

Abstract
A dominant type of spontaneous autoreactive B cell activation in murine lupus is the extrafollicular generation of plasmablasts. The factors governing such activation have been difficult to identify due to the stochastic onset and chronic nature of the response. Thus, the ability to induce a similar autoreactive B cell response with a known autoantigen in vivo would be a powerful tool in deciphering how autoimmune responses are initiated. We report here the establishment and characterization of a system to initiate autoreactive extrafollicular B cell responses that closely mirror the spontaneous response using IgG anti-chromatin Abs. We demonstrate that exogenously administered anti-chromatin Ab, presumably by forming immune complexes (ICs) with released nuclear material, drives activation of RF B cells in AM14 Tg mice. Anti-chromatin elicits autoreactive B cell activation and development into AFCs at the T-zone/red pulp border. Plasmablast generation occurs equally in BALB/c, MRL/+ and MRL/lpr mice, indicating that an autoimmune-prone genetic background is not required for the induced response. Importantly, infused IgG anti-chromatin induces somatic hypermutation (SHM) in the absence of a GC response, thus proving the extrafollicular SHM pathway. This system provides a window on the initiation of an autoantibody response and reveals authentic initiators of it.
Keywords: B cells, autoantibodies, systemic lupus erythematosus
Introduction
The activation of autoreactive B cells plays a central role in the development of systemic autoimmunity. In addition to secreting pathogenic autoantibodies, autoreactive B cells can promote the activation of autoreactive T cells, which in turn can mediate tissue damage [1–4]. In lupus patients and mice genetically predisposed to lupus, the activation of autoreactive B cells is a selective process. Certain autoantigens, including nuclear Ags and self-IgG are preferred and recurrent targets of B cell autoimmunity. Not all patients or lupus-prone mice have all the possible autoantibodies, but rather loss of tolerance to each autoantigen is stochastic [1, 5].
It is important to define the signals and autoantigens that lead to autoreactive B cell activation as well as the consequences for those B cells. Due to the heterogeneous and rare nature of autoreactive B cells, Ig transgenic (Tg) mouse models for self or pseudo-self Ags have been invaluable. Such models have revealed early B cell tolerance checkpoints and mechanisms, including clonal deletion, receptor editing, anergy and clonal ignorance [6–14]. There has been much less success in using these models to elucidate how autoreactive B cells are activated. In part, this is due to the fact that B cells in some of the Tg models are so deeply tolerized that there is little activation even on a lupus-prone background [15–17]. However, some anti-DNA models, such as the 3H9 heavy chain Tg [18] and knock-in [19], and the AM14 RF heavy (H) chain Tg mouse model [11] that is the subject of this report, have been useful in revealing the phenotype of autoreactive B cell activation. Although 3H9 anti-DNA B cells are anergized in normal hosts [9, 20], and AM14 RF B cells are not [21], both systems demonstrate spontaneous activation of autoreactive B cells and AFC formation outside of B cell follicles in the lupus-prone MRL/lpr strain [22–24]. A striking finding was that in the spleens of AM14 Tg MRL/lpr mice, RF B cells undergo somatic hypermutation at extrafollicular sites but are not found in splenic GCs [24]. AM14 Tg MRL/lpr mice have activated B cell blasts as well as plasmablasts, both of which turn over rapidly, resulting in a highly dynamic response [25, 26]. Interestingly, even with the benefit of an Ig Tg to restrict the B cell repertoire, the initiation of the response in any given animal is unpredictable [25, 26].
Despite determining the sites of activation and the nature of the responding cells, prior studies have not elucidated: what are the in vivo antigenic stimuli for RF B cells (or any autoreactive B cell); why does a specific autoantibody response initiate stochastically; and what promotes the extrafollicular pathway of somatic hypermutation rather than the more conventional GC pathway. The answers to such questions will provide important mechanistic insight and presumably could also identify potential therapeutic targets.
Investigating these questions has been problematic, in part due to the nature of spontaneous autoimmunity itself. The stochastic onset of activation, without a defined starting time point, makes it difficult to determine the order of events in the process. Similarly, it is difficult to identify the autoantigens involved or the cells and signals required for propagation of a spontaneous response. We therefore concluded that a system that would allow for an experimentally controlled initiation of an autoreactive B cell response characteristic of spontaneous systemic autoimmunity would be very helpful to address such issues. The AM14 RF system is useful for this purpose, since IgG is a known part of the autoantigen and can be readily introduced to attempt initiation of the RF response. We hypothesized that providing IgG ICs as an RF autoantigen would lead to faithful reproduction of the spontaneous activation of autoreactive B cells in lupus-prone mice. However, previous attempts using IgG2a ICs with foreign protein [24, 27] led to a GC response rather than an extrafollicular response, suggesting that a different form of IC was required to generate the latter.
A potential insight into this puzzle came from the result that, in vitro, IgG2a anti-chromatin mAbs are mitogenic for AM14 B cells [28, 29] but conventional ICs are not. The potent activation mediated by IgG2a anti-chromatin was subsequently shown to be MyD88 dependent, with a major role for Toll-like receptor 9 (TLR9) [28, 29]. The paradigm of activation through dual BCR-TLR engagement was extended in vitro to B cells that were anti-DNA and anti-Sm, two other dominant specificities of lupus, with respect to TLR9 and TLR7 [29, 30]. These studies demonstrated a unique method of activation, but only measured proliferation, not differentiation, and it was unclear how they would reflect the in vivo situation. Moreover, these cultures fail to survive beyond two days and thus are not suitable for investigating differentiation.
Based on the in vitro ability of IgG2a anti-chromatin mAbs to cause AM14 B cell proliferation, we hypothesized that they might elicit extrafollicular activation in vivo. We tested this hypothesis by exposing AM14 RF B cells in vivo to high levels of anti-chromatin antibodies and present evidence that this does indeed lead to extrafollicular B cell activation and AFC formation in a manner that faithfully reproduces the phenotype of spontaneous RF B cell activation in MRL/lpr mice. In contrast, IgGs of other specificities, whether for haptens or another self-Ag, caused no detectable AFC response. Therefore, one mechanism to generate the typical extrafollicular response is via IgG anti-chromatin, which presumably forms ICs in vivo with endogenous chromatin [31]. We next took advantage of this system to show that this RF AFC response to anti-chromatin Abs can occur in autoimmune-prone MRL/lpr, young MRL/+ and even non-autoimmune BALB/c mice. Thus, we conclude that neither an autoimmune-prone environment, nor autoimmunity-related genetic defects [32] are required for these AM14 B cells to become activated. Finally, we demonstrated that this response induces clonal expansion and somatic hypermutation, as originally found in AM14 B cells spontaneously activated in MRL/lpr mice [24], and in autoantibody-secreting cells in non-Tg mice [33]. Thus we have identified an important in vivo autoantigen for RF B cells and provide insight into what may stimulate the unique extrafollicular response that generates both RF and anti-DNA Abs in lupus-prone mice.
Results
IgG2a but not IgG2b anti-chromatin antibodies elicit an RF AFC response
To try to reproduce the spontaneous extrafollicular RF response, we acutely raised the serum concentration of IgG2a anti-chromatin, an antigen for AM14 B cells, by growing the hybridoma PL2-3 i.p. in AM14 Tg MRL/lpr mice (referred to hereafter as Tg mice, Fig. 1). PL2-3 has been shown to be mitogenic for Tg B cells in vitro by forming ICs with nuclear material from apoptotic cells in culture media [28]. Mice were sacrificed 7–8 days after hybridoma injection, and serum and spleens were harvested. Because AM14 B cells become activated spontaneously in MRL/lpr mice with age [25], we used 7 week old and younger mice, as spontaneous activation is only detectable in mice that are a number of weeks older. We previously showed that in H Tg mice, a small percentage of B cells express one of two closely related Vκ8 family light chains rearranged to Jκ4 or Jκ5, that reconstitute the RF specificity [11, 21, 23, 24]. These B cells are specifically detected by the mAb anti-idiotype 4-44, which identifies only Tg-encoded RF B cells [11, 24].
Figure 1.

Experimental design for the use of IgG2a anti-chromatin antibodies to acutely raise levels of IgG2a RF self-antigen. For hybridoma treatment: mice injected i.p. with pristane on days -10 and -3 (gray arrows), followed by hybridoma injection on Day 0 (black arrow), and sacrifice on Day 7. For protein treatment: mice were injected with 0.5mg purified antibody on Days 0, 2, and 5 (white arrows) and were sacrificed on Day 8.
Tg MRL/lpr mice treated with PL2-3 developed large numbers of 4-44+ AFCs (Fig. 2A), comparable to those generated in older mice that had undergone spontaneous activation [24, 25]. In contrast, the control hybridoma PL2-8, an IgG2b clonal relative of PL2-3, had no effect (Fig. 2A). Treatment with an IgG2a anti-desmoglein secreting antibody, NAK10 [34], also did not lead to generation of 4-44+ AFCs (Fig. 2A). This suggested that anti-nuclear antibodies that had Fc regions that could be bound by the AM14 BCR, but not other antibodies, could drive AFC generation. Consistent with this, mice treated with a different anti-nuclear IgG2a hybridoma, the anti-DNA PA4 [35], had increased numbers of AFCs (Fig. 2A).
