

Abstract
Can we consider cancer as a “metabolic disease”? Tumors are the result of a metabolic selection, forming tissues composed of heterogeneous cells that generally express an overactive metabolism as a common feature. In fact, cancer cells have to deal with increased needs for both energy and biosynthetic intermediates, in order to support their growth and invasiveness. However, their high proliferation rate often generates regions that are not sufficiently oxygenated. Therefore, their carbohydrate metabolism has to rely mostly on a glycolytic process that is uncoupled from oxidative phosphorylation. This metabolic switch, also known as the “Warburg Effect”, constitutes a fundamental adaptation of the tumor cells to a relatively hostile environment, and supports the evolution of aggressive and metastatic phenotypes. As a result, tumor glycolysis may constitute an attractive target for cancer therapy. This approach has often raised concerns that anti-glycolytic agents may cause serious side effects on normal cells. Actually, the key for a selective action against cancer cells can be found in their hyperbolic addiction to glycolysis, which may be exploited to generate new anti-cancer drugs showing minimal toxicity. In fact, there is growing evidence that supports many glycolytic enzymes and transporters as suitable candidate targets for cancer therapy. Herein we review some of the most relevant anti-glycolytic agents that have been investigated so far for the treatment of cancer.
Keywords: anticancer agents, glycolysis, inhibitors, tumor metabolism, warburg effect
Introduction
Carbohydrate metabolism in tumors: the Warburg effect
Alterations in cancer cells bioenergetics constitute an emerging hallmark of cancer. The fact that metabolism in cancer cells substantially differs from that in healthy cells had been known since many decades. In fact, normal cells rely generally on mitochondrial oxidative phosphorylation (OXPHOS) to generate energy from glucose, whereas most cancer cells instead rely on glycolysis, uncoupled from OXPHOS. About a century ago, Otto Warburg first described the importance of this peculiar glucose metabolism occurring in tumors and the relationship between cancer and altered metabolism,[1, 2] and was awarded the Nobel Prize in medicine in 1931 for his revolutionary work indicating glycolysis as the major anaerobic glucose metabolism within tumor cells (Warburg effect), although whether metabolism change is a cause or a consequence of cancer is still not clear. Cancer cells are generally more “hungry” of nutrients than normal cells in order to sustain their high proliferative rates. This is shown by: 1) higher consumption of glucose, due to the lower efficiency in energy production by anaerobic glycolysis; 2) increased extracellular acidosis, because of the high production of lactic acid and other acidic species. This metabolic change ensures an adequate and rapid supply of energy and biosynthetic intermediates from glucose, and thus high vitality, even in the absence of sufficient levels of oxygen in hypoxic regions of cancer tissues.[3] For many decades, however, tumor metabolism has received only marginal attention, because it was thought that any intervention on sugar metabolism would have had unacceptable effects on healthy cells too. In recent years, interest has been renewed, because cancer cells were found to have very strong metabolic dependencies, which are not associated to normal cells. The deregulation of cellular energetics is one of the emerging hallmarks of cancer and there are increasing evidences pointing at interventions on tumor glycolysis as a novel strategy for selective anti-cancer therapies. Therefore, drugs resulting from this approach are supposed to be highly selective against cancer cells and, therefore, devoid of important undesirable side effects, since cancer cells display an exaggerated addiction to glycolysis, when compared to normal cells.[4–7] Many of the key effectors of glycolysis (enzymes and transporters) can be considered as promising targets for therapeutic intervention against cancer.[8, 9] Some of them are overexpressed in invasive tumors and, therefore, offer a relatively safe therapeutic window for anticancer agents that target them. Presently, there are several small molecules at the preclinical/clinical stage that are reported to act as metabolic modulators in cancer cells,[10–12] and they will be systematically discussed in this review.
The glycolytic process
During glycolysis, glucose is subjected to a series of biochemical transformations devoted to demolishing its structure with production of energy (ATP), and each step is catalyzed by specific enzymes (Figure 1). In normal cells, the glycolytic process is mostly coupled to OXPHOS, so pyruvate enters mitochondria and undergoes an oxidative transformation to acetyl-CoA, which then enter the tricarboxylic acid cycle (DCA) and eventually produces CO2, together with a considerable amount of ATP. However under certain conditions, especially under oxygen deprivation, OXPHOS cannot take place, so pyruvate is instead converted to lactate by enzyme lactate dehydrogenase (LDH). This last step is fundamental because it allows to regenerate oxidized cofactor NAD+, which is needed for the regular progress of glycolysis (see conversion of glyceraldehyde-3-P to 1,3-biphosphoglycerate, Figure 1), even when there is not enough oxygen to promote NADH re-oxidation. In this case lactate is then ejected out of the cell by monocarboxylate transporter 4 (MCT4), in order to maintain the intracellular pH within acceptable levels. Extrusion of lactate from the cell is one of the main causes of extracellular acidosis occurring in these situations. This “anaerobic” glycolytic pathway is much less efficient than OXPHOS in producing energy, since only 2 molecules of ATP are produced by each glucose molecule, versus the ~36 ATP units usually produced following the tricarboxylic acid (TCA) cycle. However, it should be noticed that glycolysis generates ATP more rapidly than OXPHOS, and this offers a selective advantage to rapidly growing tumor cells.
Figure 1.
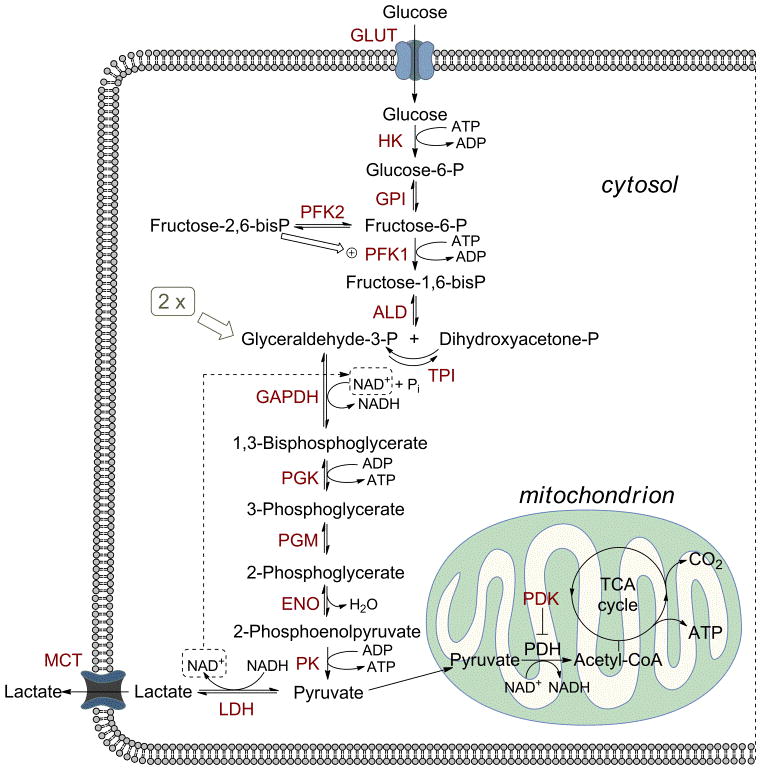
Glucose metabolism through the glycolytic flux. GLUT, glucose transporter; HK, hexokinase; GPI, glucose-6-phosphate isomerase; PFK, phosphofructokinase; ALD, aldolase; TPI, triosephosphate isomerase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PGK, phosphoglycerate kinase; PGM, phosphoglycerate mutase; ENO, enolase; PK, pyruvate kinase; LDH, lactate dehydrogenase; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; MCT, monocarboxylate transporters.
In most cancer cells, especially the most aggressive phenotypes, there is a substantial uncoupling of glycolysis from OXPHOS with consequent production of high levels of lactate (Warburg effect). This metabolic modification gives the tumor an evolutionary advantage, consisting in an adaptation to the more-or-less transient hypoxic conditions occurring during the progress of the disease. This metabolic preference is also shown by a remarkably higher uptake of glucose by cancer cells through transmembrane glucose transporters (GLUT), which has to compensate the higher energy and anabolite demand of rapidly growing cells and the poor efficiency of the glycolytic process. The diagnostic use of radiolabelled glucose analog 18F-fluorodeoxyglucose (FDG) constitutes an experimental confirmation of the selective high uptake of glucose in invasive tumors.[13] Due to this feature, it is clear that any enzyme or transporter that promotes the glycolytic flux may be considered as a potential target to block tumor progression.
Lactate: not just a by-product of glycolysis
The end-product of glycolysis, lactate, is produced in large excess in tumors. However, it does not constitute only a simple discharge product. On the contrary, it actively contributes to many aspects promoting tumor invasiveness, proliferation and survival. In fact, the extent of lactate accumulation in primary tumors was found to be inversely correlated with patient survival in many cases.[14] First of all, the active secretion of lactic acid outside the tumor cells significantly contributes to the acidification of the extracellular milieu, in addition to other mechanisms promoting tumor acidosis.[15] This renders the environment around tumor tissues more suitable for colonization and invasion by cancer cells. Moreover, lactate also actively stimulates tumor cell migration, by activation of β1-integrins, and angiogenesis, following a stimulation of VEGF production in endothelial cells.[16] Furthermore, extracellular lactic acid was found to inhibit the ability of the immune system to eradicate aberrant cells, thus contributing to the immune escape phenomenon.[17] Finally, increased survival of cancer cells to radiotherapies and to several chemotherapeutic drugs is supported by the general antioxidant properties of lactate, which inhibits the cytotoxic actions caused by reactive oxygen species (ROS) generated during these treatments.[18]
One striking aspect concerning lactate in cancer tissues is its role in a particular cell-cell shuttle system, also known as the “lactate shuttles”.[19] In fact, tumor cells are normally highly heterogeneous in their content of oxygen and lactate, and can be roughly classified into two categories: “normoxic/oxidative”, which are closer to blood vessels, or “hypoxic/glycolytic”, which are present at a farther location from the vascular network (Figure 2). These two types of cells establish a symbiotic cell-cell shuttle of lactate, which normally occurs also in skeletal muscles and in the brain, consisting in the production of lactate by the glycolytic cell and its uptake and utilization by the oxidative counterpart.[20] Basically, glucose is actively taken up through transporter GLUT into the less oxygenated cells, which employ it in the glycolytic process to produce ATP. The final conversion of pyruvate to lactate is catalyzed by lactate dehydrogenase 5 (LDH5). Monocarboxylate transporter 4 (MCT4) then extrudes lactic acid from hypoxic cells into the extracellular milieu. Then, lactate functions as a metabolic fuel in oxidative tumor cells, where it is taken up through MCT1 and oxidized to pyruvate by LDH1, thus entering the TCA cycle in mitochondria with production of energy and CO2. These observations support the central role played by lactate in the tumor functionality. Therefore, effectors responsible of its production (LDH5), cell extrusion (MCT4), cell uptake (MCT1) or utilization (LDH1) can constitute further targets for anti-cancer drugs.
Figure 2.

Roles of lactate in the symbiontic model of intercellular shuttle between “glycolytic” and “oxidative” tumor cells.
HIF-1-induced changes in glycolysis
The vast majority of human cancers display an overexpression of several glycolysis-related genes, leading to the Warburg effect. Hypoxia-inducible-factor 1 (HIF-1) is a key player in the promotion of this phenomenon shown by aggressive tumors. This factor is constituted by two subunits, HIF-1α and HIF-1β; while the β-subunit is constitutively nuclear, HIF-1α is unstable under normoxic conditions, since it is rapidly hydroxylated by enzymes belonging to the family of prolylhydroxylases (PHDs), provided there is enough oxygen to support this process. Once hydroxylated, HIF-1α is conjugated to the von Hippel-Landau (VHL) protein, then poly-ubiquitylated and eventually degraded by the proteasome.[21] Under hypoxic conditions the PHD-initiated inactivation of HIF-1α does not take place and this subunit migrates into the cell nucleus where it binds to the β-subunit. This process leads to the formation of functionally active HIF-1, that activates the transcription of a series of genes.[22, 23] The most significant, though not exclusive, target gene products of HIF-1 that are involved in the promotion of the glycolytic flux are: glucose transporters 1 and 3 (GLUT1, GLUT3), hexokinases 1 and 2 (HK1, HK2), phosphofructokinase 1 (PFK1) and 2 (PFK2, in particular PFKFB3), aldolases A and C (ALDA, ALDC), phosphoglycerate kinase 1 (PGK1), enolase 1 (ENO1), pyruvate kinase M2 (PKM2), pyruvate dehydrogenase kinases 1 and, most likely, 2 (PDK1, PDK2), lactate dehydrogenase 5 (LDH5), and monocarboxylate transporter 4 (MCT4). The HIF-1-induced overexpression of GLUT1 and GLUT3 strongly supports the remarkably higher glucose uptake found in tumor cells, in response to their increased energy and anabolite demands, as well as to the lower efficiency of the glycolytic process leading to lactate. Enhanced transcription of enzymes HK1-2, PFK1-2, ALDA-C, PGK1, ENO1 and PKM2 directly contributes to the enhancement of the glycolytic rate from glucose to pyruvate. The role played by PDK1 and PDK2 is to inhibit pyruvate dehydrogenase (PDH), an enzyme that promotes the oxidation of pyruvate to acetyl-CoA in the mitochondria, thus introducing it into the TCA cycle. Therefore, HIF-1 activates the expression of these PDKs, so that pyruvate is precluded from entering the final OXPHOS process, and contemporarily promotes the expression of LDH5, which instead converts pyruvate to lactate. The picture is completed with the enhancement of the production of MCT4, which is responsible for the extrusion of lactic acid out of the cell. These HIF-1-linked proteins are all potential targets for antiglycolytic cancer agents, whose inhibition should lead to selective damages for invasive tumor cells, where HIF-1-promoted gene transcription is more relevant, with fewer side effects expected in normal cells. Nevertheless, other targets involved in the glycolytic flux should also be considered for the development of potential antitumor drugs, since all the effectors of glycolysis were generally found to be more or less overexpressed upon HIF-1 intervention, and they all will be discussed in the following sections.
Glycolytic effectors as potential targets in cancer therapy
Glucose transporters
The entrance of glucose inside the cell occurs by facilitated diffusion and is mainly dependent on glucose transporters (GLUTs), consisting of three different classes with tissue-specific distribution and distinct affinity for glucose and other carbohydrates. Class 1 comprises four members, GLUT1-GLUT4 whose preferential substrate is glucose, while the other two classes, class 2 (GLUT5) and 3 (GLUT6, 8, 10, HMIT), are more selective for other sugars. All these classes share a same tertiary structure, characterized by 12 transmembrane domains, in which the sequence of residues is highly conserved. In particular, class 1 is 48–63 % identical in human and have been extensively characterized.[24] GLUTs result to be widely over-expressed in cancer cells with respect to normal tissues, especially in high proliferative and malignant tumors, contributing to the high glycolytic flux observed in this kind of tissues. In addition to the up-regulation of expression, the activity of GLUTs in tumors is 10–12 fold higher than that in healthy cells, demonstrating a strong dependence of cancer cells on glucose transporter for their survival.[25] In particular, GLUT1 and GLUT3, whose expression is regulated by HIF-1, can be considered the main over-expressed isoforms in a wide range of human cancers. Moreover, they are correlated with poor prognosis and radioresistance of several types of human tumors. Hence, the activation of their expression can be considered as a typical feature of the malignant phenotype.[26]
Considering the fundamental role of this transporter for glycolytic tumor cells, GLUT-inhibition may represents a very attractive way of attacking cancer by blocking its main nutrient uptake, thus leading to a reduction of the glycolytic flux and to cell death by starvation. However, it is difficult to specifically inhibit this protein only in tumors without affecting normal cells. This could explain why very few specific GLUT-inhibitors have been developed so far. Inhibition of this target by means of antibodies[27] and antisense nucleic acids,[28–30] either alone or in combination with chemotherapeutic drugs, has been reported. Furthermore, both natural and synthetic small organic molecules showing GLUT-inhibitory properties have been discovered.
Some natural products belonging to the family of flavonoids, which are polyphenolic compounds widely found in plants, exert inhibitory effects on glucose transporters (Figure 3). For example, it was reported that naringenin (1), a flavanone mainly present in grapefruit which was shown to selectively bind to estrogen receptor beta,[31] also inhibits insulin-stimulated glucose uptake in breast cancer cells, by disrupting the insulin-induced GLUT4 translocation from intracellular compartments to the plasma membrane. In fact, a concentration of 100 μM of naringenin proved to inhibit glucose uptake by approximately 50 % and cell proliferation by about 20 %.[32] Other flavonoids, such as myricetin (2), fisetin (3), quercetin (4) and its glucoside analog isoquercitrin (5), proved to inhibit GLUT2, an isoform mostly present in the intestine, with potential anti-diabetic/anti-obesity effects (IC50=13–64 μM);[33] however no data are currently available concerning their effects on GLUT1 and GLUT3, which constitute the most interesting isoforms as anticancer targets. Phloretin (6), a dihydrochalcone which can be found in apple tree leaves, acts as a competitive inhibitor of GLUT1,[34] and it is known for its ability to retard tumour growth in vitro and in vivo.[35, 36] Moreover, it was demonstrated that it sensitizes cancer cells to chemotherapeutic agents, such as daunorubicin, in colon cancer, non-small cell lung cancer and renal carcinoma cells in hypoxia.[37]
Figure 3.

