

Abstract
Microtubule stabilizers are powerful anti-mitotic compounds and represent a proven cancer treatment strategy. Several classes of compounds in clinical use or trials, such as the taxanes and epothilones, bind to the same region of β-tubulin. Determining how these molecules interact with tubulin and stabilize microtubules is important both for understanding the mechanism of action and enhancing chemotherapeutic potential, e.g. reducing side effects, increasing solubility, and overcoming resistance. Structural studies using nonpolymerized tubulin or stabilized polymers have produced different models of epothilone binding. Here, we used directed mutagenesis of the binding site on Saccharomyces cerevisiae β-tubulin to analyze interactions between Epothilone B and its biologically relevant substrate, dynamic microtubules. Five engineered amino acid changes contributed to a 125-fold increase in Epothilone B cytotoxicity independent of inherent microtubule stability. The mutagenesis of endogenous β-tubulin was done in otherwise isogenic strains. This facilitated the correlation of amino acid substitutions with altered cytotoxicity using molecular mechanics simulations. The results, which are based on the interaction between Epothilone B and dynamic microtubules, most strongly support the binding mode determined by NMR spectroscopy-based studies. This work establishes a system for discriminating between potential binding modes and among various compounds and/or analogues using a sensitive biological activity-based readout.
Keywords: epothilone, microtubule, tubulin, taxol binding site, microtubule stabilizer, drug design, antitumor agents
Introduction
Anti-mitotic compounds that bind tubulin and disrupt microtubule dynamics provide for important cancer therapies, and understanding the molecular mechanisms of these compounds holds the promise of optimizing and exploiting this clinically proven strategy. The taxanes represent an important class of anti-cancer agents, e.g. paclitaxel and docetaxel, that produce cytotoxicity by enhancing the assembly of tubulin, stabilizing microtubules, suppressing microtubule dynamics, and causing mitotic arrest and apoptosis.[1] The epothilones are a group of natural products produced by the myxobacterium Sorangium cellulosum[2] that bind to tubulin in microtubules at a site that overlaps with the paclitaxel site, and generate cytotoxicity through a similar mechanism.[3, 4] An attractive property of the epothilones is that they may retain excellent activity against cells that have become resistant to the taxanes due to over-expression of the multi-drug transporter.[3–5] Currently Epothilone B (EpoB, Fig. 1) and various epothilone analogues are in clinical trials[6] and Ixabepilone is approved for use against some cancers. However, the molecular interactions of the epothilones with tubulin/microtubules remain unclear.
Figure 1.
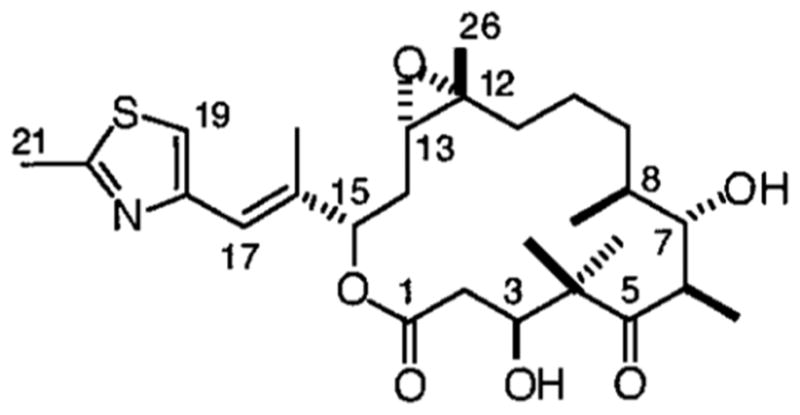
Structure of Epothilone B (EpoB).
The region of β-tubulin that binds to the taxanes and epothilones can exert powerful influence over microtubule dynamics. Moreover, the knowledge of the molecular details at this site carries significant clinical potential. Our current understanding of how taxanes and epothilones interact with β-tubulin comes mainly from the electron crystal (EC) structures of complexes between the compounds and a tubulin polymer induced by zinc ions,[7, 8] and from NMR spectroscopy,[9, 10] supported by structure-activity relationship analysis (SAR)[11] and molecular modeling efforts.[12] Although the two classes of compounds compete with each other for binding[3, 4] and bind to the same cavity in β-tubulin,[3, 4] the EC structures of the drug-tubulin polymer complexes have shown that the binding interactions are not identical.[7, 8] This is also reflected in results with wild-type yeast tubulin. EpoB binds to Saccharomyces cerevisiae tubulin, but paclitaxel does not.[13]
Two strikingly distinct models of Epothilone binding to tubulin have been generated using an NMR[10] based approach or EC[7] derived measurements at ~3 Å resolution which was not adequate to directly define the bound conformation. Thus there is a need to determine the active conformation of tubulin-bound epothilone using structure-activity approaches. To date, SAR data for the epothilones have been limited to ligand binding studies with wild-type tubulin, or spontaneously occurring tubulin mutations in tumor or culture cell lines. Alternatively, site-directed mutagenesis of endogenous tubulin would allow controlled analysis of the epothilone-binding pocket. By converting five residues in S. cerevisiae β-tubulin to those of mammalian brain tubulin, we were able to impart paclitaxel-binding activity to budding yeast tubulin.[14, 15] Here we report that the five amino acid changes that confer paclitaxel binding to yeast tubulin drastically increase the cytotoxicity of EpoB, indicating that the interactions that mediate the potency of both compounds may be more similar than previously predicted based on electron crystallography.[7] These changes, Ala19Lys, Thr23Val, Gly26Asp, Asn227His, and Tyr270Phe allowed the role of each of these residues to be individually examined. This effect was cumulatively mediated by the five substitutions. Thus, we were able to utilize molecular modeling simulations to distinguish between the current models for the binding of EpoB to tubulin.