Figure 2.

Anti-nuclear antibodies cause AFC formation and antibody secretion. (A) 4-44+ ELISPOTs from splenocytes of Tg MRL/lpr mice treated with antibody secreting hybridoma or protein ICs. Treatment groups are listed on the x-axis. PL2-3: IgG2a anti-chromatin (n=35); PL2-8: IgG2b anti-chromatin (n=11); PA4: IgG2a anti-DNA (n=4); NAK: IgG2a anti-desmoglein (n=11); protein ICs: ICs of 23.3 (IgG2a anti-NP) and NP-CGG (n=3). No hybridoma: pristane only (n=11). *p<0.05 by Mann-Whitney U test, compared to PL2-8 group (negative control). (B) 4-44+ ELISPOTs from splenocytes of Tg MRL/lpr (black bars), Tg MRL+/+ (gray bars) or Tg BALB/c mice (open bars) treated with PL2-3 (n=5-6 for each strain), PL2-8 or 23.3 hybridoma. *p<0.05 by Mann-Whitney U test, compared to combined PL2-8 data. (C) 4-44+ ELISPOTs from splenocytes of Tg MRL/lpr (n==26, 5, 7 respectively) or Tg BALB/c (n=5, 4, 8 respectively) mice treated with purified PL2-3 (black bars), PL2-8 (gray bars), or Hy1.2 (IgG2a anti-TNP, open bars) for 1 week. *p<0.001 by Mann-Whitney U test, compared to PL2-8 group (negative control).
Previously, we reported that IgG2a anti-TNP/TNP-KLH protein ICs stimulate an RF GC response in Tg MRL/lpr animals [24, 27]. In order to determine if in the same system a T-dependent antigen was able to drive differentiation of AM14 B cells into AFCs, we generated similar protein ICs from purified IgG2a anti-NP and NP-CGG and injected them in alum into Tg MRL/lpr mice. This immunization did not generate 4-44+ AFCs (Fig. 2A). Identical protein ICs injected without alum into Tg MRL/lpr mice did not lead to AM14 B cell activation as measured by FACS, ELISpot or immunohistology (data not shown).
Anti-chromatin elicits similar RF AFC responses in autoimmune-prone and non-autoimmune-prone strains
AM14 B cells become activated spontaneously in lupus-prone mice, but do not generate AFCs in non-autoimmune mice [23, 24, 27]. Autoimmune-prone mouse strains have genetic defects [32, 36, 37] pro-inflammatory cytokines [38–41], and activated T cells [4, 42–44]. Furthermore, autoimmune mice may have persistent self-antigen due to impaired clearance mechanisms [45]. Any of these factors could be important in the activation of AM14 B cells in response to anti-chromatin antibodies. In order to investigate how these strain-specific factors would affect the activation of AM14 B cells, we supplied PL2-3 hybridoma cells to Tg MRL/lpr, MRL+ and BALB/c animals. All three strains of AM14 Tg mice responded with indistinguishable AFC formation (Fig. 2B). This indicates that initial AM14 B cell activation and differentiation induced by anti-chromatin antibodies does not require an intrinsic B cell defect, a pro-inflammatory environment, or an environment with enhanced cell death or turnover. As an additional control in this series of experiments, we used an anti-NP secreting hybridoma, 23.3, that did not lead to generation of AFCs, confirming that the specificity of the IgG2a antigen is important (Fig. 2B).
Although the hybridoma experiments were controlled for specificity, they could not exclude a potential effect of the hybridoma itself. Therefore, we also tested the efficacy of purified PL2-3 antibody in Tg MRL/lpr and Tg BALB/c mice. Mice were sacrificed on day 8 after injections of 0.5mg of protein on day 0, 2, and 5. PL2-3 drove AFC formation in both MRL/lpr and BALB/c strains, while control antibodies PL2-8 and Hy1.2, an anti-TNP IgG2a, did not (Fig. 2C). Thus, although hybridoma administration provides a simple and efficient method to provide high levels of anti-chromatin, the hybridoma itself is not necessary in either strain of mice.
Anti-chromatin antibodies elicit RF plasmablasts at the extrafollicular T zone-red pulp border
Spontaneous activation of AM14 B cells in MRL/lpr mice is accompanied by development of CD22lo plasmablasts [26]. Mice given PL2-3, but not PL2-8, developed a CD22lo population of AM14+ cells (Fig. 3A, B) similar to that seen in spontaneously activated AM14 cells in MRL/lpr mice [25, 26]. There was at least a 4-fold induction of these cells upon treatment with PL2-3 hybridoma (Fig. 3B). This is a minimal estimate as the AM14+ CD22lo cells in mice treated with PL2-8 did not localize to the same region of the gate (Fig. 3B), and therefore probably represent background.
Figure 3.

Anti-chromatin antibodies cause RF B cell plasmablast differentiation as seen by FACS analysis. (A) Representative FACS plots of live spleen cells demonstrating that H Tg MRL/lpr mice treated with PL2-3, but not PL2-8 hybridoma or protein ICs, develop 4-44+/CD22 lo cells, indicative of plasmablast development [26].
(B) Combined data from multiple experiments. PL2-3, n=22; PL2-8, n=12; Protein ICs, n=6. *p=0.0007; ** p=0.0013 by Mann Whitney U test. (C) Histograms of expression of CD138, CD44, CD80 and CD86 gated on live 4-44+ CD22lo (solid line) and 4-44+ CD22hi (dashed line) subsets from mice treated with PL2-3. PNA was used to identify germinal center cells, with 4-44+CD22 hi cells from mice treated with protein ICs serving as a positive control (shaded gray).
The CD22lo cells from PL2-3-treated mice had elevated expression of CD44 and CD138 (Fig. 3C), markers associated with plasmablast differentiation [25, 26, 46]. They also expressed increased levels of CD80 and CD86, consistent with their activated status (Fig. 3C). In mice given PL2-3, there was no evidence for GC cell differentiation in either CD22hi or CD22lo cells, as all were PNA-negative. We also measured PNA expression in the mice given IgG2a anti-NP and NP-CGG ICs, described above. These mice did develop abundant PNA+ 4-44+ GC cells (Fig. 3C), although they developed neither 4-44 AFCs (Fig. 2A) nor 4-44+ CD22lo cells (Fig. 3A, B).
The localization of these activated cells was determined using immunofluorescence on splenic sections. Treatment with PL2-3 hybridoma or protein led to the appearance of brightly staining 4-44+ cells at the T cell zone-red pulp border, outside of the follicle (Fig. 4A and B), while PL2-8 treatment did not (Fig. 4C). Higher magnification revealed these cells to have abundant cytoplasm, a characteristic of plasmablasts (Fig. 4D). There were no GCs present in mice treated with PL2-3, but there were in mice treated with ICs of IgG2a anti-NP/NP-CGG (Fig. 4E–H) consistent with both ELISPOT (Fig. 2A) and FACS analysis (Fig. 3). Thus, provision of IgG2a anti-chromatin, via either hybridoma or as purified protein, led to the development of an extrafollicular plasmablast RF response without GCs, a response similar to that seen in spontaneous activation of RF B cells in the Tg MRL/lpr spleen.
Figure 4.