Natural products that inhibit GLUT.
Silybin (7, also known as silibinin), is another natural flavonoid, displaying strong inhibitory effects on proliferation and survival of different cancer cells by means of a direct interaction with GLUT1 and GLUT4. Even its oxidized form, 2,3-dehydrosilybin, inhibits cellular glucose uptake by interacting with GLUT4. Both silybin and 2,3-dehydrosilybin inhibit glucose uptake in a competitive manner with Ki values of 60 and 114 μM, respectively.[38] A phase II clinical trial aimed to assess their effectiveness in patients with prostate cancer was completed in 2008, whereas a phase I in patients with advanced hepatocellular carcinoma is currently recruiting (Table 1).[39] However, the antitumor effects associated to flavonoids cannot be entirely associated to their inhibition of glucose uptake, since at least a part of this action may also be due to their well-known anti-oxidant properties.[40] Cytochalasin B (8), a fungal toxin, is a non-competitive inhibitor of glucose transport,[41] which led to the discovery of two new derivatives (9 and 10) that efficiently inhibit GLUT1. These compounds proved to block the formation of glycolytic metabolites in cancer cells and to suppress ATP synthesis when associated to the block of mitochondrial function by other agents (e.g. antimycin A).[42]
Table 1.
Principal clinical trials of compounds targeting tumor glycolysis.
| Compound name | Target[a] | Clinical status[b] | Therapeutic application[c] |
|---|---|---|---|
|
| |||
| Silybin (7) | GLUT1, GLUT4 | Phase I-recruiting Phase II-completed (Dec 2008) |
Advanced hepatocellular carcinoma Localized prostate cancer |
| 2-Deoxyglucose (2-DG, 23) | HK (GLUT) | Phase I-completed (Jul 2008) Phase I/II-terminated (Mar 2011, slow accrual) |
Advanced solid tumors Advanced cancer and hormone refractory prostate cancer |
| Lonidamine (21) | HK (MCT1) | Phase II/III-terminated (Dec 2006, hepatic adverse effects) | Symptomatic benign prostatic hyperplasia |
| TLN-232/CAP-232 (heptapeptide) | PKM2 | Phase II-completed (Mar 2008) Phase II-terminated (Oct 2010, license termination) |
Refractory metastatic renal cell carcinoma Recurring metastatic melanoma |
| R-(<−>)-Gossypol (72)/AT-101 (acetate complex) | LDH (GAPDH) | Phase II-ecruiting Phase II-ongoing Several Phase I/II-completed (as late as Apr 2011) Phase I-terminated (Apr 2011, develop. problems) Phase I/II-terminated (Sep 2011, low accrual) Phase II-terminated (May 2010, unspecified) |
Adrenocortical and head-and-neck cancers Metastatic prostate cancer A wide variety of cancers-both as single-agent and in combined therapies Metastatic solid tumors-combination with paclitaxel and carboplatin Chronic lymphocytic leukemia-combination with lenalidomide Non-small cell lung cancer-combination with erlotinib |
| AZD3965 (small molecule-undisclosed structure) | MCT1 | Phase I-ongoing | Advanced prostate cancer and non-Hodgkin lymphoma |
| Dichloroacetate (DCA, 92) | PDK | Phase I-recruiting Phase II-recruiting Phase I-ongoing Phase II-ongoing Phase II-completed (Aug 2009) |
Recurrent head-and-neck cancer Head-and-neck carcinoma-combination with cisplatin and radiotherapy Metastatic solid tumors, brain tumor Metastatic breast cancer and Non-small cell lung cancer-combination with herceptin Malignant gliomas, glioblastoma multiforme |
The conjugation of therapeutic agents with sugars is a frequently exploited strategy to improve the uptake in cancer cells overexpressing GLUTs, although they do not inhibit these transporters. In this context, it is worth mentioning a nitric oxide-releasing agent, such as S-nitroso-N-acetyl penicillamine (SNAP), which was linked to glucose at the C2 position, thus generating 2-Glu-SNAP (11, Figure 4).[43] This glucose conjugate showed a potent cytotoxic effect on ovarian carcinoma cell lines, which resulted to be increased when compared to that of its non-conjugated analog SNAP.[44] Another representative example of this approach is constituted by gluphosphamide (12), a glucose-containing cytotoxic agents, whose action is enhanced in cancer cells overexpressing GLUTs.[45] On the other hand, direct inhibitions of GLUTs were reported for other synthetic gluco-conjugated compounds. For example, a glucose-chlorambucil derivative (13) was reported to directly inhibit [14C]glucose uptake by GLUT1 in a concentration-dependent manner with an IC50 of 65 μM. This inhibition was entirely reversible, showing that it was not due to alkylation of the GLUT1 protein by 13.[46] Some structural requirements for the inhibitory activity of 13 on GLUT1 emerge from the following observations: a) the free anomeric hydroxyl group was necessary, since its methyl glycoside was considerably less active; b) the replacement of the ester linker between the sugar and the chlorambucil portion with an amide caused a drop in the activity, probably due to a reduced accessibility to the glucose unit by the transporter, as suggested by modeling studies.
Figure 4.

Synthetic GLUT-interacting agents.
Fasentin (14), a rather simple β-ketoanilide bearing a m-trifluoromethyl group and a p-chlorine atom in its phenyl ring, was initially found to sensitize PPC-1 prostate cancer cells to FAS, a death receptor belonging to the family of tumor necrosis factor (TNF).[47] Later, its mechanism of action was identified in the ability of 14 to alter the expression of genes involved in glucose metabolism and, most importantly, to inhibit glucose uptake with an IC50 value of 68 μM.[48] This effect was demonstrated to be caused by inhibition of both GLUT1 and GLUT4 transporters. Giaccia and co-workers identified a series of amido-sulfonamido-derivatives, whose most representative example is STF-31 (15), as selective cytotoxic agents for renal carcinoma cells, that have become dependent on glycolysis as a consequence of VHL deficiency.[49] Compound 15 proved to bind to GLUT1 directly and to block glucose uptake in these highly glycolytic tumor cells, which overexpress GLUT1. A molecular docking study predicts binding of 15 within the central channel of GLUT1, where it potentially interacts with to two key-residues of GLUT1, Arg-126 and Trp-412, which are both vital for glucose transport. The anticancer activity of an undisclosed soluble analog of 15 was further demonstrated in vivo, since this derivative delayed tumor growth in animals inoculated with subcutaneous human renal carcinoma cells. This effect was associated to a marked decrease in tumor glucose uptake, as shown by FDG positron emission tomography (PET). Two synthetic pseudo-sugars (16 and 17), belonging to the class of O-protected cyclohexanepolyols, proved to efficiently inhibit glucose uptake in a dose-dependent fashion.[50] None of these two derivatives caused any inhibition of the activity of two representative glycolytic enzyme, such as HK and PK, therefore their action seems to be selectively directed to the transporters, although no further evidences were reported. Finally, the calcium channel blocker verapamil, a widely used drug for the treatment of some cardiovascular pathologies, proved to block cellular glucose uptake, most likely by interacting with both GLUT1 and GLUT4.[51] Among other inhibitors of GLUTs, it is worth mentioning dipirydamole, isobutylmethylxanthine (IBMX) and forskolin, which by the way are slightly less interesting as perspective anticancer drug because they were reported to bind with higher affinity to GLUT4 than to GLUT1,[52] similarly to what was found also for anti-HIV drug indinavir.[53]
Hexokinase
Hexokinase (HK) is the enzyme that controls the first and rate-limiting step of glycolysis, which is the irreversible phosphorylation of glucose to glucose-6-phosphate (Glu-6-P) with consumption of ATP. After this step, the phosphorylated form of glucose, which results to be negatively charged, is trapped inside the cell and can be further metabolized either by glycolysis to generate ATP, or by the pentose phosphate pathway to be utilized for biosynthetic reactions. There are four isoforms of hexokinase, HK1, HK2, HK3 and HK4 (or glucokinase, GK), with different subcellular localization, tissue expression and kinetic properties. Isoforms HK1-3 possess significantly higher affinities for glucose than GK and, unlike GK, these specific isoforms are strongly inhibited by Glu-6-P.[54] While GK is composed of a single protein domain, the other three isoforms HK1-3 consist of two nearly identical domains. The binding sites for glucose and ATP both lie in the middle of these two domains, which move toward each other to narrow the cleft right after the binding of glucose. In HK1 and HK3 the N-terminal portion of the protein does not have a catalytic role, whereas it has a regulatory function, which is sensitive to a feed-back regulation by Glu-6-P, ADP and inorganic phosphate. On the contrary, HK-2 retains the catalytic capacity in both C- and N-terminal portions, so this isoform can actually double the rate of glucose phosphorylation, thus considerably speeding up the glycolytic process.[55] This feature may explain why HK2 is the isoform that is predominantly expressed in a wide range of malignant tumors, which require an enhanced glycolytic flux.[56] In fact isoform 2, together with HK1, is overexpressed by HIF-1 stimulation, as seen before.[22, 23] HK2 plays a pivotal role in promoting tumor cell growth and survival; it can be found free in the cytosol but, more frequently, it is bound to transmembrane voltage-dependant anion channels (VDACs), located within the outer mithocondrial membrane.[57] This strategic localization blocks the cytochrome c release from mithocondria, thus protecting the cancer cell from apoptosis. Furthermore, it provides a preferred access to mitochondrially generated ATP, ensuring its immediate utilization for glucose phosphorylation, which is badly needed by tumor cells for their survival. This position makes HK2 the isoform that is less sensitive to inhibition by its product Glu-6-P and, finally, it also protect it from proteolytic degradation. In a few words, the mitochondrial bound HK2 enzyme can initiate and also maintain a high glycolytic flux rate in tumors under a wide range of adverse conditions.[58] Consequently, the marked increase of HK2 in many cancer types finds its explanation in the metabolic advantage and protection against apoptosis that this isoform confers to tumors.[59] Considering the fundamental role that HK2 plays in highly malignant tumors and the fact that this enzyme, together with GLUT1, exerts the main control of the glycolytic flux,[60] it is evident that this enzyme clearly represents an attractive target for therapeutic strategies to suppress tumor growth. A proof-of-concept is obtained by genetic silencing of HK2 via anti-sense RNA in malignant hepatoma cells. In this case a significant reduction in tumor proliferation was observed. However, after the first successful period of treatment, the tumor recovered, suggesting that the it might be able to rapidly compensate any attempt to decrease the transcription of the HK2-gene.[61] HK2 expression is mostly limited to skeletal muscles and adipose tissues, so it seems possible to target this enzyme with minimal risk of causing side effects in other healthy tissues,[61] and so far there have been several different attempts to hit cancer cells by inhibiting HK, some of which have reached clinical trials.
A first type of molecules that antagonize the HK functionality is constituted by agents able to disrupt the binding of HK to mitochondria such as clortrimazole, bifonazole and methyl jasmonate (18–20, Figure 5). Clotrimazole (18) and bifonazole (19) are imidazole derivatives commonly used in the treatment of fungal infections, which proved to efficiently induce detachment of HK from mitochondria of B16 melanoma cells, thus impairing its functionality.[62] More specifically, clotrimazole was reported to disrupt the HK-VDAC interaction in HeLa cells. This caused an increase of outer mitochondrial membrane permeabilization by opening a permeability transition pore complex (or PTPC), followed by release of apoptogenic proteins which led to cell death.[63] However, HK is not the only cellular target hit by this imidazole derivatives. In fact, studies on lung carcinoma and colon adenocarcinoma cells showed that clotrimazole, provokes also the detachment from the cytoskeleton of other two glycolytic enzymes: phosphofructokinase (PFK) and aldolase (ALD). In addition to a decreased glycolytic activity, deep changes in cytoskeleton structure and cell shape were observed, which eventually led to cell death.[64] The same kind of effects were observed in MCF-7 human breast carcinoma cells upon treatment with 18: the decrease in the viability was caused by detachment of PFK and ALD from cytoskeleton, resulting in morphological and functional alterations.[65] Furthermore, clotrimazole inhibited the proliferation of human glioblastoma multiforme cells in vitro, and significantly reduced intracranial tumour growth in rodents in vivo with prolonged survival times.[66] Another compound that binds to HK and causes its subsequent release from mitochondrial outer membrane is methyl jasmonate (20, MJ), a lipophilic cyclopentanone derivative belonging to the class of plant stress hormones. The antitumor effect of 20 was initially associated to its ability to disrupt mitochondria in cancer cells, but not mitochondria isolated from non-transformed cells or from normal lymphocytes.[67] However, the exact mechanism of action of 20 was discovered later, when this compound was found to induce the detachment of mitochondria-bound hexokinase. Here again, this event causes the opening of the PTPC, with release of cytochrome which leads to cell death.[68] The effect of 20 in detaching the enzyme from mitochondria seems to be due to an efficient interference of the large lipophilic portion of this molecule with the interactions occurring between HK and VDAC, which are mostly hydrophobic.[69] Finally, among macromolecules, a protein-lipid complex named HAMLET, showing a strong binding affinity for HK1, proved to efficiently kill cancer cells, by triggering a metabolic paralysis.[70]
Figure 5.

Molecules causing the detachment of HK from mitochondria.
Direct inhibitors of HK include several small molecules, which are characterized by the presence of either a carboxylic group, or a pseudo-glucose portion (Figure 6). Lonidamine (21) is one of the most studied and efficient inhibitor of HK. It possesses an indazole scaffold, with a carboxylate in position 3 and a 2,4-dichlorobenzyl substituent on nitrogen 1. This compound displayed a selective block of glycolysis in tumor cells, with minimal effects on normal cells, since it specifically inhibits mitochondria-bound HK, which is mostly present in tumor cells, whereas it is generally absent in healthy cells.[71] Lonidamine proved to be an efficient anti-proliferative agent even against some resistant breast cancer cells and its mechanism of action, implying a reduction of glucose utilization and of lactate/ATP-production, was confirmed.[72, 73] Its ability to potentiate the therapeutic effect of other anticancer drugs in breast, ovarian, lung, brain, head and neck tumors has been documented in several cases.[74, 75] However, a phase II clinical trial involving an association of lonidamine was with diazepam for the treatment of glioblastoma multiforme, which reported a cytostatic effect on tumor growth without any improvement of the overall survival.[76] A similar result was obtained when lonidamine was associated with epirubicine in a Phase III clinical trial for metastatic breast cancer, with no improvement of the overall survival of the patients.[77] Moreover, its efficacy was mined by pancreatic and hepatic toxicity, especially after administration by intravenous injections.[78] Therefore, analogs of 21 possessing a higher selectivity for cancer cells may constitute a significant improvement of the safety and efficacy of this drug. To this end, Giorgioni’s group reported the development of several compounds where lonidamine is conjugated to a series of sugars or polyols.[79] In particular, ester derivatives where lonidamine is linked through its COOH group to the 3-OH group of D-glucose or to the 6-OH group of D-galactose, proved to be particularly efficient in improving: 1) solubility of the drug; 2) oral bioavailability, blood-brain barrier (BBB) permeation, and selective localization in tumor tissues. In fact, both D-glucose and D-galactose are substrates of many active transporters, including those present in the intestinal tract or in the BBB, as well as those overexpressed by cancer cells (GLUT1).
Figure 6.