Results and Discussion
Mutations in the taxane-binding region of β-tubulin mediate EpoB cytotoxicity
We found that changing five amino acids in the yeast β-tubulin EpoB binding pocket to those found in bovine brain tubulin (Tub2-BBBBB) increased the cytotoxicity of EpoB for these cells by 125-fold (Table 1). The ED50 for cells with wild-type tubulin (Tub2-YYYYY) was 2,633 nM, whereas Tub2-BBBBB-containing yeast had an ED50 of just 21 nM. Thus, some or all of these five amino acid substitutions are responsible for optimizing microtubule-stabilizing interactions between EpoB and tubulin.
Table 1.
EpoB ED50 values for S. cerevisiae strains with modified β-tubulin.
| Tubulin form | ED50, nM | Relative cytotoxicity, ED50(YYYYY)/ED50 |
|---|---|---|
| Tub2-BBBBB | 21 ± 3 | 125 |
| Tub2-BBBBY | 38 ± 8 | 69 |
| Tub2-BBYBB | 46 ± 6 | 57 |
| Tub2-BYBBB | 50 ± 7 | 53 |
| Tub2-BBBYB | 276 ± 50 | 10 |
| Tub2-YBBBB | 277 ± 28 | 10 |
| Tub2-YYYYY | 2,633 ± 362 | 1 |
The cytotoxicity of epothilone B for the different yeast strains was determined as described in the methods. Each ED50 reflects the average (±SEM) of three to five experiments.
To hone in on the specific interactions responsible for the cytotoxic effects of EpoB we created variant tubulins by individually changing the five substitutions in Tub2-BBBBB back to those found in yeast tubulin. These new variants showed a range of sensitivities. Figure 2 represents the growth inhibition by EpoB from a single experiment, while Table 1 reports the results from three to five experiments for each tubulin variant. Compared to Tub2-BBBBB yeast, the largest losses in cytotoxicity occurred when either the Lys19 or the His227 of brain tubulin was back mutated to the Ala19 or Asn227 or yeast (Tub2-YBBBB or Tub2-BBBYB). These single amino acid changes were each associated with a 13-fold increase in ED50, respectively (Table 1). Yeast strains with individual brain-to-yeast back mutations Val23Thr, Asp26Gly, or Phe270Tyr, on the other hand, showed only modest decreases in cytotoxicity. The ED50 values of the Thr23, Gly26, or Tyr270 strains increased ~2-fold compared to cells with all five substitutions (50, 46, and 38 nM vs. 21 nM, respectively). Thus, amino acids Lys19 and His227 contribute significantly to effective interactions between tubulin and EpoB. However, the cumulative effect of the five mutations was much greater than any single mutation.
Figure 2.

Growth inhibition by EpoB of S. cerevisiae strains with modified β-tubulin. The graph depicts a representative experiment in which growth rates were monitored over 24 h in the presence of increasing EpoB concentrations.
Mutations in the taxane-binding region of β-tubulin do not increase inherent microtubule stability
Although all five mutated amino acids lie within the EpoB binding pocket of tubulin, it is possible that the mutations generally stabilize microtubules, either intrinsically or by disrupting interactions with cellular regulators. This could render cells more sensitive to microtubule stabilizers such as EpoB, resulting in an increase in cytotoxicity. To determine whether the five mutations had stabilized microtubules independently of EpoB binding we analyzed in vivo microtubule dynamics in cells containing Tub2-YYYYY or Tub2-BBBBB in the absence of EpoB (Fig. 3). Overall, we found microtubule dynamics in the two strains to be largely unaffected by the substitutions alone, suggesting the observed cytotoxicity is due to EpoB-tubulin interactions (Table 2). The most notable changes in the Tub2-BBBBB cells were a decrease in depolymerization rate and an increase in attenuation, which could reflect slightly stabilized microtubules. However, this was accompanied by a decrease in rescue frequency, relative to Tub2-YYYYY cells, which may reflect slightly destabilized microtubules.
Figure 3.

Cytoplasmic microtubule dynamic behavior in G1 cells containing Tub2-YYYYY (top), and Tub2-BBBBB (bottom). Two representative lifetime history plots (black & gray) depicting the length of individual microtubules over time are presented for each condition.
Table 2.