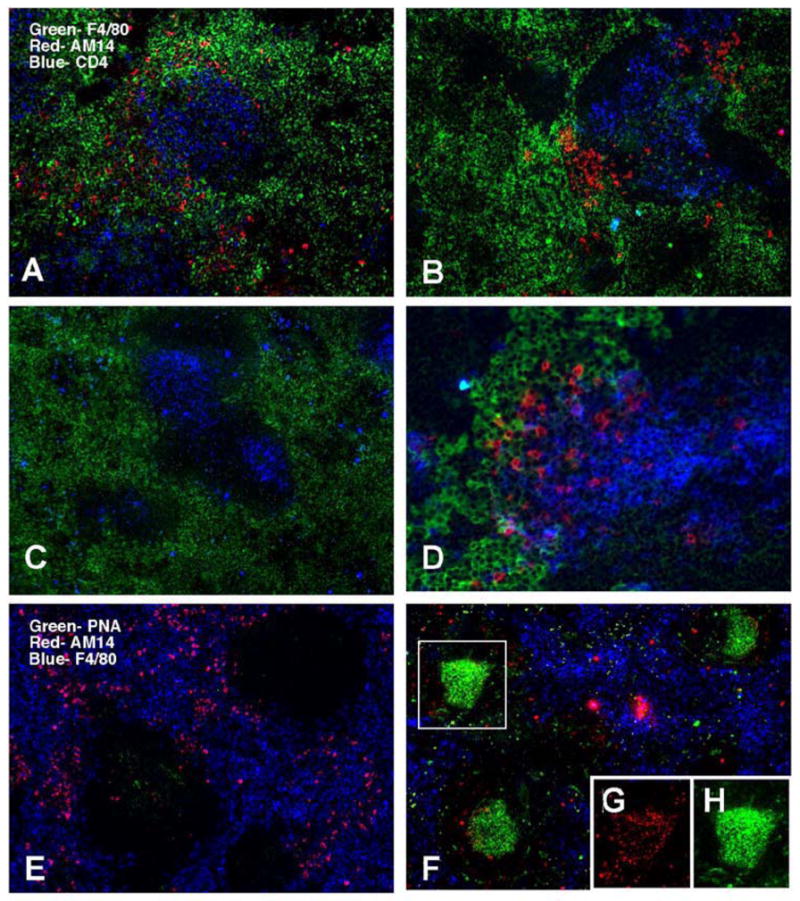
AM14 B cells activated by anti-chromatin antibodies localize to the T zone-red pulp border but not GCs in Tg in spleens of MRL/lpr mice. (A–D) AM14 idiotype (4-44) is shown in red, macrophage marker F4/80 in green, and CD4 in blue. (A–C) original magnification 100x; (D) 200x. (A) Tg AM14 MRL/lpr mouse treated for 1 week with PL2-3 hybridoma shows 4-44+ B cells at the T zone-red pulp border. (B) Same as (A) but mice were treated with PL2-3 protein. (C) Tg AM14 MRL/lpr mouse treated for 1 week with PL2-8 hybridoma had few detectable 4-44+ B cells. (D) Higher magnification of spleen of a PL2-3 treated mouse. (E-H) AM14 idiotype (4-44) is shown in red,, F4/80 in blue, and GC marker PNA in green. (E) AM14 MRL/lpr mouse treated with PL2-3 does not show germinal centers. (F) AM14 MRL/lpr mouse treated with protein ICs developed GCs. (G and H) GCs in these mice are positive for AM14 (G) and PNA (H) as shown by the single channel images of the region boxed in (F).
Stimulation with anti-chromatin antibodies induces somatic hypermutation at extrafollicular sites
SHM was originally thought to be restricted to GCs [47–49], but we previously discovered that during the spontaneous RF response in MRL/lpr mice, SHM occurred in AM14 B cells at the T zone-red pulp border [24]. This was demonstrated by microdissections of 10–30 cells at these sites. Sequences derived from these cells often shared VJ rearrangements as well as some V region mutations, but also displayed intraclonal diversity [24]. Mathematical analysis demonstrated that the mutation rate was comparable to that found in GCs [50]. Further, in those studies we could not exclude that mutation was possibly induced first at another site, such as a GC, at a previous time in these older MRL/lpr mice, even though we did show that SHM continued at the extrafollicular site [24]. In addition, until now there was not another robust example of mutation at occurring extrafollicularly, such that SHM is still widely believed to be restricted to the GC [51, 52].
Therefore, to determine whether mutation was induced, 4-44+ B cells were microdissected from splenic sections of four Tg MRL/lpr and four Tg BALB/c mice that had been treated with PL2-3 protein every other day for 2 weeks (Fig. 5A). From each microdissection, called a “pick”, the DNA was amplified to determine Vκ sequences and mutation rates. Lists of all picks from Tg MRL/lpr and Tg BALB/c mice and their number of mutations are in Tables 1 and 2. We chose to generate multiple sequences (6–18) from each pick to maximize our chances of identifying mutated sequences and being able to determine clonally related sequences in order to build genealogical trees (Fig. 5B), even though this likely resulted in some reisolation of sequences amplified from the same cell. Out of 6 total picks from Tg MRL/lpr mice, all 6 showed mutation with an average of 1.8 mutations per sequence (Table I). Interestingly, Tg BALB/c mice had mutation in 4 of 6 picks, but with a lower average of 0.4 mutations per sequence (Table II). As discussed, it is likely that our sequence collection includes multiple copies of a single sequence that do not represent different cells. Although this does not affect the genealogical tree shape, it does impact the mutations/sequence calculation. Therefore, we also calculated the number of mutations per unique sequence (Tables I and II). We found an average of 2.8 and 0.8 mutations per unique sequence for picks from Tg MRL/lpr and Tg BALB/c, respectively. The distribution of mutations per unique sequence (Fig. 5C) is significantly different between the two strains (p = 0.005). We previously calculated the PCR error rate of our procedure using Pfu Turbo as < 0.05 mutations per sequence [24]; these current sequences were generated with Pfu Ultra, which is reported to have an 3-fold lower error rate (Stratagene). Thus, the observed mutation rates are far above PCR error in both MRL/lpr and BALB/c mice.
Figure 5.

Somatic hypermutation occurs at extrafollicular sites of Tg MRL/lpr and Tg BALB/c mice given IgG2a anti-chromatin. (A) Immunohistochemistry of a representative microdissected region showing AM14 B cells (blue) in a splenic section from a Tg MRL/lpr mouse treated with PL2-3 protein every other day for 2 weeks. Photos are from before and after laser microdissection. Picks typically contained 10–30 AM14+ B cells. Note that image quality is suboptimal due to use of foil-coated slides required for microdissection. (B) Representative genealogical trees from two Tg MRL/lpr mice (trees A2, B3 and C1) and two Tg BALB/c mice (trees E1, E2, and F4) which demonstrate ongoing mutation as reflected by intraclonal diversification. Two additional clones (B2 and F3) from these same picks had no mutation and are thus fully described in the Tables and not shown. Circles at the base of trees represent germline sequences. Non-mutated sequences actually found in a pick are listed in this circle; if there were no germline isolates the circle has no sequence identifiers. Every other open circle, or “node”, represents one or more clones with the same sequence, again as indicated by sequence identifiers. Open circles are implicit nodes required by the genealogy, although no actual sequences representing those nodes were found. Tree “branches” are the lines between nodes. The positions of and types of nucleotide exchanges are listed alongside branches. (C) Histogram of the distribution of the number of mutations per unique sequence for all Tg MRL/lpr and Tg BALB/c picks. The distributions of mutation between MRL/lpr and BALB/c are significantly different (p=0.005, chi-squared test for trend).
Table I.
Summary of microdissections and mutations in Tg MRL/lpr mice treated with PL2-3 hybridomaa
| Mouse | Pick | Tree Name | Total Sequences | Total Mutations | Mutations/Sequence | Total Unique Sequences | Total Mutations Among Unique Sequences | Mutations/Unique Sequence | Number Of Independent Mutations | Trunk Mutations | Trunk Mutations/Unique Seq |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 11-8-05 C | 1 | A1 | 15 | 33 | 2.2 | 3 | 6 | 2 | 4 | 2 | 0.67 |
| 11-8-05 C | 2 | A2 | 18 | 48 | 2.67 | 3 | 11 | 3.67 | 5 | 3 | 1 |
| 11-8-05 D | 1 | B1 | 16 | 1 | 0.06 | 2 | 1 | 0.50 | 1 | 1 | 0.50 |
| 11-8-05 D | 2 | B2 | 10 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| 11-8-05 D | 2 | B3 | 2 | 2 | 0.17 | 1 | 1 | 1 | 1 | 1 | 1 |
| 10-18-06 B | 2 | C1 | 7 | 46 | 6.57 | 4 | 26 | 6.50 | 17 | 2 | 0.50 |
| 10-18-06 C | 1 | D1 | 1 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 |
| 10-18-06 C | 1 | D2 | 5 | 0 | 0.00 | 1 | 0 | 0.00 | 0 | 0 | 0 |
|
| |||||||||||
| Total | 74 | 130 | 1.8 | 16 | 45 | 2.8 | 28 | 9 | 0.6 | ||
Sequences recovered from four Tg MRL/lpr mice treated with PL2-3 were grouped by pick and assigned to a lettered genealogical tree. Identical sequences in a pick were conservatively treated as one clone, instead of as the products of multiple cells, and were assigned to one ‘unique sequence.’ Trunk mutations identify those mutations that are common to all mutated sequences and are in bold in genealogical trees in Fig. 5B.
Table II.