Structures of HK-inhibitors.
A brominated derivative of pyruvate, 3-bromopyruvate (22), had been “historically” considered as a HK2-inhibitor, although the identification of its real target in blocking glycolysis is still disputed.[80] In fact, its alkylating properties were shown to interfere with many intracellular proteins and this might contribute to the overall cytotoxic and anti-glycolytic activity of 22.[81] Anyway, the inhibition of HK activity probably occurs by means of the alkylation of thiol groups, which are important for enzymatic function. However, the anti-glycolytic activity of 22 is also supported by inhibition of another enzyme involved in the glycolytic flux (GAPDH).[82] As a matter of fact, studies conducted on human hepatocellular carcinoma (HCC) cell lines, revealed that GAPDH resulted to be substantially pyruvylated upon exposure to 3-bromopyruvate.[83] The ATP-depleting properties of 22, quantified with a Ki value of 2.4 mM for the glycolysis/HK-inhibition, proved to efficiently contribute to block the progression of aggressive tumors in vivo.[84, 85] Even multidrug-resistant cancer cells resulted to be sensitive to its action, probably because of the reduction of ATP storage, which is needed to pump the inhibitor out of the cell.[86] The inhibitory activity of 22 on GAPDH in cancer cells was later confirmed by a more recent study, and its anti-glycolytic properties were reinforced by an efficient inhibition of phosphoglycerate kinase (PGK), another glycolytic enzyme, and of mitochondrial enzyme succinate dehydrogenase (SDH).[87] Another mechanism of action likely involved in the cytotoxic activity of 22 was demonstrated to be related to its effect in increasing the intracellular amount of total reactive oxygen species (ROS) in hepatoma[88] and colon carcinoma cells,[89] where it also potentiated the antitumor efficacy of cisplatin and oxaplatin. This mechanism was exploited in a study demonstrating that gene therapy with D-amino acid oxidase (DAO), a promising therapeutic protein that induces oxidative stress and apoptosis through generation of ROS, sensitizes glioma cells to the antiglycolytic effect of 22.[90] Other combinations of 22 with various anti-cancer agents have been tested so far: cytotoxic synergistic effects resulted from association of a propyl ester of 22 with rapamycin (mTOR-inhibitor) in human lymphoma and leukemia,[91] and of 22 with geldanamycin (HSP90-inhibitor) in pancreatic cancer.[92] More recent studies on animal models of human hepatoma reported a limited antitumor efficacy of 22 in vivo, which was associated to a certain hepatotoxicity.[93] At the present, there are no human ongoing clinical trials involving 22, probably because of the lack of selective cytotoxic effects associated to its administration, as well as the low economic potential of this non patentable molecule.
One of the most widely studied HK-inhibitors is 2-deoxy-D-glucose (23, 2-DG), a glucose analog in which the hydroxyl group in position 2 is replaced by hydrogen. This compound was found to inhibit hexokinase by competition with glucose (Ki=0.25 mM).[94] Actually, 23 is phosphorylated by HK, and the product 2-deoxy-D-glucose-6-phosphate cannot be further metabolized in the glycolytic process or diffuse outside the cells, so it accumulates inside the cells and inhibits the glycolytic flux.[95] This inhibition causes the block of the production of energy by means of glycolysis, with consequent ATP depletion, cell cycle inhibition and cell death,[96] especially in hypoxic tumor cells, while in aerobic cells these effects are less pronounced.[97] Moreover, 23 leads to an approximately 35 % decrease in the amount of hexokinase bound to mitochondria, supporting the hypothesis that the distribution of hexokinase inside the cells depends on the metabolic state of the cell itself.[98] On the other hand, in normoxic cancer cells the mechanism of action of 23 does not seem to depend on the inhibition of glycolysis, but on an abnormal N-linked glycosylation of proteins.[99] 2-DG proved to be effective in vivo in combination with two chemotherapeutic drugs, adriamycin and paclitaxel, increasing the cytotoxic effects in nude mouse xenograft models of human osteosarcoma and non-small cell lung cancer. The combination of 2-DG with either adriamycin or paclitaxel evidently led to a significant reduction of the tumor growth and to an increased survival of the animals compared with the two agents alone.[100] Similarly, the association of 23 with 2-methoxyestradiol-3,17-O,O-bis-sulphamate, a microtubule disruptor, in the treatment of breast and prostate xenograft models, a reduction of the tumor volume was obtained.[101] Another example of strong synergism with 2-DG was found in the sensitization of glioblastoma cells to their treatment with histone deacetylase (HDAC) inhibitors.[102] A radiosensitizing effect of 23 in patients affected by glioblastoma multiforme was also studied: an oral dose of 200 mg kg−1 of 2-DG selectively sensitized tumor cells to a large dose of radiation (5 Gy/fraction/week) and the treatment seemed to be well-tolerated by the patients, with transient side effects similar to hypoglycemia observed in most cases, due to the reduction in the utilization of glucose also in normal tissues, especially in the brain.[103] This radiosensitizing effect is mediated by alterations in thiol metabolism caused by disruption of glycolysis, and this hypothesis was confirmed by the fact that adding N-acetyl-cysteine, a well-known thiol antioxidant, tumor cells resulted to be protected against the cytotoxic and radiosensitizing effects of 2-DG.[104] Unluckily, this compound was also shown to induce Akt activation/phosphorylation, which leads to a reduction of chemosensitizing and radiosensitizing effects of 2-DG. This indicates that 2-DG-mediated growth inhibition can be enhanced by Akt- or PI3K-inhibitors.[105] Furthermore, 23 caused an unexpected reduction of the therapeutic efficacy of radioimmunotherapy in mice bearing a human colorectal adenocarcinoma xenograft.[106] When 23 was administered to patients with glioblastoma at doses that were sufficient to limit glucose metabolism in cancer cells, relevant toxicity (brain) was observed.[103, 107] Recently, a phase I clinical study for prostate cancer was completed, defining an optimal and safe dose of 45 mg kg−1 for phase II trial.[108] However, the fact that 2-DG is a competitive general inhibitor of glucose, and glucose is present at high concentrations (about 4–6 mM) in the blood, there are still serious concerns about the real possibility that a sufficient therapeutic window may exists for this compound. Among the alternatives to 2-DG, it is worth mentioning other non-metabolizable sugars or pseudo-sugars that are supposed to act as glucose anti-metabolites. For example, D-mannoheptulose is a a seven-carbon sugar commonly found in avocado fruits that inhibits HK, thus reducing glycolysis,[109] which was recently considered as a promising non-toxic health-promoting caloric restrictor, without reducing food intake. An interesting pseudo-sugar is represented by 5-thio-D-glucose (24), which resulted to be a competitive inhibitor of HK with a Ki value of 20 mM,[110] and it demonstrated to effectively kill and radiosensitize chronically hypoxic cells in vitro.[111] Anyway, after the first promising preliminary studies, this molecule was not developed further as antitumor agent. Better successes were obtained with 2-halogen substituted D-glucose derivatives. In fact, the non-radioactive analog of radiopharmaceutical agent FDG, 2-fluoro-2-deoxy-D-glucose (25), was found to be more potent than 2-DG in killing hypoxic cells among a series of 2-halo-D-glucose analogs, comprising 2-chloro- (26) and 2-bromo-2-deoxy-D-glucose (27), which were realized in an attempt to improve the activity of their non-halogenated progenitor 2-DG.[112] The binding affinities of these derivatives for HK decrease as the size of the halogen increases: fluoro (25)>chloro (26)>bromo (27). Molecular modeling studies demonstrated that this is due to the fact that the increasing size of the halogens generates steric clashes that destabilize the binding with the enzyme active site. Moreover, it should be noticed that D-glucose maintained the highest affinity for HK, followed by its fluorinated analog 25 and 2-DG. This was explained considering that the 2-fluoro atom in 25 is quite similar in size and polarity to the 2-OH group of the natural monosaccharide, and mimics it better than the hydrogen present in the same position in 2-DG. This was further confirmed by the fact that H-bond contribution to the interaction of 25 with HK in the glucose-6-phosphate binding site is inferior to that of glucose, but superior to that of 2-DG. Similarly, the ability of these analogs to selectively induce cytotoxicity in hypoxic cells, rather than in normoxic cells, inversely correlates with the size of the halogen atom in position 2. In fact, when the halogen size decreases, the hypoxia-selectivity in the cytotoxic effect increases. Consequently, 25 is more potent in blocking glycolysis and consequently killing hypoxic cells than the other two halogenated analogs 26 and 27, and its activity resulted to be superior to its non-halogenated counterpart 2-DG.[113] Most importantly, 25 can be considered as a safer and less toxic agent than 2-DG, because it lacks an important side effect caused by 2-DG, constituted by unspecific glycosylation of proteins.[114] Rhenium(I)-tricarbonyl complexes containing D-glucose portions were also found to inhibit yeast HK. In particular, C-2 derivatized glucose complexes with extended spacers showed Ki values down to the sub-millimolar range, although no data on the effect of these compounds on human HK are available.[115]
Finally, among the HK-inhibitors we find some organic bisphosphonates, which are non-hydrolyzable analogs of inorganic diphosphate. However, their inhibitory potencies against human isoforms of HK are rather low, whereas they efficiently inhibit the isoform produced by Trypanosoma cruzi, hence they find potential applications as agents against Chagas disease.[116]
Glucose-6-phosphate isomerase
Glucose-6-phosphate isomerase (GPI), also known as phosphoglucose isomerase, catalyzes the reversible isomerization of glucose-6-phosphate into fructose-6-phosphate, which represents the second step of glycolysis. The two main isolated isoforms of this enzyme (GPI1 and GPI2), are homodimers of subunit-A of 63.2 kDa; isoform 3 (GPI3) is a heterodimer composed of one type-A subunit and a larger type-B subunit of 69.8 kDa, whereas isoform 4 (GPI4) is a homodimer composed exclusively of B-subunits.[117] This protein shows multiple activities: inside the cell it acts as a glycolytic enzyme, whereas it behaves as a cytokine when it is secreted outside the cell. In fact, molecular cloning and sequencing have identified GPI as an autocrine motility factor (AMF), which is considered capable of stimulating motility in cancer cells. The levels of GPI/AMF were found to be elevated in patients with a wide range of tumors and are strictly associated with cancer progression, angiogenesis, and metastasis.[118–122] It was also reported that the expression of GPI/AMF is up-regulated in some tumor cells by hypoxia and that hypoxia-induced GPI/AMF mRNA expression is controlled, at least in part, by HIF-1.[123] The stimulation of tumor cell motility is initiated by the binding of GPI/AMF to the AMF receptor (AMFR, gp78) on the cell surface, which is known to be a glycosylated seven transmembrane helix protein of 78 kDa with the C-terminal region expressed outside the cell. Funasaka et al. reported that GPI/AMF down-regulation by siRNA in human lung fibrosarcoma cells resulted in an increased sensitivity to oxidative stress and oxidative stress-induced cellular senescence,[124] together with the induction of a mesenchymal-to-epithelial transition, leading to a dramatic reduction of their motility and metastatic character.[125] Crystallographic studies have shown that AMF consists of three domains, and that the substrate (or inhibitor) is stored between the large and small domains, corresponding to approximately residues 117–288.[126] Then, the fundamental interactions between AMF and AMFR were detected. In particular, the N-glyco side-chain of the receptor acts as a trigger for the binding of AMF, and the interaction between the 117-C-terminal part of AMF and the extracellular core protein of its receptor results to be absolutely necessary during this process.[127] Furthermore, the role of GPI in invasiveness of tumors was confirmed by the discovery that this enzyme induces the expression of matrix metalloproteinase-3 (MMP-3) in hepatoma cells.[128]
One of the first GPI-inhibitors to be discovered is N-(bromoacetyl)ethanolamine phosphate (28, Figure 7), which is considered an active-site directed inhibitor of this enzyme. Mutagenesis data revealed that the imidazole ring of His306, present in the active site of GPI and directly involved in the isomerization step, could be the nucleophile that attacks the bromo-alkyl portion of 28, forming a covalent bond that irreversibly inactivates the enzyme. This compound affect not only the GPI enzymatic activity, but also the AMF-induced cell motility in vitro in mouse colon tumour cells.[129–131] Most of the molecules showing competitive inhibitory properties against GPI possess a sugar-phosphate structural motif, which mimics the natural substrate (glucose-6-P). The structures of these sugar-based GPI inhibitors closely resemble that of the 1,2-cis-enediol(ate) (Figure 7), which is the high energy intermediate produced in the enzyme active site during the isomerization of glucose-6-P into fructose-6-P. One of them is 5-phospho-D-arabinoate (29), a pentose phosphate mimicking the enediol(ate) intermediate, which blocks the catalytic activity of GPI obtained from rabbit muscle with a Ki value of 2 μM. Results from the cell motility assay on mouse colon tumor cells demonstrated that 29 also reduces the AMF induced-cell motility.[132, 133]
Figure 7.

Structures of GPI-inhibitors.
Another mimic of the high energy intermediate is 5-phospho-D-arabinohydroxamate (30), a synthetic sugar analog where the hydroxamate function was used due to its similarity with the 1,2-cis-enediol(ate) portion. In fact, this planar functional group closely mimics the atoms in positions 1 and 2 of the intermediate, differing only for a nitrogen atom in place of C1 and, therefore, it constitutes a better mimic than the carboxylic moiety of 29. The inhibitory properties of 30 were compared to those of the above-mentioned competitive inhibitor 29. These assays were conducted at pH 8 on PGIs obtained from three species (yeast, rabbit and Bacillus stearothermophilus). Hydroxamate 30 showed a Ki value of 98 nM on PGI from B. stearothermophilus and around 200 nM on the other two isoforms; all three values resulted markedly lower than the values obtained by inhibitor 29. These data indicates that 30 is the strongest competitive GPI-inhibitor with respect to substrate D-fructose-6-phosphate obtained to date.[134, 135] An intermediate in the pentose phosphate pathway, 6-phospho-D-gluconate (31), is a six-carbon phospho-sugar considered as a competitive GPI inhibitor, which possesses an extra CHOH portion, when compared to the above-mentioned arabinose-type inhibitors 29 and 30. Its structure is more similar to the enzyme substrate than to the 1,2-cis-enediol(ate) intermediate. In fact, it differs from the substrate glucose-6-phosphate for the presence of a carboxylic function instead of the aldehyde group. For this reason, 31 is a rather weak inhibitor, as demonstrated when its inhibitory Ki value of 42 μM on rabbit PGI is compared with those obtained by 29 and 30 (2 and 0.2 μM, respectively).[136, 137] A GPI-inhibitor of reduced size is represented by D-erythrose-4-phosphate (32), another intermediate in the pentose phosphate pathway which, differently from the other inhibitors reported above (29–31), does not bear an extra anionic portion in addition to the phosphate group. Compound 32 showed a Ki value of 0.7 μM in the inhibition of a rabbit isoform of GPI. The inhibitory potency of 32 proved to be independent from pH (in the 6.0 ÷ 9.0 range), whereas inhibitors 29–31 showed a marked pH-dependence in their inhibition, probably because of the variations of the ionization degrees induced to their hydroxamic/carboxylic groups, which consequently affected their electrostatic interactions with the enzyme active site.[138] Studies conducted on murine fibrosarcoma cells confirmed that GPI-inhibitors 31 and 32 were able to counteract the autocrine motility-induction of this protein. In particular, enzyme inhibition assays proved that 32 is more potent (residual isomerase activity of 18 % at 100 μM) than 31 (77 % at the same concentration); however, both compounds were similarly able to reduce cell motility.[139] An X-ray structure of the complex of mouse GPI with 32 showed that this inhibitor is involved in strong interactions with catalytically active residues, such as His388, which contacts O3, and Lys518, which binds both O3 and O4 of the inhibitor.[140]
Phosphofructokinase
Two types of 6-phosphofructokinase (PFK) enzymes exist in mammalian cells: 6-phosphofructo-1-kinase (PFK1), which promotes the irreversible conversion of fructose-6-phosphate to fructose-1,6-bisphosphate, and 6-phosphofructo-2-kinase (PFK2), a bifunctional enzyme that acts both as a kinase and as a phosphatase, by regulating the synthesis of fructose-2,6-biphosphate and its transformation back to fructose-6-phosphate. For this reason, PFK2 is also named fructose-2,6-bisphosphatase (FBPase).[141] PFK1 family is constituted by several hetero-tetrameric or homo-tetrameric isozymic forms of 380 kDa composed of only three subunit types, M (muscle), L (liver and kidney) and C or P (platelet), that are codified by separate genes and possess different kinetic properties. PFK1 is activated by AMP, ADP and inorganic phosphate and inhibited by ATP, citrate and fatty acids. PFK2 direct reaction produces fructose-2,6-bisphosphate, which is the most potent allosteric activator of PFK1 (Figure 1). This effect is also able to relieve the allosteric inhibition of PFK1 caused by ATP, allowing an increased glycolytic flux through the PFK catalyzed glycolytic step.[142] PFK2/FBPase comprises four isoenzymes, PFKFB1-4, that differ in their kinetic properties and are encoded by four different genes, expressing several isoforms of each isoenzyme. The pfkfb3 gene encodes both ubiquitous and constitutive PFK2, and HIF-1 inducible PFK2, termed PFKFB3, produced by alternative splicing. PFKFB3 lacks a serine phosphorylation residue that is critical for the down-regulation of its kinase activity. For this reason it possesses a very high kinase vs. phosphatase rate ratio, thus favoring the formation of fructose-2,6-bisP.[143–145] Moreover, PFKFB3 is found to be overexpressed and highly phosphorylated in aggressive tumors when compared to normal tissues.[146, 147] This kind of expression pattern leads to an increase in the production of fructose-2,6-bisP in tumors, with consequent allosteric activation of PFK1 and an overall increase in the glycolytic flux, making this enzyme a valid target for anticancer therapies.[148]
One example of PFK-inhibitors is represented by sulforaphane (33, Figure 8), an isothiocyanate derivative naturally found in cruciferous vegetables, such as broccoli, that was recently identified as the principal and very potent inducer of phase II detoxification enzymes in mouse tissues and murine hepatoma cells in vitro. Many studies demonstrated the anti-cancer properties of this compound in several cancer cell lines, mainly through apoptotic mechanisms. Among several proteins associated with apoptosis induced by 33, PFKFB4 has been considered one of the most significant targets of its mechanism of action. PFKFB4, as well as PFKFB3, proved to be down-regulated by 33 in hepatocellular carcinoma cells, thus suggesting an anti-glycolytic role for this natural compound. Moreover, 33 also decreased the expression of HIF-1α, which strongly regulates the expression of PFKFB enzymes. In conclusion, the potent induction of apoptosis by 33 does not involve a direct enzyme inhibition of PFK, but it proceeds by means of an inhibition of the PFKFB4-pathway.[149]
Figure 8.