Parameters of dynamic instability for cytoplasmic microtubules in vivo.
| Tubulin form | Tub2-BBBBB | Tub2-YYYYY |
|---|---|---|
| Polymerization Rate (μm/min) | 1.4 ± 0.7 (30) | 1.7 ± 0.7 (33) |
| Depolymerization Rate (μm/min) | 1.9 ± 0.7 (38)* | 2.6 ± 1.2 (38) |
| Catastrophe Frequency (per min) | 0.59 (28) | 0.64 (29) |
| Rescue Frequency (per min) | 1.07 (21) | 1.33 (25) |
| Time spent polymerizing, % | 24 | 29 |
| Time spent depolymerizing, % | 29 | 29 |
| Time spent attenuated, % | 46 | 42 |
| polymerization duration, s | 33 ± 13 (30) | 34 ± 16 (33) |
| depolymerization duration, s | 31 ± 15 (38) | 30 ± 15 (33) |
| attenuation duration, s | 44 ± 28 (44)* | 34 ± 16 (49) |
| total microtubules observed | 28 | 28 |
| total time observed, s | 4014 | 3858 |
Number of events is in parentheses. Rates/durations are presented as mean ± SD.
p < 0.05 compared to Tub2-YYYYY by unpaired, two-tailed t-test.
In order for inherent microtubule stability to be responsible for the graded cytotoxicity profile, each mutation would likely contribute some degree of increased microtubule stability. Previously, of 53 mutations created in an alanine-scanning analysis of yeast Tub1 (α-tubulin), only six (11%) produced detectable levels of resistance to the microtubule destabilizing compound benomyl, suggestive of microtubule stabilization.[16] However, 37 mutations (70%) increased sensitivity to benomyl revealing that, overall, tubulin mutations are more likely to destabilize rather than stabilize microtubules. Likewise, in our previous systematic analysis of cysteine residues in Tub2, 17% of mutations resulted in increased benomyl resistance while 42% caused increased benomyl sensitivity.[17] These results support our conclusion that the increased EpoB cytotoxicity is directly due to increased affinity of the drug to the mutated tubulins.
Molecular modeling correlates amino acid changes with EpoB cytotoxicity
Together, our cytotoxicity results and microtubule dynamics comparison demonstrate that the five mutated residues differentially contribute to the binding affinity of EpoB for tubulin and/or microtubules. To help interpret the changes in the EpoB ED50 values as a function of the different binding site mutations, we performed molecular mechanics simulations based on the EC and NMR structures using a yeast tubulin homology model. The binding scores for EpoB interacting with the panel of tubulin mutants, presented in Table 3, were determined using four different formulations, ChemScore,[18] DrugScore,[19] G-Score[20] and PMF.[21] The various scoring models generate different quantitative values due to the fact that each takes into account a different set of physicochemical properties. Therefore, scores for different mutants within a given formulation can be compared to understand the relative contribution of each residue to EpoB binding. Also, this allows for correlating the scores of each mutation in the EC and NMR models with the observed in vivo cytotoxicity data.
Table 3.
Approximate binding scores computed for EpoB in complex with mutants of yeast tubulin as a function of binding mode model and computational scoring function.[a]
| Tubulin form | Electron Crystallography Model[b] | NMR Model[c] | ||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Scoring model | Scoring model | |||||||
| Chem[d] | Drug[e] | G[f] | PMF[g] | Chem | Drug | G | PMF | |
| Tub2-BBBBB | −23.14 | −100.8 | −195.0 | −40.49 | −22.43 | −113.4 | −181.6 | −33.00 |
| Tub2-BBBBY | −23.02 | −101.9 | −194.8 | −36.79 | −21.62 | −111.0 | −176.8 | −27.03 |
| Tub2-BBYBB | −22.66 | −97.7 | −193.8 | −38.96 | −21.90 | −108.6 | −177.1 | −32.95 |
| Tub2-BYBBB | −22.11 | −100.5 | −197.4 | −36.48 | −20.97 | −111.9 | −177.2 | −36.21 |
| Tub2-BBBYB | −19.55 | −87.5 | −165.6 | −27.56 | −19.19 | −102.4 | −160.4 | −18.96 |
| Tub2-YBBBB | −22.60 | −99.7 | −188.3 | −39.65 | −16.80 | −96.2 | −139.7 | −11.36 |
| Tub2-YYYYY | −10.72 | −83.8 | −126.4 | −17.00 | −14.20 | −93.9 | −145.9 | −11.11 |
|
| ||||||||
| Correlation R(log[ED50]) | 0.75 | 0.51 | 0.73 | 0.68 | 0.83 | 0.76 | 0.70 | 0.77 |
Different scoring functions have different physical interpretations, but in each case a more negative score indicates stronger binding, and in each case, as shown in the bottom row, scores should approximately correlate with log[ED50].
Ref.[7]
Ref.[10]
ChemScore[18]
DrugScore[19]
G-Score[20]
PMF[21]
The scores for both models largely predicted Tub2-YYYYY to exhibit the lowest affinity for EpoB, while the bovine-like Tub2-BBBBB produced the highest ligand affinity. Additionally, the scores for Tub2-YBBBB, the mutant with the lowest sensitivity, strikingly discriminated between the EC and NMR binding modes across all four scoring models (Table 3, bold text). In the EC model the scores for this mutant were close to those for Tub2-BBBBB and ranged from the first to fourth highest ligand affinities of the tubulin variants containing four of the five brain residues. Conversely, in the NMR model the values were much closer to those for Tub2-YYYYY, and always predicted the lowest EpoB binding affinity of the six tubulin mutants. Clearly, the NMR model more correctly predicted the cytotoxicity data for Tub2-YBBBB.