Summary of microdissections and mutations in Tg BALB/c mice treated with PL2-3 hybridomaa
| Mouse | Pick | Tree Name | Total Sequences | Total Mutations | Mutations/Sequence | Total Unique Sequences | Total Mutations Among Unique Sequences | Mutations/Unique Sequence | Number Of Independent Mutations | Trunk Mutations | Trunk Mutations/Unique Seq |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 12-20-05 A | 1 | E1 | 4 | 1 | 0.25 | 2 | 1 | 0.50 | 1 | 1 | 0.5 |
| 12-20-05 A | 1B | E2 | 1 | 2 | 2.00 | 1 | 2 | 2.00 | 2 | 2 | 2 |
| 12-20-05 A | 2 | E3 | 3 | 0 | 0.00 | 1 | 0 | 0.00 | 0 | 0 | 0 |
| 12-20-05 A | 2B | E4 | 1 | 0 | 0.00 | 1 | 0 | 0.00 | 0 | 0 | 0 |
| 12-20-05 B | 2 | F1 | 3 | 2 | 0.67 | 2 | 2 | 1.00 | 2 | 0 | 0 |
| 12-20-05 B | 2B | F2 | 2 | 2 | 1.00 | 2 | 2 | 1.00 | 2 | 0 | 0 |
| 12-20-05 B | 3 | F3 | 2 | 0 | 0.00 | 1 | 0 | 0.00 | 1 | 0 | 0 |
| 12-20-05 B | 3B | F4 | 4 | 5 | 1.25 | 4 | 5 | 1.25 | 3 | 1 | 0 |
| 12-20-05 C | 1 | G1 | 9 | 0 | 0.00 | 1 | 0 | 0.00 | 0 | 0 | 0 |
|
| |||||||||||
| Total | 29 | 12 | 0.4 | 15 | 12 | 0.8 | 11 | 4 | 0.3 | ||
Sequences recovered from four Tg BALB/c mice treated with PL2-3 were grouped by pick and assigned to a lettered genealogical tree. See Table I legend.
If mutation is ongoing at a site, then clonally related sequences should differ by point mutations at least some of the time. This will generate genealogic trees with branching patterns [49, 53]. At GC mutation rates of 0.5 mutations/cell/generation, each L chain will get 0.25 mutations/generation. This means that on average we can expect identical daughters approximately 75% of the time, but different ones the rest of the time. Thus picks with eight independent sequences should have at least some diversity, though identical sequences are clearly quite compatible with high mutation rates and in fact are expected. In Fig. 5B, representative genealogic trees from PL2-3 treated Tg MRL/lpr mice and Tg BALB/c mouse are presented. In each node of the tree, all sequences containing that particular sequence are identified. Four out of eight trees from MRL/lpr mice had at least one branch, while four of the nine trees from BALB/c mice had at least one branch. Some trees had multiple branches including second level branches (Fig. 5B, trees A2, C1 and F4). Both the significant branching in the genealogical trees and the number of mutations per sequence demonstrate that mutation was occurring at a high rate at extrafollicular sites in mice treated with PL2-3. In particular, sequences that share some but not all mutations are extremely unlikely to be generated by amplification.
Discussion
In the AM14 RF response in MRL/lpr mice, plasmablasts and activated B cells blasts proliferate and undergo somatic hypermutation in the splenic T zone-red pulp border, in the absence of a detectable GC response [24]. In order to understand how and why RF B cells are activated at the T zone-red pulp border, we sought, by introducing autoantigen to RF Tg mice, to develop a system that would reproduce these events.
In this report, we have identified IgG2a anti-chromatin antibodies as an in vivo autoantigen for RF B cells. This ligand, by itself, is sufficient to generate many characteristic features of the spontaneous RF B cell response seen in MRL/lpr mice. In contrast, other potential IgG2a-containing ligands did not. In particular, traditional immunization of AM14 Tg mice with protein-containing ICs stimulated a GC response with minimal, if any, extrafollicular plasmablast formation. Thus, anti-chromatin antibodies, probably acting as ICs formed with ubiquitous endogenous chromatin [31], have a unique property of stimulating RF B cells to undergo an extrafollicular, rather than a GC, response.
It has been notoriously difficult to artificially elicit disease-associated autoantibody responses in vivo. For example, immunization with DNA is relatively ineffective in generating anti-DNA Abs [54], although bacterial DNA and DNA conjugated to certain immunogenic DNA-binding proteins can be effective [55–57]. Nonetheless, in none of these cases was it shown that the nature of the in vivo response to administered “autoantigen” corresponded to the spontaneous response. Thus, the ability to induce the extrafollicular RF response can be useful as an in vivo method to test the requirements for autoreactive B cell activation.
For example, this system gave us the unique opportunity to stimulate the same autoreactive B cells in lupus-prone and non-autoimmune mouse strains equally, in order to test the role of genetic background. Despite the fact that B cells in MRL/lpr, MRL/+ and other autoimmune-prone strains as well as lupus patients do have intrinsic defects [58–60], we found that the elicitation of RF AFCs was equally efficient in Fas-deficient and sufficient MRL strains and BALB/c mice. However, an unexpected finding was larger numbers of somatic mutations in 4-44+ B cells from Tg MRL/lpr mice compared to BALB/c mice, suggesting the intriguing possibility that regulation of mutation rate or selection could be an intrinsic strain-specific difference. These differences in mutation levels are based on our data in a single system, and whether they relate to differences in mutation rate or rather to differences in cell proliferation and survival rates (which in turn would influence mutation content) is unknown. The disparity in mutation content could be attributed to Fas-deficiency, MRL background genes, or both. It is of interest that Takahashi, et al. [61] found that GCs and memory B cells accumulated more mutations in Fas-deficient than Fas-sufficient mice on a C57Bl/6 background. We also found more mutations in spontaneous RF+ GCs of MRL/+ and MRL/lpr mice, compared to BALB/c [27].
The rapid development of RF AFCs in response to anti-chromatin antibodies could also help to explain the phenomenon that we have previously termed “conversion”, referring to the stochastic onset of a large AFC response in AM14 Tg MRL/lpr mice [25]. This onset is accompanied by rises in serum RF and circulating RF B cells, that go from background to high levels in a period of one to two weeks [25]. It is evident from the current results that such a response can actually develop within a matter of a few days from the point at which IgG2a anti-chromatin antibodies exceed a threshold level. What in turn causes an increase in IgG2a anti-chromatin (or other antigenic antibody) beyond such threshold is not yet determined. Autoimmune mice have impaired clearance of apoptotic cells [45, 62], which could eventually lead to increased available autoantigen, and exposure to apoptotic cells can lead to autoantibody generation [63]. Interestingly, we previously found a correlation between the onset of conversion in AM14 Tg MRL/lpr mice and the appearance of IgG2a anti-chromatin serum Abs (but not with anti-chromatin of other isotypes) [25].
The ability to stimulate the response at a known point in time also allowed us to better resolve the relationship between the mutating extrafollicular response and any potentially associated or prior GC response. Because we could see a full blown extrafollicular response in a week in the absence of a concurrent GC response, the extrafollicular response and its attendant SHM does not appear to require a GC response to occur first. This is a conclusion that could not be drawn firmly from our studies of the spontaneous response [24]. Importantly, we were able to show that SHM is easily detectable within a week or so after the initiation of the response, with actual onset of mutation probably several days before this time. The branching patterns and shared mutations seen among sequences derived from small clusters of cells demonstrate that mutation was occurring locally [24, 33, 64], and the presence of multiple mutations in a single V region in such young mice strongly suggests that mutation was initiated only after anti-chromatin IgG2a was provided.
One question raised by our observations is why anti-chromatin antibodies are sufficient to initiate the differentiation of RF B cells into plasmablasts. In vitro data would suggest that TLR signals are at least in part responsible. This concept is largely based on the pioneering studies by Marshak-Rothstein and colleagues, which demonstrate the in vitro activation of RF B cells by anti-chromatin antibodies occurs in a MyD88-dependent manner [28]. These signals most likely come through TLR9, and possibly TLR7 [28, 30, 65]. Our studies establish the in vivo relevance of these in vitro experiments, which relied on two-day proliferation assays and hence did not assess differentiation.
Although in vitro this type of response is TLR-dependent, it is unclear whether this is true in vivo, as other signals such as from T cells and cytokines are likely to be present and could substitute for the TLR signal. Another complicating factor in vivo is that chromatin-containing ligands may directly activate dendritic and other cell types in vivo, which in turn could promote the response [66, 67], while these cells are absent in vitro. These potential contributions to the response in vivo will ultimately have to be dissected using complex approaches. Of interest in this regard, MyD88-deficient Fas-deficient mice of mixed genetic background lacked substantial serum RF [30]; however, TLR9-deficient non-Tg MRL/lpr mice do make RF (S. Christensen and MJS, unpublished observations), illustrating the complexity of the in vivo situation.