Structures of inhibitors of the PFK/FB-pathway.
Two of the most representative anti-inflammatory drugs, salicylic acid (34) and acetylsalicylic acid (35), were also reported as compounds able to modulate PFK activity and glucose metabolism. In fact, these salicylic derivatives proved to decrease cell viability, glucose consumption, lactate production and PFK activity in MCF-7 human breast cancer cell line and their enzyme inhibitory effects were fully reversible. The inhibition of PFK was also confirmed on the purified isolated enzyme. These two molecules alter the enzyme quaternary structure, by stabilizing the dimeric (inactive) conformation of the enzyme rather than the tetrameric (active) form, and they consequently inhibit PFK activity. Furthermore, in these experiments 34 generally proved to be more effective than 35, suggesting that the inhibitory effects showed by 35 are not dependent on the acetylating ability of the compound on PFK. This difference might tentatively be ascribed to the higher solubility of 34 over 35, that can positively contribute to its interaction with the target.[150] 2,5-Anhydro-D-mannitol (36) closely resembles the β-D-fructofuranose ring structure. This compound can be phosphorylated by PFK to form 2,5-anhydro-D-mannitol-1-phosphate, which can be considered an analog of both fructose-1-P and fructose-6-P, being a C2-symmetrical molecule. The monophosphate of 36 is a substrate for PFK1 and the resulting product, 2,5-anhydro-D-mannitol-bisphosphate, is an analog of β-D-fructose-1,6-bisphosphate rather than of α-fructose-1,6-bisphosphate, so it cannot be hydrolyzed by fructose-1,6-biphosphatase, which is instead selective for the α-anomer. Hence, the bisphosphate pseudo-substrate accumulates inside the cell. Assays on rat hepatocytes showed that 36 caused a decrease in fructose-2,6-bisP intracellular concentration. Most likely, monophosphate derivative of 36 mimics fructose-6-P, but cannot be phosphorylated in the 2 position because it lacks the 2-OH group, so it acts as an inhibitor of PFK2.[151] The most important specific PFKFB3-inhibitor discovered so far is represented by 3-(3-pyridinyl)-1-(4-pyridinyl)-2-propen-1-one (37), also known as 3PO. A recent study by Clem et al. discovered 3PO by computational modeling and virtual screening of chemical databases. This study revealed that 3PO was an efficient inhibitor of PFKFB3, with a Ki value of 25 μM and a mixed inhibition mechanism. It was able to decrease both the intracellular concentration of fructose-2,6-bisP and the glucose uptake, thus suppressing the glycolytic flux in several cancer cell lines, with IC50 values ranging from 1.4 to 24 μM. Moreover, 37 efficiently counteracts tumor growth in lung, breast and hematopoietic mice xenografts at a concentration of 70 mg kg−1, with negligible effects on normal human bronchial epithelial cells.[152] Very recently, three chromene derivatives (38–40), were identified as PFKFB3-inhibitors competitive to the natural substrate fructose-6-P. Penta-hydroxy-substituted chromene-4-one 38, named N4A, displayed a Ki value of 1.29 μM. A reduction of glycolytic flux was observed upon treatment of HeLa cells (human cervical cancer) with 38, resulting in a lower amount of intracellular fructose-6-P and a diminished lactate production. X-ray studies confirmed that 38 competes with the substrate for the same binding pocket in the enzyme active site. A couple of π-cation interactions were found between the two electron-rich rings of 38 and protonated residues Arg74 and Arg98 of the enzyme. Furthermore, a series of H-bond, either direct, or water-mediated, were detected to occur between the abundant hydroxyl substituents and polar portions of the protein. In particular, the 4′-OH group of the 2-(p-phenol)-substituent of 38 establishes a strong hydrophilic interaction with protonated Arg132, in a position usually occupied by 6-phosphate group of the natural substrate fructose-6-P. An improvement of the inhibitory potency (Ki=0.24 μM) was obtained by another chromene-4-one derivative, compound 39 (or YN1), which also proved to effectively reduce glycolysis in HeLa cells. In this case, the p-phenol substituent is present in position 3 of the chromene scaffold, and places the 4′-OH group in the site normally occupied by the sugar moiety of the substrate, where it forms a strong polar interaction with Arg75 and the aryl ring established a π-cation interaction with Arg189. This new orientation of 39, with respect to 38, is due to the different position of the phenol-substituent and to the loss of two hydroxyls, which contributes to an overall higher potency of the inhibitor. A similar trend is followed by ester-containing chromene-2-one derivative 40 (or YZ9), which was identified as a more potent competitive PFKFB3-inhibitor with a remarkable Ki value of 0.094 μM, although a X-ray structural analysis of this compound within the enzyme active site has yet to be obtained. These three compounds were examined also on T47D cells (human adenocarcinoma) for assessing their anti-proliferative effects: 40 (GI50 of 2.7 μM) reduced proliferation more potently than 39 (GI50 of 8.2 μM) and 38 (GI50 of 14.2 μM).[153] A compound already described in the section of HK inhibitors, clotrimazole (18, Figure 5), should also be considered a PFK-inhibitor. In fact, it alters cytoskeleton-associated PFK, by detaching it from the cytoskeleton itself. Clotrimazole seems to induce the dimerization of this enzyme, thus stabilizing its inactive dimeric conformation. In this conformation, PFK possesses a lower affinity for actin filaments and this would explain its detachment from the cytoskeleton.[154]
Aldolase
Aldolase (ALD) catalyzes the reversible cleavage of fructose-1,6-bisphosphate into two trioses, glyceraldehyde-3-phosphate (G3P) and dihydroxyacetone-phosphate (DHAP). There are two classes of aldolases: a) class I aldolases, present in eukaryotes and higher plants, which are characterized by the formation of a protonated imine (Schiff base) intermediate between the substrates and the ε-amino group of a lysine residue (Lys229) of the enzyme active site; b) class II aldolases, produced only in prokaryotes or lower eukaryotes, such as yeast, algae and bacteria, which require a divalent metal ion as a cofactor to polarize the ketose carbonyl oxygen and stabilize the enolate intermediate (in fact, they are also called “metalloaldolases”).[155] Aldolases of class I are a family of tetrameric enzymes composed of identical subunits of 40 kDa. Three aldolase isoenzymes exist in mammals, encoded by three different genes and with different predominant tissue distributions: ALD-A in skeletal muscle, ALD-B in liver, and ALD-C in brain and CNS. ALD-A and ALD-C exhibit an 81 % sequence identity so they possess very similar kinetic properties. On the other hand, ALD-B is slightly different, sharing only a 70 % sequence identity to both aldolases A and C. Considering that ALD-A and ALD-C are more efficient in catalyzing the forward reaction (cleavage of fructose-1,6-bisP into G3P and DHAP), these two isoforms are preferentially localized in tissues displaying high glycolysis rate, such as skeletal muscle, brain and erythrocytes. On the contrary, the reverse rection is preferentially catalyzed by ALD-B, which possesses a higher affinity for G3P and DHAP and is mostly found in gluconeogenic tissues such as liver.[156] ALD levels are elevated in the serum of patients with some malignant tumors, such as human lung squamous carcinoma[157] and hepatocellular carcinoma,[158] and the cellular expression of ALD-A was found to be promoted by HIF-1.[159]
Historically, ALD-inhibitors were initially studied as anti-parasitic agents, although with limited success. These studies led to some preliminary indications about the needed structural requirements for interactions with the enzyme, suggesting that the presence of a rigid aromatic scaffold in organic phosphate derivatives, substituted with hydroxyl and/or aldehyde groups, is required for acceptable inhibition potencies.[155] Following these indications, four compounds (Figure 9), such as, resorcinol bisphosphate (41), hydroquinone bisphosphate (42), 4-hydroxybenzaldehyde phosphate (43) and 2,4-dihydroxybenzaldehyde 4-phosphate (44), were tested on rabbit muscle aldolase. The first three compounds (41–43), competitively inhibited ALD with Ki values of 40 μM, 135 μM and 500 μM, respectively. Salicylaldehyde derivative 44 (estimated Ki*~35 μM) displayed a different mechanism of action, which involves the formation of a Schiff base between the inhibitor and Lys146 of the enzyme active site, because the addition of sodium borohydride inactivated the enzyme irreversibly. This could not be proved for the other aldehyde derivative, 43, thus confirming the importance of the ortho-OH group of compound 44 in promoting the condensation of its aldehyde portion with the NH2-group of the lysine residue.[160] Later, naphthyl phosphate derivatives 45 and 46 were tested on rabbit muscle aldolase. Bisphosphate 45 behaved as a potent competitive inhibitor (Ki=0.28 μM), whereas aldehyde derivative 46 showed instead a time-dependent strong inhibition involving a Schiff base formation (Ki*~24 nM), representing the most post potent inhibitor for ALD. The phosphate groups seem to play a fundamental role in the interaction with the enzyme. In fact, one phosphate of 45 interacts with one of the two phosphate-binding sites of ALD, where the C6 portion of the substrate binds. Moreover, the dephosphorylated hydroxyl-naphthaldehyde analogue of 46 showed no activity on ALD. Similarly to salicylic derivative 44, the presence of the hydroxyl group ortho to the aldehyde in 46 proved to be essential in determining its mechanism of action (Schiff base formation).[161] Hexitol diphosphate, consisting in a diastereoisomeric mixture of glucitol bisphosphate (47) and mannitol bisphosphate (48), had been known a long time as a competitive inhibitor of class I muscle ALD (Ki~1.2 μM).[162] More recently 47 and 48 were separately tested on rabbit muscle aldolase, and they both behaved as competitive inhibitors with Ki values of 100 μM (47) and 7.3 μM (48).[163] An X-ray crystallographic study on rabbit muscle ALD revealed that the disposition of 48 within the enzyme active site nicely overlaps with that of the natural substrate, whose anionic phosphate groups form charged interactions with Lys-107 on one side and Arg-303 on the other extremity of the molecule.[164] Finally, D-lactaldehyde was found to inhibit the reverse reaction (leading to aldol products) catalyzed by ALD, probably as a consequence of Schiff base formation with the above mentioned catalytic Lys residue.[165]
Figure 9.

Structures of ALD-inhibitors.
Triosephosphate isomerase
Triosephosphate isomerase (TPI) catalyzes the reversible isomerization of dihydroxyacetone-phosphate (DHAP) to glyceraldehyde-3-phosphate, although it does not exert a direct control on the glycolytic flux. It is a homodimeric enzyme containing two 27 kDa subunits, whose expression results to be increased in several hypoxic tumors, being regulated by the HIF-1 pathway.[166] People affected by TPI-deficiency present high DHAP concentrations, particularly in red cells, and they are consequently affected by a series of blood disorders, often associated to cardiovascular and neuromuscular dysfunctions.[167] These evidences would not support TPI as a safe target for antiglycolytic cancer therapeutics. In fact, only potential antiprotozoal drugs acting as inhibitors of parasites isoforms of TPI have been described so far.[168]
Glyceraldehyde-3-phosphate dehydrogenase
The addition of a phosphate group to glyceraldeyde-3-phosphate to give 1,3-bisphosphoglycerate with the simultaneous reduction of NAD+ to NADH is catalyzed by glyceraldehyde-3-phosphate dehydrogenase (GAPDH). This enzyme is a homo-tetramer composed on identical 37 kDa subunits,[169] which is overexpressed in several malignant cancer types.[170–173] GAPDH obtained from highly dedifferentiated and rapidly growing malignant Ehrlich ascites carcinoma cells showed catalytic and physical properties that were strikingly different from those observed when this enzyme was purified from other normal sources: the tumor-derived enzyme was composed of two different subunits of 33 and 54 kDa, respectively, thus suggesting that GAPDH expressed in malignant tumors is significantly altered.[174] In particular the cancer isoform is not substantially inhibited by physiological concentrations of ATP and this may support the high glycolysis rate of cancer cells. Furthermore, non-glycolytic roles were recently attributed to GADPH, such as induction of autophagy, which may permit cellular survival when mitochondrial activity is compromised (i.e., in most glycolytic cancer cells).[175]
Similarly to 3-bromopyruvate (22, Figure 6) discussed above for its inhibitory properties against both HK and GAPDH, iodoacetate (49, Figure 10) is an alkylating agent also known as an irreversible GAPDH-inhibitor: it reacts with the sulfhydryl group of Cys149 present in the active site of the enzyme, which is essential for catalytic activity by participating to the initial formation of the thioemiacetal intermediate with the aldehyde group of the substrate.[176] Actually, 49 is a non-selective inhibitor, since it is also active on glucose-6-phosphate dehydrogenase and 6-phosphogluconate dehydrogenase, two key enzymes of the pentose phosphate pathway. The anticancer properties of 49 have not been fully elucidated. For example, Rodríguez-Enríquez et al. concluded that 49, strongly blocked glycolysis, but it did not affect tumor cell proliferation.[177] On the other hand, Bhardwaj et al. found that 49 decreased cell survival in a concentration- and time-dependent manner in pancreatic cancer cells.[178] Koningic acid (50, also known as heptelidic acid or avocettin) is a sesquiterpene antibiotic extracted from the fungus Trichoderma koningii that, similarly to iodoacetate and probably through its epoxide moiety, binds covalently to Cys149 of the GAPDH active site,[179] thus inhibiting the enzyme.[180] This compound proved to be an irreversible inhibitor of rabbit muscle GAPDH, causing an inhibition which is competitive against the natural substrate with a Ki value of 1.1 μM.[181] High-glycolytic cancer cells are killed upon exposure to a 10 μg mL−1 concentration of 50 through glucose-dependent ATP depletion, and doses of 1 mg kg−1 of the same molecule proved to be effective in vivo, suppressing the tumour growth of Erlich ascites in mice. However, treatment with this molecule also showed a certain toxicity in mice, in particular it caused hematological dysfunctions affecting erythrocytes, which are exclusively dependent on glycolysis for energy production.[182] α-Chlorohydrin (51, 3-chloropropan-1,2-diol) was reported to block glycolysis by inhibiting GAPDH in mammalian spermatozoa, although a certain inhibitory effect on both ALD and TPI was also detected.[183, 184] A study on male antifertility agents later demonstrated that the (S)-enantiomer of 51 and a chloro-analogue of DHAP (52, 1-chloro-3-hydroxypropanone), inhibited GAPDH by means of their common metabolite (S)-3-chlorolactaldehyde, which seems to be the real inhibitor of this enzyme.[185, 186] However, 51 showed a certain degree of astrocytic toxicity, although this effect does not seem to be due to energy deprivation, rather to a disruption of cellular redox state.[187] Nitroxyl (HNO), generated in situ by Angeli’s salt (Na2N2O3), was reported to irreversibly inhibit GAPDH, probably by a permanent modification of the active site cysteine residue. The reactivity of HNO with thiols is well documented, although these studies indicate that the irreversible reaction with the catalytic SH-group of the enzyme starts with the initial formation of a N-hydroxysulfenamide (R-S-NHOH) intermediate, which then rearranges to a more stable sulfinamide (R-S(O)-NH2) adduct, thus blocking the enzyme activity.[188, 189] Studies on a series of tumor xenografts demonstrated that HNO reduces tumor mass in a mouse xenograft model of breast cancer, although the overall effect does not seem to be exclusively due to GAPDH-inhibition, since HNO also decreases levels and activity of HIF-1α, reduces VEGF production, thus decreasing tumor angiogenesis and inducing apoptosis.[190] It is worth mentioning that some adenosine derivatives proved to inhibit GAPDH isoforms of pathogen agents such as Trypanosoma brucei, Trypanosoma cruzi and Leishmania mexicana,[191] although their activities on the human isoform could not be confirmed as of yet. Finally, a certain GAPDH-inhibition was associated to gossypol, a polyphenol natural product which will described later in the “Lactate dehydrogenase” section.
Figure 10.

Structures of GAPDH-inhibitors.
Phosphoglycerate kinase
Phosphoglycerate kinase (PGK) reversibly catalyzes the first energy-producing glycolytic step by transferring a phosphate from 1,3-bisphophoglycerate to ADP, thus generating ATP and 3-phosphoglycerate. Human PGK is a monomer of 48 kDa present in two isoforms: PGK1, expressed in all somatic and cancer cells and subjected to HIF-1 up-regulation,[192] and PGK2, which is only found in spermatozoids for normal motility and fertility of mammalian spermatozoa. This enzyme consists of two domains, the ADP/ATP-binding site, which is a hydrophobic cleft in the C-terminal domain, and the 3-phosphoglycerate/1,3-bisphosphoglycerate binding site in the N-terminal domain, rich in arginine and histidine residues (“basic patch”). These two domains are connected by a conserved hinge, which bends after binding to both substrates, in such a way that the two domains move toward each other in a proper position for phosphate transfer. PGK does not exert any important control of glycolysis, so its overexpression in different kinds of tumors, such as prostate,[193] gastric,[194] and hepatocellular,[195] may have different roles. In addition to its participation to glycolysis, an additional role played by PGK in tumors was associated to its extracellular secretion and regulation of the angiogenic process, by acting as a disulphide reductase enzyme.[196] Moreover, overexpression of PGK1 seems to be associated to the development of a multi-drug resistance phenotype in different cancers, by means of a MDR-1 independent mechanism.[197]
The initial interest in developing PGK-inhibitors rose thanks to their potential therapeutic use in cardiovascular and respiratory diseases, since they indirectly enhance oxygen release by hemoglobin,[198] and in parasitic affections.[199] At first, series of monophospho- and arseno-dervatives,[200] as well as bisphosphonate analogs[201] of the natural substrate 1,3-bisphophoglycerate were initially synthesized and evaluated as PGK-inhibitors. Among the most potent PGK-inhibitors, it is worth mentioning some α,α-difluoromethylenephosphonate derivatives, such as 53 and 54, which are structurally very similar to the enzyme substrate (Figure 11). Compound 53, bearing an aromatic linker between the two pharmacophoric phosphate terminals, proved to be a potent competitive inhibitor of human PGK, with an IC50 value of 0.96 μM.[202] The importance of the α,α-difluoromethylenephosphonate portion was further confirmed also in a series of amide/aliphatic-spaced derivatives. In fact PGK-inhibitors possessing this -CF2- moiety displayed a stronger binding affinity (by NMR) than their non-fluorinated counterparts, and this effect was correlated to the increased acidity of the fluorinated portions, that possibly favors the binding of the inhibitor in the “basic patch” of the enzyme. This can be exemplified by comparing tetrafluoro-substituted derivative 54 (Kd=4 μM on yeast PGK) with its non-fluorinated analog (Kd=675 μM).[203] A QSAR analysis of a wide series of these inhibitors further established the importance of the following pharmacophore portions: 1) non-hydrolyzable C–P bonds bearing anionic phosphonate groups, which mimics the phosphate moieties of the natural substrate; 2) α-halogenation (next to the phosphorous), which strongly improves the affinity of the resulting PGK-inhibitors.[204]
Figure 11.