In both models the scores for Tub2-BBBBY, Tub2-BBYBB, and Tub2-BYBBB correlated well with the cytotoxicity data. The scores were closer to those for Tub2-BBBBB than to Tub2-YYYYY. Also consistent with the cytotoxicity data is the fact that the scores for Tub2-BBBYB were farther away from Tub2-BBBBB, and fell somewhere between the scores for Tub2-BBBBB and Tub2-YYYYY. Of the two models, the binding scores produced by the NMR model generally correlated better with the in vivo cytotoxicity data across the range of tubulin variants (Table 3).
Variations in the predicted binding mode as a function of different tubulin mutants are reported in Figure 4 for models assessed from NMR data (upper two quadrants) and EC characterization (lower). These illustrations each focus on comparing the most potent complex (EpoB bound to Tub2-BBBBB) with those of two specific tubulin variants: Tub2-YBBBB (left side) and Tub2-BBBYB (right side). From these figures, one can perceive clear differences in the predicted EpoB binding modes arising from the NMR and EC models, however; there are also similarities across the two models in the types of EpoB-tubulin interactions that are predicted to occur.
Figure 4.

Comparisons between the computationally minimized structures for the complex of EpoB bound to Tub2-BBBBB tubulin (grey structures in all panels) vs. EpoB in complex with the Tub2-YBBBB (left-hand quadrants) or Tub2-BBBYB (right quadrants) tubulin mutants (shown as green-colored sticks in all panels). The two top quadrants depict complexes solved based on ligand-binding according to NMR characterization (12), while the two bottom quadrants depict complexes derived from assuming binding modes analogous to the EC prediction (9). All heteroatoms within the ligand (EpoB) and key receptor residues (tubulin) are colored according to standard CPK coloring; tubulin secondary structure is rendered as tan ribbons. Thick black arrows denote favorable interactions enjoyed by the Tub2-BBBBB complex but absent for corresponding mutants. Thin dashed arrows are used to indicate the position of residue 227.
The fairly benign nature of the back-substitutions at residues 23, 26 and 270 can be explained by the following analysis. In both the NMR and EC models, the sidechain methyls of Val23 (Tub2-BBBBB) engage in favorable lipophilic interactions with EpoB that are not fully matched by Thr23 (Tub2-BYBBB). In the NMR structure, both methyl carbons on Val23 of Tub2-BBBBB are predicted to have lipophilic interactions with 11-methylene on EpoB (4.03Å and 4.15Å respectively) whereas for Tub2-BYBBB, Thr23 has only one sidechain methyl (at a distance of 4.05 Å) to produce a comparable interaction. In the EC model, whereas both of the Val23 methyl groups have modest interactions with EpoB (one with the 21-Me at 4.20 Å; the other with 26-Me at 4.58 Å), Thr23 appears to engage in no favorable lipophilic interactions, instead only forming a possible weak H-bond between the sidechain hydroxyl proton and the thiazole N of EpoB (3.77 Å). In the EC and NMR models, the Asp26 Cα in Tub2-BBBBB approaches somewhat more closely to favorable lipophilic contacts than does Gly26 Cα in Tub2-BBYBB (4.63 vs. 4.92 Å separation from the thiazole in the EC model; 4.90 vs. 5.13 Å distance from 12-Me for the NMR complex). Furthermore, the Asp26 Cβ (not present in Tub2-BBYBB) is also available for weak lipophilic interactions in both models. The structure of the Tub2-BBBBY (Tyr270) complex with EpoB is very similar to that observed for Tub2-BBBBB (Phe270) in both models, however the very modest difference in EpoB potency upon mutation of this residue can be rationalized by slightly closer proximity between Phe270 and EpoB carbon atoms (EC: 3.91 Å; NMR: 3.73 Å) than is possible for Tyr270 (EC: 4.18 Å; NMR: 3.84 Å), with all other aspects of the complexes being very closely conserved upon mutation. Together the modeling and cytotoxicity data suggest that Phe270 does not mediate critical interactions with EpoB. Consistent with this conclusion, a human ovarian carcinoma cell line with an acquired Phe270Val mutation displayed 24-fold paclitaxel resistance yet only a 3-fold effect on EpoB activity.[22]
The predicted structure for the Epothilone binding site in the yeast tubulin homology model is marginally more spatially constricted than that resolved for bovine brain tubulin. Thus, whereas prior studies have suggested close interactions between the EpoB thiazole ring and the His227 residue in bovine brain tubulin,[7, 10, 13] such coupling may not be energetically favored in the more constrained yeast tubulin receptor. Specifically, for a receptor model based on the yeast tubulin sequence, we do not predict that the EpoB thiazole ring will engage in H-bonding with the Asn227 residue present in wild-type yeast tubulin or in either H-bonding or π-stacking with His227 present in the mutant forms. Rather, both the EC and NMR models predict lipophilic interactions between the imidazole ring of His227 and the EpoB macrocycle. In the EC model there is a 3.57 Å separation between the imidazole ring and the 4-Me on EpoB for Tub2-BBBBB, and in the NMR model there is a 3.81 Å and 3.96 Å distance to the 4-Me and 6-Me, respectively. The lipophilic interactions between the imidazole ring and the EpoB 4-Me or 6-Me are unavailable to the Asn227 residue in Tub2-BBBYB. An additional difference evident in the NMR model is further EpoB stabilization by His (as compared to Asn) via a methyl/π interaction with EpoB 16-Me (3.21 Å separation).