A related question is why activation takes place either exclusively or preferentially at the T zone-red pulp border. This is the normal site for certain types of T-independent B cell responses, although these are usually quite transient and are generally not known to undergo high rate somatic hypermutation [68–70]. As antigens such as TNP-Ficoll, which are not known to contain a TLR-ligand, generate an extrafollicular response [68, 69, 71], again it is not clear what the non-exclusive role is for this family of receptors. Some data indicate that dendritic cells (DCs) promote plasmablast development and survival in T-independent extrafollicular responses [67, 72]. Thus, by analogy, DCs potentially could serve key roles directing the extrafollicular response. Indeed, during the spontaneous RF response in MRL/lpr mice [24], DCs, which are abundant at this site in MRL/lpr animals [73], are seen juxtaposed with RF B cells. DCs are also a source of BAFF, which has recently been shown to enhance antibody secretion [74], and APRIL, both of which promote plasmablast development [72] possibly via TACI [75–77]. DCs located at the T zone-red pulp border could be activated via TLRs, as well as Fc receptors (FcRs), which can bind anti-chromatin ICs [66]. DCs can present undegraded Ag to B cells [78] and it was recently shown in LN that B cells can recognize unprocessed Ag displayed on DCs before ever entering the follicle [79]. Against the concept that DCs are important are the recent findings that CD11chi DCs are dispensable for plasmablast responses to T-independent type 2 antigens [80] and vesicular stomatitis virus infection [81]. However, these situations may differ substantially from a chronic autoimmune response.
Since IgG2a anti-chromatin by itself was sufficient to stimulate RF B cell proliferation and AFC differentiation, one might expect accompanying RF responses in autoimmune mice and patients with conditions associated with anti-nuclear antibodies (ANAs) [1]. Indeed, there are good examples of this. Although it is commonly believed that RFs are rare in lupus, in fact a significant fraction of lupus patients have detectable RF (27% in a recent authoritative study of 352 patients, ref. [82]). Other diseases have a high prevalence of both ANAs and RF, including Sjogren’s syndrome and chronic hepatitis C [83–87]. Furthermore, we have recently found a high prevalence of elevated RF levels in other lupus-prone mouse strains (manuscript in preparation). Although IgG anti-chromatin can stimulate RF B cells, it is certain that RF autoantibodies can be generated via multiple pathways [88, 89]. In systemic autoimmune syndromes there are likely other immunogenic forms of ICs. These ICs may include ligands for TLRs other than TLR9, including TLR7 as recently shown in vitro by Lau, et al. [30]. They may also include ligands for other non-TLR innate immune receptors, though this remains to be determined.
In summary we have shown that IgG anti-chromatin Abs are sufficient to elicit a robust and rapid extrafollicular RF autoantibody response closely resembling the spontaneous RF response in MRL/lpr mice; other types of IgGs led to a GC response or no response. Such a system is much more tenable than the stochastic and unpredictable activation of autoreactive B cells found in autoimmune mice [5, 25]. This system enabled us to demonstrate that an autoimmune-prone genetic background is not required for the response to take place. It also allowed us to establish that mutation occurs at this site without the need for a prior GC response. In the future, by combining IgG anti-chromatin Ab infusion with mutant mice and cell transfer approaches, we and others should be able to further dissect the mechanisms of autoreactive B cell activation and propagation.
Materials and Methods
Mice
The AM14 heavy conventional IgM-only Tg [11] was backcrossed at least 10 generations onto the MRL/lpr, MRL/+ and BALB/c backgrounds. All mice were housed under specific pathogen-free conditions. PCR to detect the H chain Tg was performed as described [11]. All studies were approved by the Yale Institutional Animal Care and Use Committee.
FACS Analysis
Splenocytes were prepared and flow cytometric analysis was performed as described [23].
ELISPOT Analysis
ELISPOT analysis was performed as previously described [21, 23].
Anti-Chromatin Hybridomas
The IgG2a and IgG2b anti-chromatin hybridomas, PL2-3 and PL2-8 [90], were obtained from Mark Monestier. For in vivo hybridoma growth, mice were injected with 0.25 ml of pristane (Sigma) on days 0 and 7. Hybridoma cells were injected (107 cells) i.p. on day 10, and mice were sacrificed on day 17 or 18.
Protein Purification and Administration
PL2-3 protein was purified from hybridoma cells grown in serum free media or from ascites grown in Rag-1-deficient mice. Ascites fluid was collected and clarified by centrifugation for 4 minutes at 7000 rpm. Protein from hybridoma supernatants was purified on a Protein G column (Pharmacia) while protein from ascites was purified by ammonium sulfate precipitation followed by purification via QAE-Sephadex (Pharmacia). Purified protein was then concentrated, filtered, and tested by SDS-page for purity and by anti-chromatin ELISA for quantification. PL2-3 protein was injected (0.5mg) i.p., every other day. No gross differences were observed in the potency of protein purified from either source and they were used interchangeably.
Antibody Reagents
Abs prepared in our laboratory as described [11] were: 4-44-biotin (anti-Id), 4-44FITC, 4-44-Alexa 647, 4-44-Alexa 488, 4-44-Alexa 568, 160A1 (anti-CD80)-Alexa 488 and Pgp-1 (anti-CD44)-Alexa 488. The antibodies GL7 (anti-CD86)-FITC, anti-CD22.2-FITC, anti-CD22.2-PE, and anti-CD138 (conjugated to PE and unlabeled) were obtained from BD Pharmingen. BM8-Alexa647 (anti-F4/80) and A3-1-biotin (anti-F4/80) were obtained from Caltag. Streptavidin-Alexa 647 and streptavidin-PE (Molecular Probes) were used to detect biotinylated reagents. PNA-FITC was obtained from Vector.
Histology/Immunofluorescence
Sections were prepared as described [91] and stained with antibodies described above and specified in figure legends. Nuclei were identified with 4′,6′-diamidino-2-phenylindole (Molecular Probes). Fluorescent images were captured on an Olympus BX-40 microscope using a SPOT-RT Slider (Scanalytics) digital camera. For immunohistochemistry, 4-44-FITC or 4-44-biotin antibodies along with anti-FITC-alkaline phosphatase (Molecular Probes) and streptavidin-HRP (Southern Biotechnology Associates) were developed with Fast Blue BB or 3-amino-9-ethyl-carbazole (Sigma-Aldrich), as described previously [21].
Laser Capture Microdissection
Sections were cut from OCT-embedded spleens onto Leica PEN-membrane 2.0 μm slides. Staining was completed on these sections to identify extrafollicular 4-44+ B cells. 4-44+ B cell clusters (10–30 cells) were microdissected using a Leica Instruments LMD6000 and excised cells were digested overnight at 37°C–55°C in 10 μl of 0.8 mg/ml proteinase K, 50 mM Tris, pH 8, 50 mM KCl, 0.63 mM EDTA, 0.22% Igepal, and 0.22% Tween 20.
Sequencing
Sequencing was performed as described [24] except with Pfu Ultra (Stratagene) rather than Pfu Turbo. In brief, Vκ8-Jκ4/5 rearranged sequences were amplified by nested PCR: external primers 5′-AGCTATGCATTTTCACTGTC-3′ and 5′-AGCCCTCTCCATTTTCTC-3′, and internal primers 5′-TTGTGATGACACAGTCTCC-3′ and either 5′-AAGTTACCCAAACAGAACC-3′ (Jκ4) or 5′-TGTACTTACGTTTCAGCTCC-3′(Jκ5). Amplified DNA was cloned using the TOPO-Zero blunt cloning kit (Invitrogen Life Technologies) and further amplified by picking transformed colonies directly into a PCR reaction with primers: M13 forward (5′-GTAAAACGACGGCCAG-3′) and M13 reverse (5′-CAGGAAACAGCTATGAC-3′) provided in the kit. The PCR product was purified using the QIAquick PCR purification kit (Qiagen), mixed with the sequencing primer T3 (5′-ATTAACCCTCACTAAAGGGA-3′) and sequenced by the Keck Biotechnology Resource Laboratory at Yale University.
Sequence Analysis
Sequences of each pick were aligned to the appropriate germline V gene (i.e. the most similar one) found in the IMGT Ig sequence database [92]. They were always Vκ8-19 or Vκ8-24 combined with Jκ4 or Jκ5. (Note that Vκ8-24 has a different allele in MRL and BALB/c mice).
From these aligned sequences, phylogenetic trees were calculated using maximal parsimony criteria [93]. We developed and used a set of computer algorithms to handle the specific circumstances of Ig hypermutation analysis (manuscript in preparation). By combinatorial matching of the end regions of Vκ and Jκ, the algorithm determined all possible V-J junctions, including those potentially generated by P-nucleotides. This allowed us to differentiate between junctional diversity and somatic hypermutation in the region of the V-J join, as those bases that could not be accounted for by any combination of the germline sequences were considered to be mutations. Junctional diversity was then used to separate independent clones that may have been found in the same microdissection and which nonetheless used the Vκ and Jκ. The computer algorithm was also used to identify cases of independent parallel mutations, which could be attributed to hybridization in the PCR amplification process. Among all the sequences we found only one independent parallel mutation, indicating that there were few if any such PCR artifacts. Since isolated independent parallel mutations occur with reasonably high frequency, particularly in hotspots, this observed single instance was not discarded in view of the absence of any other possibility of PCR hybridization in that or any other sequence set.