Structural derivation of some potent bisphosphonate-based PGK-inhibitors from enzyme natural substrate 1,3-bisphosphoglycerate.
Phosphoglycerate mutase
Phosphoglycerate mutase (PGM) converts 3-phosphoglycerate (3-PG) to 2-phosphoglycerate (2-PG), transferring the phosphate group from C-3 to C-2. This transfer occurs through the formation of a 2,3-bisphosphoglycerate intermediate (2,3-BPG), which is generated upon donation of one phosphoryl groups by a phosphohistidine complex present in the enzyme active site. In mammals, there are three dimeric isoforms of PGM, which result from homodimeric and heterodimeric combinations of two very similar subunits of about 32 KDa coded by separate genes: the M (muscle-derived form) and the B (brain, where this isoform was originally isolated) subunits. The MM-isoform is almost exclusively found in smooth muscle, the MB-isoform in cardiac and skeletal muscle and the BB-isoform is ubiquitous.[205] In particular, the BB-type, also named PGM1, is up-regulated in many human cancers,[206] especially in hypoxic conditions, supporting that PGM1 may play an important role in malignancy.[207] Moreover, overexpression of PGM1 led to the immortalization and indefinite proliferation of mouse embryonic fibroblasts, whereas reduction of its expression by RNA interference induced a senescent phenotype in these cells.[208] Very recently, it was demonstrated that PGM1 activity is under control of Sirtuin 1 (Sirt1), belonging to the group of NAD+-dependent deacetylase enzymes. Under glucose restriction, the levels of Sirt1 are markedly increased and this mediates the deacetylation of PGM1 lysine residues in the C-terminal site of the enzyme, thus down-regulating its catalytic activity and reducing the overall glycolytic flux.[209]
So far PGM has not been considered as a highly relevant enzyme of the glycolytic process, so very few PGM-inhibitors have been developed (Figure 12). One of them is benzene hexacarboxylic acid (55), a very simple polyanionic compound at physiological pH, which acts on PGM by blocking the entrance of the active site cleft and by establishing multiple interactions with the basic residues present in the enzyme catalytic site. It competitively inhibits some yeast isoforms of PGM with Ki values of 6 μM (Saccharomyces cervisiae) and 22 μM (Schizosaccharomyces pombe). Unfortunately, its highly charged structure causes a very low cell permeability, thus preventing its use as a drug.[210] A synthetic peptide consisting of 31 aminoacids, PGMtide (MRQIKIWFPNRRMKWKKHHHHHHPWLIRHGE), was reported to possess good membrane permeability in cancer cells, and to inhibit PGM activity, leading to growth arrest of several tumor cell lines, without affecting normal cells. An analysis of the metabolic activity of one of the tested cell lines (MCF-7), showed that the glycolytic flux resulted to be indeed inhibited, and this effect was consistent with the block of PGM activity.[211] Probably, the most relevant results obtained in the development of PGM-inhibitors were achieved with the discovery of MJE3 (56). This compound was identified from a protein-reactive small molecule library, inspired to natural products, as the only compound able to block the proliferation of the invasive human breast cancer cell line MDA-MB-231 by 60 %. MJE3 was found to uniquely label PGM on a conserved lysine residue (Lys100) of the enzyme active site, consequently providing a strong evidence that covalent inhibition of PGM is the likely mechanism for the antiproliferative effect of this compound. MJE inhibited PGM activity in MDA-MB-231 cells with an IC50 value of 33 μM, similar to the value found for its antiproliferative effect (19 μM). Interestingly, MJE3 labeling and inhibition of PGM was observed only in intact cells, and this finding was explained by the presence of a labile ester group in the structure, probably the benzyl ester, which could be selectively hydrolyzed in situ to release a carboxylic acid derivative of MJE3, MJE51, that is the real active molecule, but that it did not directly react with the enzyme in cell-based assays due to its poor cellular uptake. The structural requirements for PGM inhibition were identified by synthesizing a series of analogs of MJE3 and testing their reactivity with PGM in human melanoma cell line. In particular, the features necessary for specific interaction with the desired target included the stereochemical orientation of the spiroepoxide moiety, the rigid cyclohexane scaffold, the indole ring and the carboxylic acid/ester group. In particular, the presence of the epoxide portion was essential for the biological activity of 56, hence the reactivity of this group seems to be directly involved in the covalent bond formation with Lys100. Molecular modeling studies further confirmed the observed SAR, suggesting that both indole ring and the carboxylic group established key interactions with the enzyme.[212]
Figure 12.

Structures of PGM-inhibitors.
Enolase
Enolase (ENO) catalyzes the reversible dehydration of 2-phosphoglycerate (2-PGA) to phosphoenolpyruvate (PEP), and it participates in both glycolysis and gluconeogenesis. Three dimeric isoenzymes, containing three different kinds of subunits of about 82–100 kDa, are present in humans: ENO1 (αα or non-neuronal ENO, NNE) which is found in liver, kidney, spleen and adipose tissue; ENO2 (γγ or neuron-specific ENO, NSE); ENO3 (ββ or muscle-specific ENO, MSE). ENO1-3 are tissue-specific, but possess very similar kinetic properties. Enolase is a metalloenzyme which requires two divalent metal cations for each active site for catalytic activity, prevalently Mg2+, and X-ray analysis confirmed their interactions with the substrate.[213] ENO1 is a HIF-1 target and its expression was found to be increased in lung cancer.[214] Furthermore, overexpression of ENO1 was also detected in tamoxifen-resistant breast cancer, and in head-and-neck cancer; in both cases, knockdown of ENO1 expression significantly reduced the chemoresistance and the invasive ability of these tumors.[215]
In spite of the promising results obtained by genetic silencing of ENO1 in cancer cells,[215] there has been a poor interest in the development of small molecules able to inhibit enolase. The most representative example of enolase inhibitor is constituted by phosphonoacetohydroxamic acid (57),[216] whose structure nicely overlaps with the high energy aci-carboxylate intermediate, involved in the transformation of 2-PGA into PEP (Figure 13). X-Ray studies showed that 57 binds both metal ions in the enzyme active site thanks to a double chelation.[217] A side product of many metabolic pathway, methylglyoxal (MG), was also reported to inhibit human muscle-specific enolase (ENO3), with a mechanism involving the formation of a covalent adduct between the highly reactive aldehyde group of MG and lysine residues of the enzyme.[218] A similar behavior was found with several other reactive carbonyl derivatives, such as 4-hydroxy-2-nonenal, trans-2-nonenal, and acrolein.[219] Finally, sodium fluoride was reported as an inorganic inhibitor of human neuronal enolase, where it was found to tightly bind the Mg2+ ions together with inorganic phosphate (Pi), hence forming stable complexes in the enzyme active site, which may be represented by the formula enolase·Mg2·F2·Pi.[220]
Figure 13.

Structural comparison of ENO-inhibitor 57 with the aci-carboxylate intermediate of the transformation of 2-PGA into PEP.
Pyruvate kinase
Pyruvate kinase (PK) irreversibly catalyzes the second energy-producing glycolytic step, consisting in the conversion of PEP into pyruvate, with concomitant transfer of a phosphate group from the substrate to ADP, yielding ATP. In mammals, there are four homo-tetrameric PK isoforms of 55 kDa subunits: PKL, mainly found in gluconeogenic tissues such as liver and kidney, PKR present in erythrocytes, PKM1 localized in tissues in which large amounts of energy have to be rapidly provided such as brain, heart and skeletal muscle, and PKM2 specific of cells with a high rate of nucleic acid synthesis, such as embryonic cells, adult stem cells, leukocytes, platelets and especially cancer cells. The first two isoforms, PK-L and R, are encoded by the same gene but under control of different promoters, similarly to the other two isoforms PKM1 and M2, which are encoded by the same gene but produced by alternative splicing of exons 9 and 10. During tissue differentiation in embryogenesis, fetal isoform PKM2 is progressively replaced by respective tissue-specific PK isoforms. On the contrary, a re-expression of PKM2 occurs during tumorigenesis, whereas PKM1 is down-regulated. Therefore, PKM2 has been indicated as a tumor-specific pyruvate kinase and a valid diagnostic biomarker for various cancers.[221] A structural analysis of PKM2 revealed that it needs two metal ions, one divalent and one monovalent (Mg2+ and K+, respectively) for its enzymatic activity.[222] This isoform is up-regulated by HIF-1 and, in its turn, acts as a co-activator of this factor in a positive feedback loop, stimulating HIF-1 transcriptional activity of target genes, in particular those encoding some glycolytic enzymes.[223] In contrast to the 96 % homologous PKM1, which exists only in a highly active tetrameric form, PKM2 can switch under physiological conditions between a nearly inactive dimer and an active tetramer with high affinity to PEP. The transition between the two conformations is subjected to a complex modulation by the allosteric activator fructose 1,6-bisphosphate (FBP, an upstream glycolytic intermediate, produced by PFK1), or by interaction with some oncoproteins (HPV-16 E7 and pp60v-src kinase).[224] Furthermore, direct phosphorylation of PKM2 tyrosine residue 105 inhibits the formation of the active tetramer in cancer cells.[225] It has been proposed that the regulation of dimeric/tetrameric PKM2 in tumor cells allows a balance between the glycolytic ATP production (highly active tetrameric form) and the synthesis of cell building blocks, such as proteins, fatty acids and lipids, which are necessary for rapidly proliferating cells (nearly inactive dimeric form). Hence, tumors can adapt to variations in environmental nutrient concentrations thanks to this “metabolic sensor” function of PKM2. There are evidences that the predominance of inactive dimeric PKM2 in tumors leads to accumulation of upstream glycolytic intermediates which are channeled to anabolic processes.[226] However, it was also found that PKM2 is crucial for aerobic glycolysis and provides a growth advantage to tumors. In fact, PKM2 knock-down by shRNA and its replacement with the M1 isoform in human cancer cell lines led to reduced lactate production and increased oxygen consumption in vitro, and reduced ability to form tumors in vivo, confirming that the switch to M2 isoform is necessary to acquire the metabolic phenotype of the Warburg effect, although the exact role of PKM2 in cancer still needs to be completely clarified.[227] The resulting paradoxical situation, consisting in a decreased pyruvate kinase activity together with an elevated PKM2 expression in rapidly dividing cancer cells, has been recently explained by Vander Heiden et al.,[228] who suggested an alternative glycolytic pathway to convert PEP into pyruvate in cancer cells. As a matter of fact, PEP can lose its phosphate group by phosphorylating residue His11 of enzyme PGM (see above), thus producing pyruvate even when PK activity is nearly absent. The presence of phosphorylated-His11 in PGM positively correlated with the expression of PKM2 in cancer cell lines and tumor tissues, supporting this theory. Interestingly, PKM2 also proved to be involved in a form of caspase- and Bcl-2-independent apoptosis when it is translocated to the nucleus, and this effect did not depend upon the kinase catalytic activity of the enzyme.[229] As a further complication of this picture, the recent appearance of a study by Bluemlein et al.[230] completely changed the previously established role of PKM2: an absolute quantification of PKM1 and PKM2 was carried out in several human tumors and cancer cell lines by mass spectrometry. This precise quantitative analysis showed that the M2 isoform is the predominant pyruvate kinase isoform not only in all the tested tumor samples, but also in control tissues and normal cell lines, without finding evidence for a real isoform shift from PKM1 to PKM2 in tumors. This puzzling situation make it very difficult to predict if PKM2-inhibitors or activators should be used as potential anticancer agents and, as we will see below, both approaches have been considered as suitable by different research groups.
Obviously, the concept of PKM2 being a tumor-specific isoform of pyruvate kinase has been very attractive, although recently questioned,[230] for the development of agents able to inhibit this glycolytic enzyme (Figure 14). Among natural products that can be classified as PKM2-inhibitors we find two naphthoquinone derivatives, shikonin (57) and its enantiomer alkannin (58). They both proved to be equally potent inhibitors of isoform PKM2, with IC50 values of 0.8 μM (57) and 0.9 μM (58), representing the most potent and specific inhibitors to PKM2 reported so far. They decreased both glucose consumption and lactate production in cells predominantly expressing the PKM2 isoform (MCF-7 and A549 tumor cells), and these effects were consistent with the significantly lower PKM2 activity observed in cell lysate, thus causing a block of the glycolytic flux.[231] However, both enantiomers also display several additional biological effects, which go far beyond PK-inhibition.[232] Two other naphthoquinone-based natural products, such as vitamins K3 (59) and K5 (60), were already known for their potential use as anti-cancer adjuvants, and have been recently tested as PKM2-inhibitors. Compound 60 proved to be the most potent PKM2-inhibitor, with an IC50 of 45 μM, and a moderate selectivity over the M1-isoform. Both vitamins caused a reduction of glycolytic flux and of overall survival in HeLa cells.[233] The selectivity shown by naphthoquinone derivatives 57-60 for PKM2 over PKM1 and PKL, was associated to their possible interaction with the allosteric FBP-site, since this is only present in the M2-isoform.[232, 233] The PK-substrate PEP has been widely modified to obtain inhibitors of PEP-utilizing enzymes. In particular, efficient PEP-mimics were obtained upon insertion of substituents in position C3 (R1, Figure 14), whereas the carboxylic and phosphate functionalities seemed to be essential for the binding with the enzyme (they are involved in chelation to the catalytic metal ions in the enzyme active site). Some halogenated PEP-analogues, such as 61 and 62, proved to be potent inhibitors of rabbit muscle PK, with IC50 values of 0.1 μM and 0.05 μM, respectively. The Z-configuration of the double bond resulted to be fundamental for the inhibition potency. In fact the E-isomer of 62 (not shown in figure) was much less active (IC50=120 μM) than its Z-isomer. Compound 63, which differs from PEP for the presence of a phosphonate group in place of the carboxylic acid, was found to be inactive in the presence of Mg2+, but when this ion was substituted by Mn2+ it behaved as an efficient PK-inhibitor with an IC50 value of 30 μM.[234] A high-throughput screening (HTS) performed on a library of small organic molecules led to the discovery of pyrrole-substituted salicylate derivative 64 showing good inhibition activity on human recombinant PKM2 (IC50=10 μM), and limited effects on PKM1 (34 % inhibition at 30 μM). Experiments on human non-small cell lung carcinoma cell line H1299, confirmed that compound 64 caused an evident decrease in the cellular glycolytic activity and an increase in cell death. The possible occurrence of relevant off-target effects was excluded by demonstrating that the increase in cell death in PKM2-expressing cells was sensibly more pronounced when compared to that obtained in PKM1-expressing cells.[235] Several non-small molecules were also found to inhibit PK. Cyclic polylactates are among the first synthetic PK-inhibitors reported in the literature, which proved to interfere with the glycolytic activity of tumor cells, to reduce tumor growth, and to prolong the survival of tumor-bearing mice in vivo. Nevertheless, these compounds were not selective, since they also displayed a certain non-competitive inhibition of lactate dehydrogenase (LDH, see next section).[236] A synthetic heptapeptide containing a five-residue cyclic structure (D-Phe-Cys-Tyr-D-Trp-Lys-Cys-Thr-NH2), structurally related to somatostatin, named TT-232 (also known as TLN-232 or CAP-232), was found to induce translocation of cytosolic pyruvate kinase M2 into the nucleus and this event provokes the induction of apoptotic cell death.[229] However, the anticancer effects of TT-232 do not depend exclusively on its interference with the cellular localization of PKM2, but are the result of interactions with multiple targets, including somatostatin receptors and tyrosine kinases.[237] A phase II clinical trial with this peptide was completed in 2008 on patients with refractory metastatic renal cell carcinoma, showing efficacy and absence of toxicity but, unfortunately, a subsequent phase II study on patients affected by recurring metastatic melanoma was stopped for legal issues (“license termination”) in 2010.[238] Synthetic peptide aptamers, which selectively bind PKM2, were reported to induce PKM2 dimerization and, therefore, inactivation, thus reducing the intracellular ATP/ADP ratio in tumor cells. Moreover, they moderately reduced the growth of immortalized mouse embryonic fibroblasts NIH3T3 and of human U-2OS osteosarcoma cells, although they did not induce apoptosis. Studies with these PKM2-binding aptamers confirmed the role of PK as a “metabolic sensor”: at normal glucose concentrations, when glycolysis is the main energy source, inhibition of PKM2 by peptide aptamers induced a significant decrease of the population doubling and of cell proliferation rates, together with an increase in cell size (as a parameter for biosynthetic capacity); on the other hand, when glucose is limited, the rates of population doubling and cell proliferation increased, but at the expense of cell size that decreased.[239] An antibody-like molecule, such as TEM8-Fc, composed of the protective antigen binding domain of human tumour endothelial marker 8 (TEM8) linked to Fc portion of human immunoglobulin G1, proved to suppress the growth and invasion of human tumors xenograft in rodent models. A direct interaction of TEM8-Fc with PKM2 was considered to be crucial for the antitumor effect associated to this agent.[240] Short hairpin RNA (shRNA) targeting PKM2 displayed interesting anticancer effects in vivo. In fact combination of genetic silencing of PKM2 by shRNA and treatment with cisplatin or docetaxel resulted in inhibition of tumor growth in a human A549 lung cancer xenograft model.[241]
Figure 14.