Ala19 in Tub2-YBBBB does not have significant interactions with EpoB in either the NMR or EC models, however the mutation to Lys19 affords favorable lipophilic contact in both cases. Specifically, in the case of the EC model the methyl carbon located off the thiazole ring approaches within 4.39 Å of Cγ on the Lys19 side chain, while in the NMR model, the unsubstituted thiazole carbon is within 3.83 Å of Lys19 Cγ. In the latter case, there may also be a modest electrostatic coupling between the lysine cationic N and the thiazole S (4.22 Å separation).
From this analysis, it is apparent that both the NMR and EC models predict that similarly favorable interactions, between EpoB and its high potency complex with Tub2-BBBBB, are disrupted in the various back-mutational forms and the low potency Tub2-YYYYY receptor. This is likely the reason that both the EC and NMR models produced consistently high levels of correlation between computed binding scores and experimentally observed ED50 data (Table 3). While the predicted models we have generated herein do not lend definitive corroboration to the NMR relative to the EC model, the NMR model better explains the cytotoxicity results of the Tub2-YBBBB mutant and produced higher correlations with observed in vivo results.
Summary
Determining the biologically relevant interactions between the taxanes/epothilones and microtubules is an important goal. Current models of these interactions are based largely on artificial systems (nonpolymerized tubulin[9, 10] and zinc-stabilized tubulin sheets[7, 8]). Using in vivo microtubules as substrates we demonstrated that amino acids critical for paclitaxel binding also strongly promote the cytotoxicity of EpoB, indicating that similar interactions may mediate the toxicity of both compounds. However, the three substitutions that were most critical for paclitaxel activity were least important for EpoB activity (Thr23Val, Gly26Asp, Try270Phe) while the two substitutions dispensable to paclitaxel activity made the largest contributions to EpoB activity (Ala19Lys, Asn227His).[23] Interestingly, this trend illustrates that fundamental differences in the binding interactions of the two compounds also exist. Paclitaxel and EpoB have been shown to stimulate the in vitro assembly of Tub2-BBBBB with similar activities.[14] Therefore, it is interesting that yeast containing the modified tubulin were ~300-fold more sensitive to EpoB than reported for paclitaxel, 21 nM versus 6500 nM,[23] respectively. Similar differences in human cells result from inefficient export of EpoB by the ABC-transport protein P-glycoprotein.[3] Correspondingly, perhaps paclitaxel, and not EpoB, is a substrate for any transport activity remaining in the drug sensitized AD1-8 yeast strain.
Substoichiometric concentrations of both microtubule stabilizers[24] and inhibitors[25] are known to suppress microtubule dynamics and stabilize the polymer, highlighting the importance of understanding their interactions with dynamic microtubules. Moreover, assays utilizing microtubule substrates in isogenic cells expressing endogenous levels of tubulin will reflect important transitional conformations or specific interactions that may occur at the ends of dynamic microtubules. Site directed mutagenesis of the taxane/epothilone-binding pocket will likely reveal interactions critical to the active conformation and biological activity of these important classes of compounds.
Experimental Section
Yeast strains and cytotoxicity
Yeast growth, genetic manipulations, and lithium acetate transformation were performed using standard techniques.[26] Yeast strains used or created in this study are listed in Table 4.
Table 4.
Yeast strains used in this study.
| Strain | Relevant genotype |
|---|---|
| AD1-8[27] | MATa, PDR1-3, ura3, his1, Dyor1::hisG, Dsnq2::hisG, Dpdr5::hisG, Dpdr10::hisG, Dpdr11::hisG, Dycf1::hisG, Dpdr3::hisG, Dpdr15::hisG |
| MGY612 | tub2-YYYYY-His6-URA3, otherwise isogenic to AD1-8 |
| MGY613 | tub2-BBBBB-His6-URA3, otherwise isogenic to AD1-8 |
| MGY614 | tub2-BBBBY-His6-URA3, otherwise isogenic to AD1-8 |
| MGY615 | tub2-BBBYB-His6-URA3, otherwise isogenic to AD1-8 |
| MGY616 | tub2-BBYBB-His6-URA3, otherwise isogenic to AD1-8 |
| MGY617 | tub2-BYBBB-His6-URA3, otherwise isogenic to AD1-8 |
| MGY618 | tub2-YBBBB-His6-URA3, otherwise isogenic to AD1-8 |
| MGY981 | MATa, tub2-YYYYY-His6-URA3, LEU2::GFP-TUB1::leu2D1 |
| MGY982 | MATa, tub2-BBBBB-His6-URA3, LEU2::GFP-TUB1::leu2D1 |
To determine the in vivo efficacy of EpoB the Saccharomyces cerevisiae strain AD12345678 (AD1-8)[27] which is deficient in seven drug transporters as well as one transcription factor (pdr3) was transformed with a yeast genomic DNA fragment containing the β-tubulin (TUB2) coding sequence with different subsets of the five amino acid changes and a C-terminal His6 tag as previously described (Table 5).[15, 17] This transformation strategy replaced the endogenous β-tubulin locus with the mutated version. The sensitivity of each strain to EpoB was measured by comparing the growth of an equal number of cells after 24–27 hours in rich media (YPD) at 30° C[15, 23] or by determining the doubling time from the increase in O.D. 600 nm measured every 15 min in a Tecan Safire II plate reader maintained at 30° C with continuous orbital mixing. The concentration of EpoB that inhibited growth rate by 50%, the ED50, was determined from fitting the log(EpoB) versus normalized growth rates by nonlinear regression using Prism software (Graphpad).