Acknowledgments
We gratefully acknowledge Dr. Masayuki Amagai for generously providing access to his collection of anti-desmoglein mAbs. We thank Michelle Horniak, Terrence Hunt, Melissa Baez and Denise Kent for outstanding animal husbandry. We would like to thank Ann Marshak-Rothstein for critical reading of this manuscript and the members of the Shlomchik lab autoimmunity group for many useful discussions. Supported by NIH grants P01-AI36529 and P01-AR050256.
Abbreviations
- RF
rheumatoid factor
- AFC
antibody-forming cell
- IC
immune complex
- PNA
peanut agglutinin
- SHM
somatic hypermutation
Footnotes
Conflict of interest: The authors declare no conflict of interest.
References
- 1.Tan EM. Antinuclear antibodies: diagnostic markers for autoimmune diseases and probes for cell biology. Adv Immunol. 1989;44:93–151. doi: 10.1016/s0065-2776(08)60641-0. [DOI] [PubMed] [Google Scholar]
- 2.Chan OT, Hannum LG, Haberman AM, Madaio MP, Shlomchik MJ. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J Exp Med. 1999;189:1639–1648. doi: 10.1084/jem.189.10.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shlomchik MJ, Craft J, Mamula MJ. From T to B and back again: positive feedback in systemic autoimmune disease. Nature Reviews Immunology. 2001;1:147–153. doi: 10.1038/35100573. [DOI] [PubMed] [Google Scholar]
- 4.Chan O, Shlomchik MJ. A new role for B cells in systemic autoimmunity: B cells promote spontaneous T cell activation in MRL-lpr/lpr mice. J Immunol. 1998;160:51–59. [PubMed] [Google Scholar]
- 5.Eisenberg RA, Craven SY, Warren RW, Cohen PL. Stochastic control of anti-Sm autoantibodies in MRL/Mp-lpr/lpr mice. J Clin Invest. 1987;80:691–697. doi: 10.1172/JCI113123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nemazee D, Russell D, Arnold B, Haemmerling G, Allison J, Miller JF, Morahan G, Buerki K. Clonal deletion of autospecific B lymphocytes. Immunol Rev. 1991;122:117–132. doi: 10.1111/j.1600-065x.1991.tb00600.x. [DOI] [PubMed] [Google Scholar]
- 7.Tiegs SL, Russell DM, Nemazee D. Receptor editing in self-reactive bone marrow B cells. J Exp Med. 1993;177:1009–1020. doi: 10.1084/jem.177.4.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, Pritchard-Briscoe H, Wotherspoon JS, Loblay RH, Raphael K, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature. 1988;334:676–682. doi: 10.1038/334676a0. [DOI] [PubMed] [Google Scholar]
- 9.Erikson J, Radic MZ, Camper SA, Hardy RR, Carmack C, Weigert M. Expression of anti-DNA immunoglobulin transgenes in non-autoimmune mice. Nature. 1991;349:331–334. doi: 10.1038/349331a0. [DOI] [PubMed] [Google Scholar]
- 10.Santulli-Marotto S, Qian Y, Ferguson S, Clarke SH. Anti-Sm B cell differentiation in Ig transgenic MRL/Mp-lpr/lpr mice: altered differentiation and an accelerated response. J Immunol. 2001;166:5292–5299. doi: 10.4049/jimmunol.166.8.5292. [DOI] [PubMed] [Google Scholar]
- 11.Shlomchik MJ, Zharhary D, Saunders T, Camper S, Weigert M. A Rheumatoid factor transgenic mouse model of autoantibody regulation. Int Immunol. 1993;5:1329–1341. doi: 10.1093/intimm/5.10.1329. [DOI] [PubMed] [Google Scholar]
- 12.Chen c, Nagy Z, Radic MZ, Hardy RR, Huszar D, Camper SA, Weigert M. The site and stage of anti-DNA B-cell deletion. Nature. 1995;373:252–255. doi: 10.1038/373252a0. [DOI] [PubMed] [Google Scholar]
- 13.Chen C, Prak EL, Weigert M. Editing disease-associated autoantibodies. Immunity. 1997;6:97–105. doi: 10.1016/s1074-7613(00)80673-1. [DOI] [PubMed] [Google Scholar]
- 14.Li H, Jiang Y, Prak EL, Radic M, Weigert M. Editors and editing of anti-DNA receptors. Immunity. 2001;15:947–957. doi: 10.1016/s1074-7613(01)00251-5. [DOI] [PubMed] [Google Scholar]
- 15.Rubio CF, Kench J, Russell DM, Yawger R, Nemazee D. Analysis of central B cell tolerance in autoimmune-prone MRL/lpr mice bearing autoantibody transgenes. J Immunol. 1996;157:65–71. [PubMed] [Google Scholar]
- 16.Rathmell JC, Goodnow CC. Effects of the lpr mutation on elimination and inactivation of self-reactive B cells. J Immunol. 1994;153:2831–2842. [PubMed] [Google Scholar]
- 17.Steeves MA, Marion TN. Tolerance to DNA in (NZB x NZW)F1 mice that inherit an anti-DNA V(H) as a conventional micro H chain transgene but not as a V(H) knock-in transgene. J Immunol. 2004;172:6568–6577. doi: 10.4049/jimmunol.172.11.6568. [DOI] [PubMed] [Google Scholar]
- 18.Radic MZ, Mascelli MA, Erikson J, Shan H, Weigert M. Ig H and L chain contributions to autoimmune specificities. J Immunol. 1991;146:176–182. [PubMed] [Google Scholar]
- 19.Chen C, Nagy Z, Prak EL, Weigert M. Immunoglobulin heavy chain gene replacement: a mechanism of receptor editing. Immunity. 1995;3:747–755. doi: 10.1016/1074-7613(95)90064-0. [DOI] [PubMed] [Google Scholar]
- 20.Mandik-Nayak L, Bui A, Noorchashm H, Eaton A, Erikson J. Regulation of anti-double-stranded DNA B cells in nonautoimmune mice: localization to the T-B interface of the splenic follicle. J Exp Med. 1997;186:1257–1267. doi: 10.1084/jem.186.8.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hannum LG, Ni D, Haberman AM, Weigert MG, Shlomchik MJ. A disease-related RF autoantibody is not tolerized in a normal mouse: implications for the origins of autoantibodies in autoimmune disease. J Exp Med. 1996;184:1269–1278. doi: 10.1084/jem.184.4.1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandik-Nayak L, Seo SJ, Sokol C, Potts KM, Bui A, Erikson J. MRL-lpr/lpr mice exhibit a defect in maintaining developmental arrest and follicular exclusion of anti-double-stranded DNA B cells. J Exp Med. 1999;189:1799–1814. doi: 10.1084/jem.189.11.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, Shlomchik MJ. Autoantigen-specific B cell activation in Fas-deficient rheumatoid factor immunoglobulin transgenic mice. J Exp Med. 1999;190:639–649. doi: 10.1084/jem.190.5.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.William J, Euler C, Christensen S, Shlomchik MJ. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science. 2002;297:2066–2070. doi: 10.1126/science.1073924. [DOI] [PubMed] [Google Scholar]
- 25.William J, Euler C, Leadbetter E, Marshak-Rothstein A, Shlomchik MJ. Visualizing the onset and evolution of an autoantibody response in systemic autoimmunity. J Immunol. 2005;174:6872–6878. doi: 10.4049/jimmunol.174.11.6872. [DOI] [PubMed] [Google Scholar]
- 26.William J, Euler C, Shlomchik MJ. Short-lived plasmablasts dominate the early spontaneous rheumatoid factor response: differentiation pathways, hypermutating cell types, and affinity maturation outside the germinal center. J Immunol. 2005;174:6879–6887. doi: 10.4049/jimmunol.174.11.6879. [DOI] [PubMed] [Google Scholar]
- 27.William J, Euler C, Primarolo N, Shlomchik MJ. B cell tolerance checkpoints that restrict pathways of antigen-driven differentiation. J Immunol. 2006;176:2142–2151. doi: 10.4049/jimmunol.176.4.2142. [DOI] [PubMed] [Google Scholar]
- 28.Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 29.Viglianti GA, Lau CM, Hanley TM, Miko BA, Shlomchik MJ, Marshak-Rothstein A. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19:837–847. doi: 10.1016/s1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]
- 30.Lau CM, Broughton C, Tabor AS, Akira S, Flavell RA, Mamula MJ, Christensen SR, Shlomchik MJ, Viglianti GA, Rifkin IR, Marshak-Rothstein A. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang N, Reich CF, 3rd, Monestier M, Pisetsky DS. The expression of plasma nucleosomes in mice undergoing in vivo apoptosis. Clin Immunol. 2003;106:139–147. doi: 10.1016/s1521-6616(02)00027-x. [DOI] [PubMed] [Google Scholar]