Structures of PKM2-inhibitors.
The search for selective PKM2-inhibitors has been recently paralleled by the development of activators of this enzyme isoform. The research group of C. J. Thomas considered that down-regulation of PKM2 activity eventually ends up in channeling all the glycolytic intermediates into anabolic pathways, which are required for rapid cellular growth. Therefore, they supported the therapeutic strategy of activating PKM2, with the aim to restore a normal metabolic state, which is characteristic of healthy cells. A quantitative HTS has led to the identification and to a thorough SAR exploration of three chemical classes, such as N,N′-diarylsulfonamides, thieno-pyrrole-pyridazinones, and tetrahydroquinoline-6-sulfonamides (Figure 15).[242–244] In particular, lead N,N′-diarylsulfonamide 65 showed an AC50 on human PKM2 of 0.111 μM, and a maximum response of 92 % at concentration of 57 μM (relative to activation by FBP). The mode of action of 65 strictly resembled that of FBP, consisting in an allosteric activation of PKM2, which results in an increased affinity for the PEP substrate, with no relevant effects on ADP binding. Moreover, it proved to be highly selective for the PKM2 isoform, versus PKM1, PKL, and PKR isozymes. Subsequent SAR exploration within this chemical class led to the development of compound 66, which maintained the same benzodioxine group of 65 on one side, but possessed a 3-aniline moiety at the other molecular terminal, and an extension of the central piperazine core to a 1,3-diazepane ring. Compound 66 displayed the best combination of good activity/selectivity on PKM2 (AC50=38 nM, 82 % activation at 57 μM) and appropriate aqueous solubility (29.0 μg mL−1).[242] Similarly, pyridazinone derivative 67 showed a good and selective activation potency value (AC50=63 nM) with a maximum response at 57 μM of 122 % relative to FBP. Its optimization led to improved compounds in terms of activity and solubility, such as the methylsulfoxide-substituted derivative 68, showing activation properties (AC50=73 nM, 99 % max. response) comparable to those of parent pyridazinone 67, but with an improved aqueous solubility (37.4 μg mL−1).[243] The first example of tetrahydroquinoline-6-sulfonamides (69) displayed a reduced activation potency on PKM2 (AC50=790 nM), whereas its difluoro-chlorinated derivative 70 possesses a much better AC50 of 90 nM, and is completely selective for PKM2. In addition to its higher potency relative to 69, derivative 70 also showed better overall properties, such as cell membrane permeability and microsomal stability in both mouse and human liver microsomes.[244]
Figure 15.

Structures of PKM2-activators.
Lactate dehydrogenase
Lactate dehydrogenase (LDH) is the last glycolytic enzyme that catalyzes the reduction of pyruvate to lactate, the end-product of glycolysis. This reaction is coupled to the oxidation of the cofactor NADH to NAD+. Since oxidized cofactor NAD+ is required in the glycolytic step catalyzed by GAPDH (Figure 1), the LDH-catalyzed regeneration of NAD+ allows glycolysis to continue even when OXPHOS in mitochondria is compromised. In human cells, there are five main homo- or hetero-tetrameric LDH isoforms, composed by different associations of two kinds of subunits, the M- (muscle) and the H- (heart) types, encoded by two different genes LDH-A and LDH-B, respectively. These isoforms are: homotetramers LDH5 (M4) and LDH1 (H4) (which are also respectively called “LDH-A” and “LDH-B”), and heterotetramers LDH2 (M1H3), LDH3 (M2H2) and LDH4 (M3H1). The higher the number of H-subunits an LDH contains, the lower its ability to catalyze the forward reaction (pyruvate to lactate). Therefore LDH5, composed of four M-subunits, possesses the highest efficiency among all others to convert pyruvate to lactate under anaerobic conditions, such as in skeletal muscle, liver and also hypoxic tumors. On the other hand, LDH1, composed of four H-subunits, possesses higher affinity to lactate and it is primarily involved in the conversion of lactate to pyruvate in aerobic tissues, such as heart, kidney, spleen and brain, as well as in some oxygenated tumor portions (Figure 2).[20] LDH5 expression, similarly to several other glycolytic enzymes, is transcriptionally up-regulated by HIF-1 and it was found to be correlated with aggressive phenotypes and poor prognosis in several kinds of tumors, thus being considered as a marker of malignant cancers.[245] Conversely, LDH-H subunit production is silenced in glycolytic cancer cells, a process involving hypermethylation of the LDH-B gene promoter, as demonstrated in prostate cancer.[246] Early literature data already indicated that up-regulation of LDH-A under c-Myc control ensures an efficient aerobic glycolysis in tumor cells, conferring growth advantage,[247] whereas this enzyme does not seem to be so necessary to healthy cells in normal conditions that generally use the aerobic oxidation pathway. A definitive confirmation of the key role of LDH-A in tumor maintenance was obtained by inhibition of LDH-A expression by shRNA, which decreased the ability of tumors to proliferate under hypoxic conditions and stimulated mitochondrial respiration.[248] Moreover, increased expression of LDH-A plays an important role in resistance of human breast cancer cells against Taxol and Trastuzumab: this observation was proved by genetically downregulating LDH-A, which led to a significantly increased sensitivity of resistant cells to both anticancer agents.[249] Furthermore, LDH-A knockdown by shRNA, in the background of fumarate hydratase deficient cells, results in significant reduction of tumor growth in a xenograft mouse model of renal cancer.[250] The increased lactate production operated by LDH5 also contributes to the development of an extracellular acidosis, a factor which finally facilitates tumour invasion and metastasis,[15] although it should be remembered that lactic acid is not just a “corrosive” waste product, instead it actively participates in angiogenesis, migration, immune- and radio-resistance, as well as in energy production thanks to the “lactate shuttle” (Figure 2).[19, 20] Therefore, LDH5 has been recently attracted a great deal of attention as a potential tumor target. As regards possible side effects caused by LDH5-inhibition, it was reported that people suffering of a hereditary deficiency of the ldh-A gene, leading to a complete lack of production of the LDH-A subunit, or patients with reduced LDH5 activity in muscles, show myoglobinuria only after intense anaerobic exercise (exertional myoglobinuria) due to muscle damage, without displaying any symptoms under ordinary circumstances.[251] Therefore, few side effects should be expected upon selective inhibition of LDH5 in cancer patients.
The field of LDH-inhibitors has been quite unexplored, because very few clinical applications were so far envisioned. As described in the previous section, cyclic polylactates showed a non-competitive inhibition of LDH, together with a marked inhibition of PK.[236] Among small molecules, some inhibitors of LDH were initially developed as antiprotozoal agents to specifically treat malarial infections by Plasmodium falciparum. It is well established that lactate dehydrogenase from P. falciparum (pfLDH) is a key enzyme for the survival of the malarial parasite and, as a consequence, several small organic molecules have been designed and synthesized with the aim of targeting this antimalarial target, although many pfLDH inhibitors were not very selective and showed a certain inhibition even on human LDH isoforms, including LDH5.[252] Since nowadays LDH5 is an emerging target in oncology, some of these inhibitors have been recently considered as the basis for the development of anticancer agents (Figure 16). Oxamate (71), a structural isoster of enzyme substrate pyruvate, represents a well-known inhibitor of both A and B subunits of LDH, and it efficiently competes with the natural substrate for enzyme binding. However, the anti-glycolytic action of this molecule, possessing a simple and small structure, is unselective for this enzyme. In fact it was found to inhibit aspartate aminotransferase (AAT), a key enzyme required for the malate-aspartate NADH shuttle, with a Ki of 28 μM, thus resulting a better inhibitor of AAT than LDH5 (Ki=136 μM, competitive vs. pyruvate).[253] There are several disadvantages associate to oxamate, such as its weak inhibitory potency, as shown by its high Ki value, and its poor penetration inside cells, which implies the use of high concentrations necessary to inhibit aerobic glycolysis and the proliferation of tumour cells in vitro.[254] Unfortunately, all these factors preclude its further development as a therapeutic agent.
Figure 16.

Structures of LDH-inhibitors.
Gossypol is a natural polyphenolic aldehyde derivative found in cotton seeds. It exhibits atropoisomerism, since rotation around the single bond linking the two naphthalene portions is sterically hindered and, therefore, it exists as two enantiomers. Some preliminary cytotoxicity studies on a series of cancer cell cultures derived from melanoma, lung, breast, cervix, and leukemia, showed that (R)-(−)-gossypol (72) induced a dose-dependent cytotoxic effect in all cell lines with a mean IC50 of 20 μM, and it was significantly more potent than the (S)-(+)-enantiomer.[255] In spite of the importance of the chirality on the bioactivity of gossypol, the enzyme inhibition assays that will be described hereafter have been performed with atropoisomeric mixtures. Gossypol is a well-known non-selective LDH-inhibitor competitive with NADH, displaying Ki values of 1.9 and 1.4 μM on LDH-A and LDH-B, respectively.[256] However, it also inhibits other NAD+-dependent enzymes such as GAPDH (see the “Glyceraldehyde-3-phosphate dehydrogenase” section above). It was initially identified as a potential male antifertility agent. Being a highly reactive redox reagent and a Schiff base-forming agent, as well as a polydentate metal-complexing ligand, gossypol has a complex and broad biological activity spectrum influencing many cellular functions, such as macromolecular synthesis, ion transport, membrane properties, calcium homeostasis, glycolysis, respiration and glucose uptake. Gossypol decreased levels of ATP in MCF-7 cancer cell line, but markedly affected the levels of pyridine nucleotides and glycerylphosphocholine, due to its numerous side effects on different enzymes and membranes.[257] Moreover, it showed a cytotoxic action on several cancer cell lines in vitro. In particular melanoma and colon carcinoma appeared to be the most sensitive lines to gossypol at the concentration of 5 μM. It was hypothesized that gossypol could exert these effects by inhibition of LDH as well as GAPDH, both being involved in the glycolytic production of ATP in cancer cells.[258] Among 2,3-dihydroxy-1-naphtoic acid derivatives that are structurally related to gossypol, compound FX-11 (73) was further selected as a potential anticancer agent, due to its ability to inhibit preferentially human LDH5, although it had been initially designed as an antimalarial agent.[259] More recently, this compound was reported to exert an efficient inhibition of LDH5, which is competitive vs. NADH, with a Ki value of 8 μM. Furthermore, it was demonstrated that FX-11-induced inhibition of LDH5 negatively affected cellular energy supply, by depleting ATP levels, diminished cellular production of lactate, induced oxidative stress and caused cell death. FX-11 also reduced growth in tumor xenograft models, such as human lymphoma and pancreatic cancer.[260] The greenish-yellow mordant dye galloflavin (74), a product of oxidation of gallic acid, was recently identified as an indiscriminate inhibitor of both human isoforms LDH-A (Ki=5.46 μM vs. pyruvate and 56.0 μM vs. NADH) and LDH-B (Ki=15.1 μM vs. pyruvate and 23.2 μM vs. NADH). Further in vitro studies on PLC/PRF/5 human hepatocellular carcinoma cell line showed that galloflavin reduced lactate production, ATP levels and cell growth.[261] Several NAD+/NADH analogues have been reported in the literature as inhibitors of various dehydrogenases and, in this context, it is worth mentioning the recently discovered LDH-inhibitor NADH-GA (75), which formed by an addition of glycolic acid (GA) aldehyde group to position C4 of the reduced nicotinamide ring of NADH. In this derivative, the absolute configuration of the newly formed chiral centers was not discussed so we assume that all stereoisomers were assayed as a mixture. Compound 75 proved to be a potent inhibitor of bovine heart and rabbit muscle LDH isoforms, with a competitive behavior with respect to NADH. Unlike the previously reported NAD+/NADH derivatives, 75 showed a good stability and was further studied for its promising cardioprotective effect, although no data relative to cancer cell proliferation assays were reported.[262] A fragment-based click-chemistry-supported approach led to the synthesis of a small library of bifunctional inhibitors of LDH5. Considering the close proximity of the substrate- and the cofactor-binding sites in the structure of the enzyme, an “adenosine-like” fragment such as the bis(indolyl)maleimide moiety, mimicking NADH, was linked to a “pyruvate-like fragment”, consisting in a carboxylate portion. A 1,2,3-triazole ring was chosen as the linking portion so that click-chemistry could be exploited in the production of these bifunctional candidates. Enzymatic assays revealed that compound 76 is a selective inhibitor towards the desired isoform (human liver LDH5) over other tested LDH isoforms, showing an IC50 value of 14.8 μM.[263] N-Hydroxyindole-based compounds (NHIs) have been discovered as new and efficient LDH5-inhibitors with potential therapeutic applications.[264] They represent a class of heterocyclic derivatives bearing a hydroxyl group on the nitrogen atom in position 1, together with a carboxylate in position 2 of the indole scaffold (77–79, Figure 16). They were designed on the basis of the “OH-COOH” pharmacophore motif often present in previously reported LDH-inhibitors, since the active site normally hosts an α-hydroxy- (lactate) or α-keto-acid (pyruvate). An extensive SAR made on the first class of aryl-substituted NHIs (77) revealed that aryl substituents are generally beneficial, in particular when they are placed in position 6 and, to a lesser degree, in position 5; moreover, the presence of electron-withdrawing substituents (X, Figure 16) in the aryl groups, such as Cl, CF3, CF3O, or of additional aryl portions, such as phenyl, α- and β-naphthyl, generally improves the inhibition potency.[265] The insertion of triazole linkers (78) commonly gives weaker inhibitors,[266] whereas good inhibition levels were obtained when N-methyl-sulfanilide portions were introduced in position 6 of the indole scaffold (79).[267] Some of the compounds represented by structure 77 are among the most potent hLDH5-inhibitors reported thus far (Kis in the low micromolar range), and generally exhibit remarkable levels of isoform-5 selectivity. They have also proved to sensibly decrease cellular lactate production and cause a marked reduction of cancer cell proliferation.[265] Researchers at AstraZeneca UK have recently reported an elegant fragment-based discovery of very potent inhibitors of hLDH5 belonging to the class of malonate-type diacid derivatives. Within this class, compound 80 displayed a remarkable IC50 of 0.27 μM, which currently represent the lowest value reached on hLDH5,[268] to the best of our knowledge.
Monocarboxylate transporters
At the end of glycolysis, lactate should be excreted out from glycolytic cells and transported into the extracellular environment, to avoid intracellular acidification that leads to cell death. Lactate transport across the plasma membrane is usually necessary not only for pH regulation, but it is also fundamental for allowing its uptake by those cells utilizing it for gluconeogenesis (liver and kidney) or as a respiratory fuel (heart).[269] As described above, in cancer tissues lactate actively participate to the promotion of several processes involved in tumor progression and invasion (lactate shuttle, Figure 2).[19] Four major types of pH regulators have been so far identified: the proton pump, the sodium-proton exchanger family (NHE), the bicarbonate transporter family (BCT) and the monocarboxylate transporter family (MCT).[270] Monocarboxylate transporters (MCTs) are passive lactate-proton symporters of the SLC16A (Solute Carrier) gene family and represent the main responsible of lactate extrusion, which is favored by an acidic cytosolic pH or an alkaline extracellular pH, but they are also involved in the transport of pyruvate and ketone bodies across the plasma membrane.[271] They are transporters with 12 transmembrane domains, which locate the N-and C-termini within the cytoplasm. Different MCT isoforms differ mainly in the length of the C-terminus and in the size of the intracellular loop between transmembrane domains 6 and 7. Among the populated family of MCTs, isoforms MCT1-MCT4 are the best characterized and the most studied proton-dependent transporters of monocarboxylic acids.[272] MCT4 has the lowest affinity for lactate among other monocarboxylates, and is the only member of this family to be subjected to HIF-1α regulation, so it is adapted for allowing the rapid lactate efflux produced in hypoxic glycolytic cells (Figure 2); in fact MCT4 is expressed preferentially in tissues dependent on glycolysis for the metabolism of glucose, such as skeletal muscle.[273] MCT2 and MCT3 have the highest affinity for lactate and they are responsible of the internalization of lactate into very specific tissues such as liver, kidney, brain and retina. Finally, MCT1, the most ubiquitous isoform of this family, possesses an intermediate affinity for lactate and it promotes lactate uptake by oxidative tumor cells, in the metabolic symbiotic mechanism previously described (Figure 2). Interestingly, MCT1 and MCT4 are composed of a catalytic part (MCT) and an ancillary glycoprotein (CD147, also named basigin or EMMPRIN) and the association of MCT with its ancillary protein in the endoplasmic reticulum seems to be required for functional expression and lactate transport at the cell surface. If this association does not occur, MCTs are targeted for degradation. In particular, in metastatic cancer cells, such as the highly metastatic breast cancer cell line MDA-MB-231, both CD147, identified as an extracellular matrix metalloproteinase (MMP) inducer, and MCT4 were found to be overexpressed, establishing an evident relationship between lactate transport and metastasis, as well as a synergistic activity of the MCT/CD147 complex in promoting cell migration.[274] Moreover, MCT1 and MCT4 were found to be overexpressed in several tumors, such as colorectal carcinoma and breast carcinoma,[275] where most likely they counteract lactic acidosis and maintain an appropriate intracellular pH necessary for cell proliferation. In fact, the block of lactate extrusion by MCT4 causes a lowering of intracellular pH, and the resulting increased acidity is detrimental for cancer cell viability. Moreover, MCT1 captures the lactate produced by hypoxic/glycolytic cells and extruded by MCT4, to fuel oxidative metabolism in oxygenated tumor cells (Figure 2). Therefore, inhibition of MCT1 may also contribute to an antiproliferative action. Genetic silencing of MCT1 and MCT2 by means of siRNA caused a reduction of viability in malignant glioma U-87 cells, meaning that growth of glycolytic tumors can be arrested through inhibition of the lactate transport.[276] Targeting lactate trafficking not only should block glycolytic energy supply, but it should also be a way of interfering with cancer metastasis. In particular, both MCT1[277] and MCT4[278] are currently being considered as very promising targets in oncology. Furthermore, any intervention that disrupts the interaction of these transporters with basigin may potentially lead to the development of new anticancer agents.[279]
Cinnamic acid derivatives currently represent the most studied class of MCT-inhibitors (Figure 17), especially α-cyano-4-hydroxycinnamate (α-CHC, 81), which was initially discovered for its effect on mitochondrial pyruvate transport,[280] as well as on lactate transport in Ehrlich ascites tumor cells, displaying a competitive inhibition of L-lactate transport with a Ki of 0.5 mM.[281] A more recent evaluation of the MCT1-inhibitory potency of α-CHC, revealed a Ki value of 166 μM, although this compound was not particularly selective for this isoform, since it also inhibited MCT2 (Ki=24 μM) and MCT4 (K0.5 1 mM).[282] Inhibition of MCT1 by α-CHC resulted in a retardation of tumor growth in mouse models, since hypoxic/glycolytic cells died from glucose starvation and re-sensitization of the remaining tumor cells to radiations in vivo, without relevant side effects.[20] Several studies reported an efficient reduction of intracellular pH at low extracellular pH after treatment of human melanoma cells with α-CHC, which resulted in an impaired survival of the cancer cells.[283] Treatment of malignant glioma cells with 81 resulted in alteration of their glycolytic metabolism, leading to an increased radiosensitivity of those cells.[284] When the same compound was administered to murine models of glioblastoma multiforme, it adversely impacted both invasive and proliferative abilities of that highly malignant cancer and it provoked tumor necrosis without damaging the normal surrounding tissues.[285] Stilbene disulfonates 4,4′-di-isothiocyanostilbene-2,2′-disulfonic acid (DIDS, 82) and 4,4′-dibenzamidostilbene-2,2′-disulfonic acid (DBDS, 83), are competitive inhibitors of several anion channels, including MCTs with respect to lactate, with approximate Ki values of 39.5 and 22.4 μM, respectively (tested in in rat erythrocytes), but in the case of DIDS the inhibition becomes irreversible after prolonged incubation, probably due to the reactive isothiocyanate groups that are able to covalently bind the transporters.[286]
Figure 17.