Table 5.
Specific mutations introduced to the epothilone binding pocket of yeast β-tubulin Tub2.
| Tubulin form | Specific mutations |
|---|---|
| Tub2-BBBBB | A19K, T23V, G26D, N227H, Y270F, C-term His6 |
| Tub2-BBBBY | A19K, T23V, G26D, N227H, C-term His6 |
| Tub2-BBBYB | A19K, T23V, G26D, Y270F, C-term His6 |
| Tub2-BBYBB | A19K, T23V, N227H, Y270F, C-term His6 |
| Tub2-BYBBB | A19K, G26D, N227H, Y270F, C-term His6 |
| Tub2-YBBBB | T23V, G26D, N227H, Y270F, C-term His6 |
| Tub2-YYYYY | C-term His6 |
Fluorescence microscopy and microtubule dynamics analysis
In vivo analyses of microtubule dynamics in G1 (unbudded) cells were performed essentially as described previously.[28] A control cell harboring an exogenous copy of GFP-Tub1 (α-tubulin; pMG3)[28] under the TUB1 promoter was then transformed with fragments to replace the endogenous TUB2 locus with either tub2-YYYYY-His6 or tub2-BBBBB-His6 (Table 5). Cells were grown to midlog phase in SD-complete media (0.67% yeast nitrogen base without amino acids, 2% glucose, and supplemented with amino acids)[26] and placed onto a microscope slide padded with 1% agarose in the same medium. Coverslips were sealed using VALAP and epifluorescence time-lapse images of G1 cells were obtained on a Zeiss AxioImager M2 microscope with a Piezo-electric driven Z-stage, 63X 1.4 NA objective, Semrock filters, and a Coolsnap HQ2 CCD camera (Photometrics, Inc.) driven by SlideBook software. Image series consisted of 8–10 Z-planes spaced 0.75 μm apart obtained every six seconds for at least 6 minutes. The three dimensional length of individual astral microtubules was subsequently determined at each timepoint using SlideBook software. Polymerization and depolymerization events were defined as a line through at least four data points (24 s) that spanned a range of > 0.4 μm with an R2 value of ≥ 0.84. Periods of attenuation were defined as persisting at least four data points with net length changes of < ± 0.2 μm. Data points that did not fit these criteria were discarded. The percentage of time spent in each phase was determined by dividing the sum of the time in each phase by the total classified time for all microtubules analyzed. Catastrophes were defined as a transition into depolymerization following polymerization or attenuation. Rescues were defined as transition out of depolymerization into polymerization or attenuation. Only the time spent polymerizing or attenuated was considered to determine the catastrophe frequency. Similarly, only time spent depolymerizing was considered to calculate rescue frequency. Cells containing Tub2-YYYYY-His6 or Tub2-BBBBB-His6 (MGY981 or MGY982) were imaged on three separate days. Cells were grown and maintained at 23° C during imaging. P values were determined by Student’s t test.
Molecular modeling
Molecular mechanics simulations were based on a model of the structure of yeast beta tubulin generated via the SwissModel[29] tool (according to default first-approach mode specifications) based on the epothilone A/bovine brain tubulin electron diffraction crystal structure.[7] Recognizing uncertainty in the real epothilone binding mode, we modeled one scenario that assumed an EpoB binding mode as represented in the original crystallographic structure and a second scenario designed to emulate the binding mode obtained by NMR experiments of Carlomagno et al.[10] To prepare an EpoB-tubulin structure for evaluating the effect of receptor mutations within the context of the original crystal structure, we used SYBYL 8.1 (2009, Tripos, Inc., St. Louis MO) to modify the co-crystallized epothilone A into EpoB (i.e., by substituting a methyl group for the proton on C12 of epothilone A), and created estimated structures for the relevant mutants by substituting the corresponding amino acids (Ala/Lys19, Thr/Val23, Gly/Asp26, Asn/His227, and Phe/Tyr270) via the Biopolymer module in SYBYL. We constructed initial models representing the NMR-predicted scenario in a similar manner with the exception that we modified the α-β-γ-δ-torsion of His227 from 172.2° to 267.6° to support a potential π-stacking interaction with the EpoB thiazole (instead of the previously assumed H-bond), and we repositioned the ligand to encourage lipophilic interactions between the EpoB 4-Me groups and tubulin residues Leu215, Leu228 and Leu273, and between the 6-Me group and Ala231 and Phe270. This repositioning was accomplished by a short (1 ps) low temperature (200K) molecular dynamics simulation in SYBYL via the Tripos molecular force field[30] and Gasteiger-Marsili electrostatics[31] using weak (10 kcal/mol Å) constraints that held the interacting carbon pairs (as mentioned above) within 3–5 Å of each other. Together the seven tubulin variants (Tub2-BBBBB, Tub2-YBBBB, Tub2-BYBBB, Tub2-BBYBB, Tub2-BBBYB, Tub2-BBBBY and Tub2-YYYYY) and two EpoB binding modes produced 14 EpoB/tubulin models.