- 32.Fairhurst AM, Wandstrat AE, Wakeland EK. Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Adv Immunol. 2006;92:1–69. doi: 10.1016/S0065-2776(06)92001-X. [DOI] [PubMed] [Google Scholar]
- 33.Shlomchik MJ, Marshak-Rothstein A, Wolfowicz CB, Rothstein TL, Weigert MG. The role of clonal selection and somatic mutation in autoimmunity. Nature. 1987;328:805. doi: 10.1038/328805a0. [DOI] [PubMed] [Google Scholar]
- 34.Tsunoda K, Ota T, Aoki M, Yamada T, Nagai T, Nakagawa T, Koyasu S, Nishikawa T, Amagai M. Induction of pemphigus phenotype by a mouse monoclonal antibody against the amino-terminal adhesive interface of desmoglein 3. J Immunol. 2003;170:2170–2178. doi: 10.4049/jimmunol.170.4.2170. [DOI] [PubMed] [Google Scholar]
- 35.Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J Immunol. 2004;172:6692–6700. doi: 10.4049/jimmunol.172.11.6692. [DOI] [PubMed] [Google Scholar]
- 36.Wakeland EK, Liu K, Graham RR, Behrens TW. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 2001;15:397–408. doi: 10.1016/s1074-7613(01)00201-1. [DOI] [PubMed] [Google Scholar]
- 37.Kotzin B. Systemic lupus erythematosus. Cell. 1996;85:303–306. doi: 10.1016/s0092-8674(00)81108-3. [DOI] [PubMed] [Google Scholar]
- 38.Alleva DG, Pavlovich RP, Grant C, Kaser SB, Beller DI. Aberrant macrophage cytokine production is a conserved feature among autoimmune-prone mouse strains: elevated interleukin (IL)-12 and an imbalance in tumor necrosis factor-alpha and IL-10 define a unique cytokine profile in macrophages from young nonobese diabetic mice. Diabetes. 2000;49:1106–1115. doi: 10.2337/diabetes.49.7.1106. [DOI] [PubMed] [Google Scholar]
- 39.Esfandiari E, McInnes IB, Lindop G, Huang FP, Field M, Komai-Koma M, Wei X, Liew FY. A proinflammatory role of IL-18 in the development of spontaneous autoimmune disease. J Immunol. 2001;167:5338–5347. doi: 10.4049/jimmunol.167.9.5338. [DOI] [PubMed] [Google Scholar]
- 40.Perez de Lema G, Maier H, Nieto E, Vielhauer V, Luckow B, Mampaso F, Schlondorff D. Chemokine expression precedes inflammatory cell infiltration and chemokine receptor and cytokine expression during the initiation of murine lupus nephritis. J Am Soc Nephrol. 2001;12:1369–1382. doi: 10.1681/ASN.V1271369. [DOI] [PubMed] [Google Scholar]
- 41.Lemay S, Mao C, Singh AK. Cytokine gene expression in the MRL/lpr model of lupus nephritis. Kidney Int. 1996;50:85–93. doi: 10.1038/ki.1996.290. [DOI] [PubMed] [Google Scholar]
- 42.Giese T, Davidson WF. Evidence for early onset, polyclonal activation of T cell subsets in mice homozygous for lpr. Journal of Immunology. 1992;149:3097–3010. [PubMed] [Google Scholar]
- 43.Enghard P, Langnickel D, Riemekasten G. T cell cytokine imbalance towards production of IFN-gamma and IL-10 in NZB/W F1 lupus-prone mice is associated with autoantibody levels and nephritis. Scand J Rheumatol. 2006;35:209–216. doi: 10.1080/03009740500417791. [DOI] [PubMed] [Google Scholar]
- 44.Tsokos GC, Mitchell JP, Juang YT. T cell abnormalities in human and mouse lupus: intrinsic and extrinsic. Curr Opin Rheumatol. 2003;15:542–547. doi: 10.1097/00002281-200309000-00004. [DOI] [PubMed] [Google Scholar]
- 45.Licht R, Dieker JW, Jacobs CW, Tax WJ, Berden JH. Decreased phagocytosis of apoptotic cells in diseased SLE mice. J Autoimmun. 2004;22:139–145. doi: 10.1016/j.jaut.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 46.Finke D, Baribaud F, Diggelmann H, Acha-Orbea H. Extrafollicular plasmablast B cells play a key role in carrying retroviral infection to peripheral organs. J Immunol. 2001;166:6266–6275. doi: 10.4049/jimmunol.166.10.6266. [DOI] [PubMed] [Google Scholar]
- 47.Berek C, Berger A, Apel M. Maturation of the immune response in germinal centers. Cell. 1991;67:1121–1129. doi: 10.1016/0092-8674(91)90289-b. [DOI] [PubMed] [Google Scholar]
- 48.Pascual V, Liu YJ, Magalski A, de Bouteiller O, Banchereau J, Capra JD. Analysis of somatic mutation in five B cell subsets of human tonsil. J Exp Med. 1994;180:329–339. doi: 10.1084/jem.180.1.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354:389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 50.Kleinstein SH, Singh JP. Why are there so few key mutant clones? The influence of stochastic selection and blocking on affinity maturation in the germinal center. Int Immunol. 2003;15:871–884. doi: 10.1093/intimm/dxg085.sgm. [DOI] [PubMed] [Google Scholar]
- 51.de Yebenes VG, Ramiro AR. Activation-induced deaminase: light and dark sides. Trends Mol Med. 2006;12:432–439. doi: 10.1016/j.molmed.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 52.Li Z, Woo CJ, Iglesias-Ussel MD, Ronai D, Scharff MD. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev. 2004;18:1–11. doi: 10.1101/gad.1161904. [DOI] [PubMed] [Google Scholar]
- 53.Clarke SH, Huppi K, Ruezinsky D, Staudt L, Gerhard W, Weigert M. Inter- and intraclonal diversity in the antibody response to influenza hemagglutinin. J Exp Med. 1985;161:687–704. doi: 10.1084/jem.161.4.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Madaio MP, Hodder S, Schwartz RS, Stollar BD. Responsiveness of autoimmune and normal mice to nucleic acid antigens. J Immunol. 1984;132:872–876. [PubMed] [Google Scholar]
- 55.Gilkeson GS, Grudier JP, Karounos DG, Pisetsky DS. Induction of anti-double stranded DNA antibodies in normal mice by immunization with bacterial DNA. J Immunol. 1989;142:1482–1486. [PubMed] [Google Scholar]
- 56.Desai DD, Krishnan MR, Swindle JT, Marion TN. Antigen-specific induction of antibodies against native mammalian DNA in nonautoimmune mice. J Immunol. 1993;151:1614–1626. [PubMed] [Google Scholar]
- 57.Pisetsky DS. The immunologic properties of DNA. J Immunol. 1996;156:421–423. [PubMed] [Google Scholar]
- 58.Sobel ES, Mohan C, Morel L, Schiffenbauer J, Wakeland EK. Genetic dissection of SLE pathogenesis: adoptive transfer of Sle1 mediates the loss of tolerance by bone marrow-derived B cells. J Immunol. 1999;162:2415–2421. [PubMed] [Google Scholar]
- 59.Kumar KR, Li L, Yan M, Bhaskarabhatla M, Mobley AB, Nguyen C, Mooney JM, Schatzle JD, Wakeland EK, Mohan C. Regulation of B cell tolerance by the lupus susceptibility gene Ly108. Science. 2006;312:1665–1669. doi: 10.1126/science.1125893. [DOI] [PubMed] [Google Scholar]
- 60.Liossis SNC, Kovacs B, Dennis G, Kammer GM, Tsokos GC. B cells from patients with systemic lupus erythematosus display abnormal antigen receptor-mediated early signal transduction events. J Clin Invest. 1996;98:2549–2557. doi: 10.1172/JCI119073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Takahashi Y, Ohta H, Takemori T. Fas is required for clonal selection in germinal centers and the subsequent establishment of the memory B cell repertoire. Immunity. 2001;14:181–192. doi: 10.1016/s1074-7613(01)00100-5. [DOI] [PubMed] [Google Scholar]
- 62.Potter PK, Cortes-Hernandez J, Quartier P, Botto M, Walport MJ. Lupus-Prone Mice Have an Abnormal Response to Thioglycolate and an Impaired Clearance of Apoptotic Cells. J Immunol. 2003;170:3223–3232. doi: 10.4049/jimmunol.170.6.3223. [DOI] [PubMed] [Google Scholar]
- 63.Mevorach D, Zhou JL, Song X, Elkon KB. Systemic exposure to irradiated apoptotic cells induces autoantibody production. J Exp Med. 1998;188:387–392. doi: 10.1084/jem.188.2.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kleinstein SH, Louzoun Y, Shlomchik MJ. Estimating hypermutation rates from clonal tree data. J Immunol. 2003;171:4639–4649. doi: 10.4049/jimmunol.171.9.4639. [DOI] [PubMed] [Google Scholar]