Structures of MCT-inhibitors.
Organomercurial derivative p-chloromercuribenzenesulfonic acid (pCMBS, 84) proved to inhibit MCT1 with a Ki value of 112 μM.[287] This compound is known to rapidly react with thiol groups in proteins and its inhibitory activity on anion transporters has been linked to this property. In fact, basigin (CD147), the ancillary glycoprotein of both MCT1 and MCT4, was found to be the real target for pCMBS through its cysteine residues.[288] As a consequence, 84 efficiently inhibits MCT4 too, with a K0.5 of 21 μM.[282] A synthetic anhydride of L-(+)-lactic acid, iso-butylcarbonyl lactyl anhydride (iBCLA, 85) proved to interfere with the lactate transport, although its direct inhibition of MCTs was not demonstrated. Treatment of Ehrlich ascites tumor cells with 85 caused a decrease in extracellular lactate levels and in intracellular pH, effects which are consistent with inhibition of lactate transport. Considering that exposure of iBCLA-treated Ehrlich ascites cells to hydroxylamine or dithiothreitol reversed the effect on lactate transport, it was initially hypothesized that iBCLA acylates essential sulfhydryl groups on the transporter,[289] although it was later deduced that iBCLA does not directly bind thiols, but rather amino groups that are near a disulfide linkage in the transporter.[290]
Among natural compounds, GLUT-inhibitors (Figure 3) quercetin (4) and phloretin (6) exerted appreciable inhibitory activities on MCTs. Quercetin (50 μM) caused a significant 3–4-fold increase of intracellular lactate and a decrease of intracellular pH to 6.9 in glioma cells.[291] Phloretin seems to be a non-selective inhibitor of MCT1 (Ki=5 μM), MCT2 (Ki=14 μM) and MCT4 (K0.5=41 μM).[282] Studies using 31P and 13C magnetic resonance spectroscopy of perfused cancer cells reported a relevant effect on lactate transport and on intracellular pH exerted by HK-inhibitor lonidamine (21, Figure 6), which resulted in an accumulation of lactate inside the cells, due to inhibition of lactate efflux.[292] A positive correlation of the level of MCT1 expression by neuroblastoma cells with the lonidamine response in terms of decrease in intracellular pH, further demonstrate the inhibitory effect of lonidamine on this transporter, which turned out to induce cancer cell death.[293]
Due to the fundamental role played by MCT1 in regulating lactate flux in T-lymphocytes, some specific and highly potent MCT1-inhibitors (86–89) were developed by AstraZeneca as immunosuppressors.[294] These derivatives displayed a very strong binding to MCT1, with Ki values of 0.28 nM (86), 0.10 nM (87) and 0.33 nM (88). However, these three derivatives suffered from a high lipophilicity, a very low solubility, high clearance and plasma protein binding, and they also interfered with CYP2C9 activity.[295] Nevertheless, compound 86, also known as AR-C117977, showed efficient immunosuppressive activities both in vitro and in vivo.[296] The structural optimization of these compounds led to the production of a less lipophilic and more water soluble derivative, AR-C155858 (89). This compound proved to efficiently bind to MCT1 with a Ki value of 1.2 nM (human erythrocytes), and to potently inhibit both MCT1 and MCT2, by binding to an intracellular site of these transporters.[297] Later, this class of compounds was considered for possible applications in the field of oncology and an MCT1-inhibitor, named AZD3965, whose structure has not been publically disclosed yet, currently is in Phase I clinical trial for advanced solid tumors, such as prostate cancer and non-Hodgkin lymphoma expressing MCT1 (CRUKD/12/004).[298]
Unlike other MCTs, MCT4 is instead inhibited by a large number of statin drugs. In fact, lipophilic statins, such as atorvastatin (90), significantly inhibited [14C]-labeled L-lactic acid uptake in a concentration-dependent manner, with IC50 values ranging from 30 to 90 μM; on the other hand, more hydrophilic statins, such as pravastatin, only exerted a very weak inhibition on the transporter.[299] This leads to an extracellular accumulation of lactate that could also explain why statins may cause muscle damages and, in some rare cases, lethal rhabdomyolysis.[300] Another statin, simvastatin (91), thanks to its MCT4-inhibitory properties was independently found to inhibit cellular invasion in lung cancer cells. In this study over-expression levels of MCT1 and MCT4 positively correlated with invasion activity of the cancer cells. Furthermore, genetic repression of both transporters by means of siRNA, unspecific inhibition by DIDS (82), as well as MCT1 or MCT4 selective inhibition by quercetin (4) or simvastatin (91), respectively, caused a marked reduction of tumor cell invasiveness, thus suggesting that both lactate transporters are actively involved in malignant cancer invasion and metastasis.[301]
Pyruvate dehydrogenase kinase
Pyruvate dehydrogenase kinase (PDK) plays an indirect role in glycolysis, as shown in Figure 2. Under normoxic conditions, pyruvate is mostly taken up by mitochondria and subjected to oxidative decarboxylation by pyruvate dehydrogenase (PDH) to produce acetyl-CoA, which enters the Krebs cycle to finally produce ATP and CO2. PDH activity is subjected to a regulation by pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP): PDK phosphorylates serine residues (Ser-264, site 1; Ser-271, site 2; Ser-203, site 3) on the α-subunit of PDH, thereby inactivating PDH, whereas PDP dephosphorylates, and thus activates, PDH. The activities of the these two regulatory enzymes determine the proportion of PDH in its active dephosphorylated state.[302] Phosphorylative inactivation of PDH by PDK blocks the conversion of pyruvate to acetyl-CoA and the initiation of oxidative phosphorylation, thus favoring glycolysis. In addition to this effect, PDH inhibition prevents the excessive production of toxic reactive oxygen species (ROS) in mitochondria when oxygen is nearly absent. Hence, PDK serves as a gatekeeper to ensure efficient blockage of PDH activity. Four isoenzymes of PDK (PDK1-4), encoded by distinct genes, have been identified in mammals: they are dimers with subunits of 46 kDa, that share 65 % sequence identity and sequence differences reflect variations in tissue distribution, kinetic properties and regulation. PDK1 is present mostly in the heart, PDK2 is the most ubiquitous isoform, PDK3 is predominantly found in testis and lung, whereas heart and skeletal muscle have the highest amounts of PDK4.[303] PDKs have different specificity toward the three phosphorylation sites of the PDH α-subunit: all the three sites can be phosphorylated by PDK1, whereas the other isoforms can only act on site 1 and site 2.[304] PDK1 and PDK3 expression in cancer are induced by HIF-1, thus HIF-1 not only promotes glycolysis by enhancing the expression of glycolytic genes as seen before, but it also compromises the Krebs cycle by directly trans-activating PDK enzymes. In particular, production of PDK1 induced by HIF-1 reduces pyruvate entry into the Krebs cycle and represses mitochondrial function and oxygen consumption. The resulting decreased OXPHOS causes a shift to a glycolytic metabolism and prevents the generation of ROS, allowing cancer cell survival in hypoxic conditions.[305] Consequently, PDK isoforms have been recently considered as new attractive targets for their importance in the metabolic switch of cancer cells. Inhibition of PDK may attenuate glycolysis and enhance mitochondrial respiration by increasing PDH activity. This therapeutic strategy found its validation in experiments where PDK1 was down-regulated by means of shRNA in human head-and-neck squamous cancer: under hypoxic conditions, levels of pyruvate and lactate decreased to the same extent of those found in normoxia, indicating that PDK1 plays an important role in maintaining glycolysis. Moreover, up-regulation of this enzyme in this type of cancer was found to be associated with a poor outcome and an aggressive phenotype.[306] Knockdown of PDK1 via shRNA decreased invasiveness of head-and-neck squamous cancer cells in vitro and led to a dramatic reduction of tumor growth in xenograft models.[307] Besides its role in cancer, PDK inhibition has been also considered a valuable strategy in treating diabetes and heart diseases.[303b]
Several classes of compounds inhibiting PDK have been reported in the literature (Figure 18), suggesting that quite different binding sites in the target enzyme may be exploited by inhibitors.[308] PDK has been successfully targeted by dichloroacetate (92, DCA), an analog of pyruvate, whose ability to reduce lactate production was already well-known for its approved clinical use in the treatment of hereditary lactic acidosis and of genetic mitochondrial diseases in humans.[309] Subsequently, DCA has been identified as a PDK inhibitor for its ability to stimulate PDH activity in a perfused rat heart model.[310] This orally available compound was lately discovered to possess promising anti-cancer properties, thanks to its inhibitory action on PDK. Since mitochondria in tumors are hyperpolarized compared to healthy cells and this condition is related to resistance to apoptosis, it was demonstrated that this state can be reversed by DCA. This molecule caused an enhanced efflux of pro-apoptotic factors from mitochondria and increased ROS production, thus inducing apoptosis in cancer cells, without affecting mitochondria of non-cancerous cells. Finally, it proved to be effective also in A549 xenograft tumor models.[311] Actually, DCA was reported to show even better activity against cancer models in vivo than in vitro,[312] and these evidences promoted the start of several clinical trials. The first formal human clinical trial of DCA on patients affected by glioblastoma multiforme was completed in 2009, showing some promising efficacy of DCA against this uncurable cancer, supported by its good blood-brain-barrier permeability, and several Phase I/II clinical trials are currently ongoing or recruiting (Table 1).[238, 313] Palliative use of DCA in the presence of a metastatic carcinoma caused a beneficial pain reduction, as well as a certain stabilization of the growth of metastases.[314] As for side effects, DCA seems to be a relatively safe anticancer agent, causing only a certain dose-dependent reversible peripheral nerve toxicity.[315] At the molecular level, DCA binds at the pyruvate binding site in the N-terminal regulatory (R) domain of PDK, establishing strong attractive interaction with arginine residues (Arg154 in PDK2), with various levels of potencies on each enzyme subtype ranging around the millimolar concentration (PDK1: Ki=1 mM; PDK2: Ki=0.2 mM; PDK3: Ki=8 mM; PDK4, Ki=0.5 mM).[303a, 316] Moreover, DCA increased oxygen consumption rate in human colon carcinoma cells,[317] and reversed the glycolytic phenotype and reduced metastatic growth in breast cancer.[318] This compound showed cytotoxicity in cervical cancer cells, exhibiting a synergism with cisplatin in tumour growth inhibition,[319] induced apoptosis in endometrial cancer cells,[320] and sensitized prostate cancer cells to irradiation.[321] DCA has been recently introduced into a platinum complex, generating a new anticancer agent named mitaplatin (93), a molecule in which two DCA units are appended to the axial positions of a six-coordinate PtIV center. Mitaplatin is a dual prodrug whose activation involves the reduction of platinum(IV) by the negative intracellular redox potential, promoting the release of the PtII-based anticancer drug cisplatin and two DCA molecules. It selectively induced cancer cell death in vitro, being more cytotoxic than DCA alone, but with IC50 values comparable to cisplatin.[322] Mitaplatin was effective also on cisplatin-resistant cells and it has been hypothesized that mitaplatin circumvents cisplatin resistance by inducing mitochondrial dysfunction. The enhanced lipophilicity of mitaplatin should increase its ability to cross the plasma membrane, allowing its accumulation inside cells. This event would lead to a higher intracellular platinum dose that can damage mitochondria, which thereby sensitizes the resistant cells and activate a downstream mitochondria-dependent cell death.[323] Together with DCA, only some other chlorinated carboxylic acids, such as 2-chloropropionate and 2,2-dichloropropionate, were initially known as PDK inhibitors.[324] Chlorinated acetophenones are among the first PDK inhibitors ever reported, which were considered as attractive therapeutic agents for their potential use in diabetes or ischemic conditions, while their anticancer activity was not taken into account. They were discovered by using a high-throughput screening of α-substituted methylketones. Among them, compound 94 proved to be more potent than DCA in inhibiting PDK (IC50=1 μM, in those assays IC50 for DCA was found to be in the range of 100–150 μM), but similarly to DCA it was uncompetitive with respect to ATP and it proved to be selective for PDK, since it was inactive on several other tested kinases. The results obtained with compound 94 and related analogues suggested that the carboxylic function was not absolutely necessary for activity on PDK, but one or two chlorine atoms in α to the C=O group were strictly required.[325]
Figure 18.

