, Mao M, Li B, Cavet G, Linsley PS. Nat Biotechnol. 2003;21:635. doi: 10.1038/nbt831. [DOI] [PubMed] [Google Scholar]
- 50.Zhang S, Qin C, Safe SH. Environ Health Perspect. 2003;111:1877. doi: 10.1289/ehp.6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sin N, Kim KB, Elofsson M, Meng L, Auth H, Kwok BH, Crews CM. Bioorg Med Chem Lett. 1999;9:2283. doi: 10.1016/s0960-894x(99)00376-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.