To gauge the structural effects of each tubulin mutation on the EpoB binding modes, we subjected each of the initial EpoB/tubulin models to molecular mechanics optimization. For each complex, we permitted an unlimited number of optimization steps, employed very fine criteria (maximum displacement of 0.001 Å; maximum ΔE of 0.0005 kcal/mol; gradient of 0.005 kcal/(mol Å)), and requested gradient recomputation after every geometry step. To achieve these convergence criteria in a reasonable time, we assumed that the mutations would have only a modest effect on the crystallographically resolved atomic coordinates of conserved residues and thus relaxed only the ligand, plus residues 19, 23, 26, 227 and 270. Based on results from our prior study on taxol interacting with mutations of β-tubulin,[23] we chose to compare computational estimates for the relative EpoB potency in these different receptors via the ChemScore,[18] DrugScore,[19] G-Score[20] and PMF[21] formalisms for estimating the binding score.
Acknowledgments
We thank Travis Foland for excellent technical assistance. R.S.R was supported by a National Institutes of Health postdoctoral training fellowship (T32 HL094282). This work was funded by: a National Institutes of Health grant CA105305 to R.H.H., a National Center for Research Resources (Kansas IDeA Network for Biomedical Research Excellence; NCRR award #P20 RR016475) grant to G.H.L., and a National Institutes of Health grant (R01GM094313), Cancer Research Foundation Young Investigator Grant, and American Cancer Society Institutional Research Grant (#IRG-58-004-48) to M.L.G.
References
- 1.Jordan MA. Curr Med Chem Anticancer Agents. 2002;2(1):1–17. doi: 10.2174/1568011023354290. [DOI] [PubMed] [Google Scholar]
- 2.Gerth K, Bedorf N, Hofle G, Irschik H, Reichenbach H. J Antibiot (Tokyo) 1996;49(6):560–563. doi: 10.7164/antibiotics.49.560. [DOI] [PubMed] [Google Scholar]
- 3.Kowalski RJ, Giannakakou P, Hamel E. J Biol Chem. 1997;272(4):2534–2541. doi: 10.1074/jbc.272.4.2534. [DOI] [PubMed] [Google Scholar]
- 4.Bollag DM, McQueney PA, Zhu J, Hensens O, Koupal L, Liesch J, Goetz M, Lazarides E, Woods CM. Cancer Res. 1995;55(11):2325–2333. [PubMed] [Google Scholar]
- 5.Altmann KH, Wartmann M, O’Reilly T. Biochim Biophys Acta. 2000;1470(3):M79–91. doi: 10.1016/s0304-419x(00)00009-3. [DOI] [PubMed] [Google Scholar]; O’Reilly T, Wartmann M, Brueggen J, Allegrini PR, Floersheimer A, Maira M, McSheehy PM. Cancer Chemother Pharmacol. 2008;62(6):1045–1054. doi: 10.1007/s00280-008-0695-9. [DOI] [PubMed] [Google Scholar]
- 6.Melichar B, Casado E, Bridgewater J, Bennouna J, Campone M, Vitek P, Delord JP, Cerman J, Jr, Salazar R, Dvorak J, Sguotti C, Urban P, Viraswami-Appanna K, Tan E, Tabernero J. Br J Cancer. 2011;105(11):1646–1653. doi: 10.1038/bjc.2011.438. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bystricky B, Chau I. Expert Opin Investig Drugs. 2011;20(1):107–117. doi: 10.1517/13543784.2011.542148. [DOI] [PubMed] [Google Scholar]; Ferrandina G, Mariani M, Andreoli M, Shahabi S, Scambia G, Ferlini C. Curr Pharm Des. 2012;18(19):2793–2803. doi: 10.2174/138161212800626238. [DOI] [PubMed] [Google Scholar]; Lee JJ, Swain SM. Clin Cancer Res. 2008;14(6):1618–1624. doi: 10.1158/1078-0432.CCR-07-2201. [DOI] [PubMed] [Google Scholar]
- 7.Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. Science. 2004;305(5685):866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- 8.Snyder JP, Nettles JH, Cornett B, Downing KH, Nogales E. Proc Natl Acad Sci U S A. 2001;98(9):5312–5316. doi: 10.1073/pnas.051309398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar A, Heise H, Blommers MJ, Krastel P, Schmitt E, Petersen F, Jeganathan S, Mandelkow EM, Carlomagno T, Griesinger C, Baldus M. Angew Chem Int Ed Engl. 2010;49(41):7504–7507. doi: 10.1002/anie.201001946. [DOI] [PubMed] [Google Scholar]