- 65.Marshak-Rothstein A, Busconi L, Lau CM, Tabor AS, Leadbetter EA, Akira S, Krieg AM, Lipford GB, Viglianti GA, Rifkin IR. Comparison of CpG s-ODNs, chromatin immune complexes, and dsDNA fragment immune complexes in the TLR9-dependent activation of rheumatoid factor B cells. J Endotoxin Res. 2004;10 :247–251. doi: 10.1179/096805104225005850. [DOI] [PubMed] [Google Scholar]
- 66.Boule MW, Broughton C, Mackay F, Akira S, Marshak-Rothstein A, Rifkin IR. Toll-like Receptor 9-Dependent and -Independent Dendritic Cell Activation by Chromatin-Immunoglobulin G Complexes. J Exp Med. 2004;199:1631–1640. doi: 10.1084/jem.20031942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Garcia De Vinuesa C, Gulbranson-Judge A, Khan M, O’Leary P, Cascalho M, Wabl M, Klaus GG, Owen MJ, MacLennan IC. Dendritic cells associated with plasmablast survival. Eur J Immunol. 1999;29:3712–3721. doi: 10.1002/(SICI)1521-4141(199911)29:11<3712::AID-IMMU3712>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 68.Liu YJ, Zhang J, Lane PJ, Chan EY, MacLennan IC. Sites of specific B cell activation in primary and secondary responses to T cell-dependent and T cell-independent antigens. European Journal of Immunology. 1991;21:2951–2962. doi: 10.1002/eji.1830211209. [DOI] [PubMed] [Google Scholar]
- 69.Toellner KM, Jenkinson WE, Taylor DR, Khan M, Sze DM, Sansom DM, Vinuesa CG, MacLennan IC. Low-level hypermutation in T cell-independent germinal centers compared with high mutation rates associated with T cell-dependent germinal centers. J Exp Med. 2002;195:383–389. doi: 10.1084/jem.20011112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.MacLennan ICM, Toellner KM, Cunningham AF, Serre K, Sze DMY, Zuniga E, Cook MC, Vinuesa CG. Extrafollicular antibody responses. Immunological Reviews. 2003 Aug;194:8–18. doi: 10.1034/j.1600-065x.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- 71.Garcia de Vinuesa C, O’Leary P, Sze DM, Toellner KM, MacLennan IC. T-independent type 2 antigens induce B cell proliferation in multiple splenic sites, but exponential growth is confined to extrafollicular foci. Eur J Immunol. 1999;29:1314–1323. doi: 10.1002/(SICI)1521-4141(199904)29:04<1314::AID-IMMU1314>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 72.MacLennan I, Vinuesa C. Dendritic cells, BAFF, and APRIL: innate players in adaptive antibody responses. Immunity. 2002;17:235–238. doi: 10.1016/s1074-7613(02)00398-9. [DOI] [PubMed] [Google Scholar]
- 73.Fields ML, Sokol CL, Eaton-Bassiri A, Seo S, Madaio MP, Erikson J. Fas/Fas ligand deficiency results in altered localization of anti-double-stranded DNA B cells and dendritic cells. J Immunol. 2001;167:2370–2378. doi: 10.4049/jimmunol.167.4.2370. [DOI] [PubMed] [Google Scholar]
- 74.Sakurai D, Kanno Y, Hase H, Kojima H, Okumura K, Kobata T. TACI attenuates antibody production costimulated by BAFF-R and CD40. Eur J Immunol. 2007;37:110–118. doi: 10.1002/eji.200636623. [DOI] [PubMed] [Google Scholar]
- 75.von Bulow GU, van Deursen JM, Bram RJ. Regulation of the T-independent humoral response by TACI. Immunity. 2001;14:573–582. doi: 10.1016/s1074-7613(01)00130-3. [DOI] [PubMed] [Google Scholar]
- 76.Hondowicz BD, Alexander ST, Quinn WJ, III, Pagan AJ, Metzgar MH, Cancro MP, Erikson J. The role of BLyS/BLyS receptors in anti-chromatin B cell regulation. Int Immunol. 2007:dxm011. doi: 10.1093/intimm/dxm011. [DOI] [PubMed] [Google Scholar]
- 77.Balazs M, Martin F, Zhou T, Kearney J. Blood dendritic cells interact with splenic marginal zone B cells to initiate T-independent immune responses. Immunity. 2002;17:341–352. doi: 10.1016/s1074-7613(02)00389-8. [DOI] [PubMed] [Google Scholar]
- 78.Bergtold A, Desai DD, Gavhane A, Clynes R. Cell Surface Recycling of Internalized Antigen Permits Dendritic Cell Priming of B Cells. Immunity. 2005;23:503–514. doi: 10.1016/j.immuni.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 79.Qi H, Egen JG, Huang AY, Germain RN. Extrafollicular activation of lymph node B cells by antigen-bearing dendritic cells. Science. 2006;312:1672–1676. doi: 10.1126/science.1125703. [DOI] [PubMed] [Google Scholar]
- 80.Hebel K, Griewank K, Inamine A, Chang HD, Muller-Hilke B, Fillatreau S, Manz RA, Radbruch A, Jung S. Plasma cell differentiation in T-independent type 2 immune responses is independent of CD11c(high) dendritic cells. Eur J Immunol. 2006;36:2912–2919. doi: 10.1002/eji.200636356. [DOI] [PubMed] [Google Scholar]
- 81.Scandella E, Fink K, Junt T, Senn BM, Lattmann E, Forster R, Hengartner H, Ludewig B. Dendritic Cell-Independent B Cell Activation During Acute Virus Infection: A Role for Early CCR7-Driven B-T Helper Cell Collaboration. J Immunol. 2007;178:1468–1476. doi: 10.4049/jimmunol.178.3.1468. [DOI] [PubMed] [Google Scholar]
- 82.Witte T, Hartung K, Sachse C, Matthias T, Fricke M, Kalden JR, Lakomek HJ, Peter HH, Schmidt RE. Rheumatoid factors in systemic lupus erythematosus: association with clinical and laboratory parameters. SLE study group. Rheumatol Int. 2000;19:107–111. doi: 10.1007/s002960050112. [DOI] [PubMed] [Google Scholar]
- 83.Dunne JV, Carson DA, Spiegelberg HL, Alspaugh MA, Vaughan JH. IgA rheumatoid factor in the sera and saliva of patients with rheumatoid arthritis and Sjogren’s syndrome. Ann Rheum Dis. 1979;38 :161–165. doi: 10.1136/ard.38.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Markusse HM, Otten HG, Vroom TM, Smeets TJ, Fokkens N, Breedveld FC. Rheumatoid factor isotypes in serum and salivary fluid of patients with primary Sjogren’s syndrome. Clin Immunol Immunopathol. 1993;66:26–32. doi: 10.1006/clin.1993.1004. [DOI] [PubMed] [Google Scholar]
- 85.Agnello V, Chung RT, Kaplan LM. A role for hepatitis C virus infection in type II cryoglobulinemia. N Engl J Med. 1992;327:1490–1495. doi: 10.1056/NEJM199211193272104. [DOI] [PubMed] [Google Scholar]
- 86.Pechere-Bertschi A, Perrin L, de Saussure P, Widmann JJ, Giostra E, Schifferli JA. Hepatitis C: a possible etiology for cryoglobulinaemia type II. Clin Exp Immunol. 1992;89:419–422. doi: 10.1111/j.1365-2249.1992.tb06973.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Clifford BD, Donahue D, Smith L, Cable E, Luttig B, Manns M, Bonkovsky HL. High prevalence of serological markers of autoimmunity in patients with chronic hepatitis C. Hepatology. 1995;21:613–619. [PubMed] [Google Scholar]
- 88.Nemazee D. Immune complexes can trigger specific, T cell-dependent, autoanti-IgG antibody production in mice. J Exp Med. 1985;161:242–256. doi: 10.1084/jem.161.1.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Van Snick J, Coulie P. Rheumatoid factors and secondary immune responses in the mouse. I. Frequent occurrence of hybridomas secreting IgM anti-IgG1 autoantibodies after immunization with protein antigens. Eur J Immunol. 1983;13:895. doi: 10.1002/eji.1830131106. [DOI] [PubMed] [Google Scholar]
- 90.Losman MJ, Fasy TM, Novick KE, Monestier M. Monoclonal autoantibodies to subnucleosomes from a MRL/Mp(−)+/+ mouse. Oligoclonality of the antibody response and recognition of a determinant composed of histones H2A, H2B, and DNA. J Immunol. 1992;148:1561–1569. [PubMed] [Google Scholar]
- 91.Hannum LG, Haberman AM, Anderson SM, Shlomchik MJ. Germinal center initiation, variable gene region hypermutation, and mutant B cell selection without detectable immune complexes on follicular dendritic cells. J Exp Med. 2000;192:931–942. doi: 10.1084/jem.192.7.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lefranc MP, Giudicelli V, Kaas Q, Duprat E, Jabado-Michaloud J, Scaviner D, Ginestoux C, Clement O, Chaume D, Lefranc G. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2005;33:D593–597. doi: 10.1093/nar/gki065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9:1657–1659. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]