- 10.Carlomagno T, Blommers MJ, Meiler J, Jahnke W, Schupp T, Petersen F, Schinzer D, Altmann KH, Griesinger C. Angew Chem Int Ed Engl. 2003;42(22):2511–2515. doi: 10.1002/anie.200351276. [DOI] [PubMed] [Google Scholar]
- 11.Reese M, Sanchez-Pedregal VM, Kubicek K, Meiler J, Blommers MJ, Griesinger C, Carlomagno T. Angew Chem Int Ed Engl. 2007;46(11):1864–1868. doi: 10.1002/anie.200604505. [DOI] [PubMed] [Google Scholar]
- 12.Giannakakou P, Gussio R, Nogales E, Downing KH, Zaharevitz D, Bollbuck B, Poy G, Sackett D, Nicolaou KC, Fojo T. Proc Natl Acad Sci U S A. 2000;97(6):2904–2909. doi: 10.1073/pnas.040546297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bode CJ, Gupta ML, Jr, Reiff EA, Suprenant KA, Georg GI, Himes RH. Biochemistry. 2002;41(12):3870–3874. doi: 10.1021/bi0121611. [DOI] [PubMed] [Google Scholar]
- 14.Gupta ML, Jr, Bode CJ, Georg GI, Himes RH. Proc Natl Acad Sci U S A. 2003;100(11):6394–6397. doi: 10.1073/pnas.1131967100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foland TB, Dentler WL, Suprenant KA, Gupta ML, Jr, Himes RH. Yeast. 2005;22(12):971–978. doi: 10.1002/yea.1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Richards KL, Anders KR, Nogales E, Schwartz K, Downing KH, Botstein D. Mol Biol Cell. 2000;11(5):1887–1903. doi: 10.1091/mbc.11.5.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta ML, Jr, Bode CJ, Dougherty CA, Marquez RT, Himes RH. Cell Motil Cytoskeleton. 2001;49(2):67–77. doi: 10.1002/cm.1021. [DOI] [PubMed] [Google Scholar]
- 18.Eldridge MD, Murray CW, Auton TR, Paolini GV, Mee RP. J Comput Aided Mol Des. 1997;11(5):425–445. doi: 10.1023/a:1007996124545. [DOI] [PubMed] [Google Scholar]
- 19.Velec HF, Gohlke H, Klebe G. J Med Chem. 2005;48(20):6296–6303. doi: 10.1021/jm050436v. [DOI] [PubMed] [Google Scholar]
- 20.Jones G, Willett P, Glen RC. J Mol Biol. 1995;245(1):43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 21.Muegge I, Martin YC, Hajduk PJ, Fesik SW. J Med Chem. 1999;42(14):2498–2503. doi: 10.1021/jm990073x. [DOI] [PubMed] [Google Scholar]
- 22.Giannakakou P, Sackett DL, Kang YK, Zhan Z, Buters JT, Fojo T, Poruchynsky MS. J Biol Chem. 1997;272(27):17118–17125. doi: 10.1074/jbc.272.27.17118. [DOI] [PubMed] [Google Scholar]
- 23.Entwistle RA, Winefield RD, Foland TB, Lushington GH, Himes RH. FEBS Lett. 2008;582(16):2467–2470. doi: 10.1016/j.febslet.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jordan MA, Toso RJ, Thrower D, Wilson L. Proc Natl Acad Sci U S A. 1993;90(20):9552–9556. doi: 10.1073/pnas.90.20.9552. [DOI] [PMC free article] [PubMed] [Google Scholar]; Derry WB, Wilson L, Jordan MA. Biochemistry. 1995;34(7):2203–2211. doi: 10.1021/bi00007a014. [DOI] [PubMed] [Google Scholar]
- 25.Gupta K, Bishop J, Peck A, Brown J, Wilson L, Panda D. Biochemistry. 2004;43(21):6645–6655. doi: 10.1021/bi036112v. [DOI] [PubMed] [Google Scholar]
- 26.Guthrie C, Fink G, Abelson J, Simon M, editors. Guide to Yeast Genetics and Molecular Biology. Academic Press; 1990. [Google Scholar]
- 27.Decottignies A, Grant AM, Nichols JW, de Wet H, McIntosh DB, Goffeau A. J Biol Chem. 1998;273(20):12612–12622. doi: 10.1074/jbc.273.20.12612. [DOI] [PubMed] [Google Scholar]
- 28.Gupta ML, Jr, Bode CJ, Thrower DA, Pearson CG, Suprenant KA, Bloom KS, Himes RH. Mol Biol Cell. 2002;13(8):2919–2932. doi: 10.1091/mbc.E02-01-0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnold K, Bordoli L, Kopp J, Schwede T. Bioinformatics. 2006;22(2):195–201. doi: 10.1093/bioinformatics/bti770. [DOI] [PubMed] [Google Scholar]
- 30.Clark M, Cramer RD, III, van Opdenhosch N. J Comp Chem. 1989;10:982–1012. [Google Scholar]
- 31.Gasteiger J, Marsili M. J Tetrahedron Lett. 1978;34:3181–3184. [Google Scholar]