Structures of PDK-inhibitors.
Other chemical classes of PDK-inhibitors cover quite a heterogeneous variety of structures. For example, natural compound radicicol (also known as monorden, 95) showed a good inhibition activity on PDK2, with the apparent Ki value of 23.3 μM (competitive with respect to ATP),[326] but its action is not selective for PDK, since it also inhibits Heat Shock Protein 90 (Hsp90) and TopoVI, by interactions with the ATP-binding pockets of these two proteins too.[327] A large library of amides of (R)-3,3,3-trifluoro-2-hydroxy-2-methylpropionic acid were initially synthesized at Novartis as potential anti-diabetic agents. Some of them, such as compound 96 (also known as Nov3r), constitute orally bioavailable nanomolar PDK inhibitors, which are remarkably more potent than DCA. In fact, amide 96 showed an IC50 of 16.5 nM on PDK, enhanced the oxidation of [14C]-lactate to 14CO2 in human fibroblasts (EC50=57 nM), lowered lactate levels to 70 % of control after low oral doses (10 μmol kg−1) in rats, although data on cancer cell lines were not reported.[328] Subsequently, several anilides of (R)-3,3,3-trifluoro-2-hydroxy-2-methylpropionic acid were developed, leading to sulfonamide derivative 97, which showed an IC50 value of 13 nM, but even in this case anticancer activity of the compounds were not explored.[329] AstraZeneca then reported anilide AZD7545 (98), initially developed for the treatment of type II diabetes. An X-ray analysis revealed that this compound binds to the lipoyl-binding pocket in the N-terminal domain of PDK1, which is different from the binding pockets respectively hosting weaker inhibitors such as DCA (92) or radicicol (95).[330] AZD7545 displayed a further increased PDH activity with an EC50 of 5.2 nM and a preferential inhibitory activity on PDK2 with an IC50=of 6.4 nM, which was shown to be non-competitive versus ATP. Its activity on other tested isoforms was either reduced (PDK1: IC50=36.8 nM) or abolished (inactive on PDK4). Furthermore, it showed an EC50 of 105 nM in the cell-based assay in which it was observed an increase in the rate of conversion of [14C]-lactate into 14CO2 in human skin fibroblasts. AZD7545 increased the percentage of active PDH ex vivo in tissue extracts and moreover, decreased blood glucose levels in vivo in animal models of diabetes after oral administration. The isoenzyme selectivity positively correlated with the ability of this compound to elevate PDH activity in vivo: in rat liver, where PDK2 is the major isoenzyme, AZD7545 reached a near complete activation of PDH, whereas in skeletal muscle and heart, which mainly express PDK4, the activation was only partial.[331] Progesterone-like oxime 99 gave an IC50 of 0.840 μM in the enzymatic assay on PDK and increased the conversion of [14C]-lactate into 14CO2 in normal human dermal fibroblasts, which is indicative of an increased oxygen consumption as a measure of activation of PDH; acute oral dosing of 300 μmol kg−1 of 99 to mouse models lowered blood levels of lactate, although this dose is much greater than that necessary for the occurrence of its main antiprogestational effects.[332] Very recently, pyrazole derivative M77976 (100), identified by in vitro screening, was crystalized with human PDK4 and it was found to bind to the ATP binding site of this enzyme and to inhibit it with an IC50 value of 648 μM (under the same conditions, radicicol 95 displayed an IC50 of about 1 mM).[333]
Other mechanisms involved in tumor glycolysis
Several additional mechanisms, that are directly or indirectly involved in the glycolytic process, have been so far considered for anticancer therapeutic interventions. Some of them are implicated in the mitochondrial dysfunction associated to the Warburg effect. This is the case of mitochondrial uncoupling protein UCP2, which was found to be overexpressed in breast cancer cells. This protein belongs to the family of uncoupling proteins which are positioned in the inner mitochondrial membrane. In particular, UCP2 promotes the irreversible injury to OXPHOS, thus increasing reliance on glycolysis, decreasing apoptotic sensitivity, and favoring tumor development. Therefore, UCP2 has been considered as a promising therapeutic target and the use of a UCP2-inhibitor, such as genepin, proved to decrease proliferation and invasiveness of MCF7 breast cancer cells, whereas normal breast epithelial cells (MCF12A) were not affected.[334] Mitochondrial OXPHOS impairment can also be due to changes occurring to the TCA cycle, such as those associated to isocitrate dehydrogenase (IDH), the enzyme that catalyzed the transformation of isocitrate to α-ketoglutarate (αKG). In fact, mutations of both cytosolic (IDH1) and mitochondrial (IDH2) isoforms of this enzyme were found in malignant gliomas and myeloid leukemias. These mutations led to a gain-of-function of both IDHs, whose mutated forms produce high amounts of (R)-(−)-2-hydroxyglutarate (2HG) from αKG.[335] The overproduction of 2-HG was found to inhibit histone demethylation, which eventually led to a block of the differentiation of non-transformed cells.[336] All these evidences have indicated mutant IDH1 and IDH2 as potential therapeutic targets, and inhibitors of these isoforms are being pursued by academic and industrial research groups.[337] Another proposed way to counteract cancer within this context consists in the administration of citrate. This tricarboxylic acid is a natural product present in numerous nutrients and constitutes a fundamental cellular intermediate of the Krebs cycle, thus actively participating to OXPHOS. Moreover, citrate displays a certain inhibitory activity on several processes involved in energy production in the cell, including enzyme PFK. Thanks to this multiple actions, it seems to efficiently contribute to block tumor progression when administered to patients affected by neoplastic diseases at high oral doses (up to 30 grams a day, with co-administration of anti-acid omeprazole to avoid gastric side-effect).[338] In vitro assays further proved the pro-apoptotic action of citrate in two human gastric carcinoma cell lines.[339] Finally, another approach is based on the concept that lactate is an active player in the processes of tumor growth, invasion, angiogenesis, radio-resistance and immune escape (Figure 2) and, therefore, its sequestration could lead to an anticancer effect. To this purpose, oligomers of poly-D-lactic acid (PDLA) were identified as agents that can trap L-lactic acid by forming stable complexes with it. These oligomers are also able to cross cell membrane and to trap intracellular lactate, so that cells cannot expel hydrogen ions produced during glycolysis. This leads to a cytotoxic intracellular acidosis that, together with the trapping of lactate and the consequential repression of its pro-invasive actions outside the cells, may eventually lead to a overall growth inhibition of the tumor.[340]
Summary
Up to now, many synthetic and natural inhibitors of various stages of tumor glycolysis have been identified. The discovery of highly potent inhibitors of the several enzymes/transporters involved in glycolysis has so far proved to be quite challenging, with only very few examples of them showing sub-micromolar activities. The general reason for this problem may be identified in the fact that these protein targets are devoted to the transformation of very polar, generally negatively charged, small substrates. This makes their active sites rather small and characterized by the presence of several cationic residues. Therefore, the design of substrate mimics that can potently inhibit these glycolytic effectors is often focused on the production of quite small and highly polar (mostly anionic) compounds, which are usually penalized by the lack of a sufficient number of strong interactions with their target and by unfavorable desolvation energy.[341] Furthermore, permeability through cell membrane is frequently hampered by excessive polarity or explicit negative charges present in the inhibitors, which then compromise their activity in cell-based assays and in vivo. Furthermore, cross-inhibition of several glycolytic promoters is possible, due to the structural similarities that are present in their natural substrates, which are often mimicked by the inhibitors. So a careful verification of highly probable off-target interferences within the various glycolytic steps should be generally considered. For example, phloretin (6) and quercetin (4) affect both GLUTs and MCTs transporters; HK-inhibitor lonidamine (21) also inhibit MCT1; gossypol (72) inhibits both GAPDH and LDH. Furthermore, the anti-glycolytic activity of alkylating agent 3-bromopyruvate (22) was found to be the overall result of its inhibitory action on HK, GAPDH, PGK and SDH: in cases like this, cross-inhibitions might lead to an amplified anti-glycolytic action, but they also risk to seriously affect carbohydrate metabolism in normal cells. A study conducted on animal models of head-and-neck (FaDu, UT-SCC-5) and human colorectal adenocarcinoma (WiDr) with PDK-inhibitor DCA (92), LDH-inhibitor oxamate (71), and MCT-inhibitor α-CHC (81), has recently challenged the possibility that these anti-glycolytic agents may effectively counteract neoplastic diseases in vivo,[342] although it should be noticed that the compounds chosen for this study generally show low potencies against their respective targets and poor cell membrane permeabilities (especially oxamate). Nevertheless, in recent times, an increasing amount of research efforts in this field has been recorded and is highlighted by the high numbers of both preclinical and clinical studies currently ongoing with inhibitors of glycolysis. In fact, over the past few years, a first series of anti-glycolytic agents have entered clinical trials (Table 1) and many others are expected to follow them in the near future. So far, the glycolytic targets that have been considered for clinical studies can only be found either at the top of the glycolytic flux, such as GLUTs and HK, or at the bottom of the same pathway, including PK, LDH and MCT. In addition, PDK affects the destiny of pyruvate, thus playing a crucial role in the lower part of the glycolytic process. Therefore, enzymes placed at the two extremities of this metabolic process presently seem to possess higher chances to become real targets for future therapeutic agents, than those involved in intermediate stages of glycolysis.
Outlook
Selective anti-glycolytic targets in perspective
The identification of new therapies that kill tumor cells while sparing healthy tissues is still a major challenge of cancer research. The Warburg effect is one of the most important metabolic alterations in malignant tumor cells that can be selectively targeted for therapy. Therefore, it is now considered for therapeutic strategies which may interfere selectively with the carbohydrate metabolism in cancer tissues, with minimal perturbation of healthy regions. In fact, glycolytic activity in cancer cells is found to be up to 200-fold higher than that in normal cells. So far, it is not clear if any of the antiglycolytic agents in clinical trials (Table 1) will meet the mandatory requirements for final clinical utilization, and the whole scientific community is eager to know if this type of approach will be successful. In fact, one major problem associated to agents interfering with tumor glycolysis is that they also target normal cells, whose glycolytic activity is fundamental. This would confer a potentially safer profile to therapeutic interventions targeting enzyme isoforms or transporters that are overexpressed by invasive cancer cells. In the light of these considerations, inhibition of LDH5, PDK or MCT1/4 might guarantee a satisfactory therapeutic window for clinical success. Fetal isoform PKM2 would also constitute a valid target, provided its real role in cancer progression will be unequivocally established. Similarly, selective inhibitors of mutant IDH1 and IDH2 might afford promising candidates. Upon a closer look at the whole glycolytic process (Figure 1), a tactical checkpoint where the flux may be ideally blocked is surely constituted by LDH5, which is placed at the strategic “bifurcation point” of glycolysis, where pyruvate destiny is decided. Considering the observations reported for natural “human LDH-A knockout” (due to a hereditary genetic deficiency), a selective block of this enzyme should not give rise to important side-effects. Inhibition of LDH5 would force pyruvate towards OXPHOS and might benefit from concomitant re-activation of PDH, operated by PDK-inhibition. In the light of these observations, a combination of PDK- and LDH-inhibitors has a great potential of restoring mitochondrial oxidation and counteracting invasive glycolytic cancer cells.
Should we eat less, exercise more and reexamine classical cytotoxic therapies? Indications from theoretical models
The development of the glycolytic phenotype can be viewed as an evolutionary process of cancer, which normally preceeds invasiveness. This phenomenon was recently explained by means of mathematical models based on “game theory”, a principle which predicts payoffs of certain choices in competitive scenarios, such as that of rapidly growing cancer tissues.[343] Irina Kareva developed one of these models by considering the “prisoner’s dilemma” to explain the competitive advantage that glycolytic cells gain, in spite of the energetical inefficiency of glycolysis, over aerobic cells.[344] The concept is that metabolic payoff is convenient when a certain number of cells opt for a cooperative glycolytic metabolism. In fact, once the core population of these cells is large enough, the “glycolytic invasion” takes place thanks to the generation of favorable environmental situations (massive production of lactic acid, niche construction, etc.).[344] This model highlights three fundamental concepts that find more-or-less good confirmations in experimental reports. 1) An increased inflow of extracellular carbon (nutrient availability, glucose concentration) can dramatically accelerate the expansion of the glycolytic cellular fraction, although it does not initially induce the metabolic switch. The link between obesity and cancer mortality is well known and could find several explanations, including production of tumor-promoting hormones from adipose tissue, or large availability of nutrients, which provide high amounts of glucose that are needed by the glycolytic cancer cells. Diabetes also constitutes a risk factor. In this case hyperglycemia, as well as high levels of insulin and insulin-like growth factor (IGF), may contribute to feed the glycolytic phenotype.[345] Therefore, drugs able to decrease glycaemia and insulin levels in the blood, such as metformin, have great potentials for cancer treatment.[346] Metabolic nutritional therapies, comprising fasting, calorie restriction and ketogenic diets, constitute other simple ways to obtain a reduction in glucose availability, and in some cases they proved to be helpful in the management of neoplastic diseases.[347] Exercising leads to a reduction of nutrient reservoirs producing weight loss and enlarging energy-demanding muscle tissues. Muscle cells constitute tough competitors for nutrients to the glycolytic cancer cells, so physical activity may actually reduce the mortality rate from cancer.[348] 2) A decreased oxygen availability contributes to the progression of the glycolytic phenotype. Hypoxic portions of tumors normally generate cell fractions that are characterized by the highest levels of malignancy. This could also explain why some anti-angiogenic therapies, by producing large poorly oxygenated areas, were reported to trigger tumor relapse and resistance after an initial good response.[349] 3) An increased cell turnover, such as that induced by cytotoxic therapies, causes a speed up of glycolytic expansion and, ultimately, might paradoxically promote cancer progression. Of course, this is only a theoretical model, which considers indiscriminate cell killing agents. However, some cytotoxic chemotherapies applied to certain particularly aggressive forms of cancer, after an initial tumor response, then produce a fast relapse of the neoplastic disease that becomes resistant (more glycolytic and invasive). All these problems could be mitigated by the use of anti-glycolytic agents, which would counteract the benefits obtained by the glycolytic phenotype in the evolutionary process.
In conclusion, deregulated cancer energetics is a vital hallmark of cancer, whose fundamental role is progressively emerging from recent studies in the field of oncology. Therefore, drugs that might be found within the several classes of compounds inhibiting the glycolytic process, when used either in mono-therapies or in combination with existing chemo- and radio-therapy protocols, may fill a gap in cancer therapy, which is still inadequate on the metastatic front. Lastly, the strong dependence on glycolysis that cancer stem cells displayed in several bioenergetics studies render the therapeutic potential of anti-glycolytic agents even more appealing.[350]
Acknowledgments
Support by the University of Pisa and by the U.S. National Institute of Health (Grant NIH-R01-GM098453) is gratefully acknowledged. The authors thank Dr. Tiziano Tuccinardi for graphical assistance.
Biography
Carlotta Granchi completed her graduate studies in Chemistry and Pharmaceutical Technology in 2007 and received her PhD in Medicinal Chemistry in 2011 at the University of Pisa (IT). During her doctorate, in 2009 she spent a research period in the group of Paul J. Hergenrother at the Department of Chemistry of the University of Illinois at Urbana-Champaign (USA). She is currently a postdoctoral research fellow under the supervision of Filippo Minutolo at the Department of Pharmaceutical Sciences of the University of Pisa. Her research is focused on the design and synthesis of small molecules able to interfere with the peculiar metabolism of invasive tumors.
Filippo Minutolo studied Chemistry and Pharmaceutical Technology at the University of Pisa (IT) until 1992. In 1993, he got an ENI fellowship to attend a triennial graduate school at the Scuola Normale Superiore in Pisa, enclosing a visiting research period (1994–1995) in the group of Ben L. Feringa at the University of Groningen (NL). In 1996 he received his PhD in Chemistry and then got a postdoctoral appointment (1997–1999) from the University of Illinois at Urbana-Champaign (USA), where he worked in the research group of John A. Katzenellenbogen. In 2000 he became a researcher at the University of Pisa and since 2006 he has held an associate professorship in Medicinal Chemistry. His main research interests include drug discovery in the fields of anti-cancer agents and nuclear receptors ligands.
References
- 1.Warburg O, Posener K, Negelein E. Biochem Z. 1924;152:319–344. [Google Scholar]
- 2.Warburg O. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Cairns RA, Harris IS, Mak TW. Nat Rev Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 4.Tennant DA, Durán RV, Gottlieb E. Nat Rev Cancer. 2010;10:267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 5.Vander Heiden MG. Nat Rev Drug Discovery. 2011;10:671–684. doi: 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- 6.Kroemer G, Pouyssegur J. Cancer Cell. 2008;13:472–482. doi: 10.1016/j.ccr.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 8.Porporato PE, Dhup S, Dadhich RK, Copetti T, Sonveaux P. Front Pharmacol. 2011;49:1–18. doi: 10.3389/fphar.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Y, Liu H, Riker AI, Fodstad O, Ledoux SP, Wilson GL, Tan M. Front Biosci. 2011;16:1844–1860. doi: 10.2741/3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shroff GK, Kuna R, Dirsipam K, Yalagala R, Mitra P. Anti-Cancer Agents Med Chem. 2011;11:272–279. [Google Scholar]
- 11.Scatena R, Bottoni P, Pontoglio A, Mastrototaro L, Giardina B. Expert Opin Invest Drugs. 2008;17:1533–1545. doi: 10.1517/13543784.17.10.1533. [DOI] [PubMed] [Google Scholar]
- 12.Sattler UGA, Hirschhaeuser F, Mueller-Klieser WF. Curr Med Chem. 2010;17:96–108. doi: 10.2174/092986710790112657. [DOI] [PubMed] [Google Scholar]
- 13.Ben-Haim S, Ell P. J Nucl Med. 2009;50:88–99. doi: 10.2967/jnumed.108.054205. [DOI] [PubMed] [Google Scholar]
- 14.Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, Mueller-Klieser W. Cancer Res. 2000;60:916–921. [PubMed] [Google Scholar]
- 15.Neri D, Supuran CT. Nat Rev Drug Discovery. 2011;10:767–777. doi: 10.1038/nrd3554. [DOI] [PubMed] [Google Scholar]
- 16.Hirschhaeuser F, Sattler UGA, Mueller-Klieser W. Cancer Res. 2011;71:6921–6925. doi: 10.1158/0008-5472.CAN-11-1457. [DOI] [PubMed] [Google Scholar]
- 17.Singer K, Gottfried E, Kreutz M, Mackensen A. Cancer Immunol Immunother. 2011;60:425–431. doi: 10.1007/s00262-010-0967-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sattler UGA, Mueller-Klieser W. Int J Radiat Biol. 2009;85:963–971. doi: 10.3109/09553000903258889. [DOI] [PubMed] [Google Scholar]
- 19.Draoui N, Feron O. Dis Model Mech. 2011;4:727–732. doi: 10.1242/dmm.007724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sonveaux P, Végran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Gallez B, Wahl ML, Feron O, Dewhirst MW. J Clin Invest. 2008;118:3930–3942. doi: 10.1172/JCI36843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu F, White SB, Zhao Q, Lee FS. Proc Natl Acad Sci USA. 2001;98:9630–9635. doi: 10.1073/pnas.181341498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semenza GL. Curr Opin Genet Dev. 2010;20:51–56. doi: 10.1016/j.gde.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denko NC. Nat Rev Cancer. 2008;8:705–713. doi: 10.1038/nrc2468. [DOI] [PubMed] [Google Scholar]
- 24.Zhao FQ, Keating AF. Curr Genomics. 2007;8:113–128. doi: 10.2174/138920207780368187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macheda ML, Rogers S, Best JD. J Cell Physiol. 2005;202:654–662. doi: 10.1002/jcp.20166. [DOI] [PubMed] [Google Scholar]
- 26.Younes M, Brown RW, Stephenson M, Gondo M, Cagle PT. Cancer. 1997;80:1046–1051. doi: 10.1002/(sici)1097-0142(19970915)80:6<1046::aid-cncr6>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 27.Rastogi S, Banerjee S, Chellappan S, Simon GR. Cancer Lett. 2007;257:244–251. doi: 10.1016/j.canlet.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 28.Chan JY, Kong SK, Choy YM, Lee CY, Fung KP. Life Sci. 1999;65:63–70. doi: 10.1016/s0024-3205(99)00219-2. [DOI] [PubMed] [Google Scholar]
- 29.Chan KK, Chan JYW, Chung KKW, Fung KP. J Cell Biochem. 2004;93:1134–1142. doi: 10.1002/jcb.20270. [DOI] [PubMed] [Google Scholar]
- 30.Chen CP, Li XX, Zhang LR, Min JM, Chan JYW, Fung KP, Wang SQ, Zhang LH. Bioconjugate Chem. 2002;13:525–529. doi: 10.1021/bc015540f. [DOI] [PubMed] [Google Scholar]
- 31.Minutolo F, Macchia M, Katzenellenbogen BS, Katzenellenbogen JA. Med Res Rev. 2011;31:364–442. doi: 10.1002/med.20186. [DOI] [PubMed] [Google Scholar]
- 32.Harmon AW, Patel YM. Breast Cancer Res Treat. 2004;85:103–110. doi: 10.1023/B:BREA.0000025397.56192.e2. [DOI] [PubMed] [Google Scholar]
- 33.Kwon O, Eck P, Chen S, Corpe CP, Lee JH, Kruhlak M, Levine M. FASEB J. 2007;21:366–377. doi: 10.1096/fj.06-6620com. [DOI] [PubMed] [Google Scholar]
- 34.a) Tsujihara K, Hongu M, Saito K, Inamasu M, Arakawa K, Oku A, Matsumoto M. Chem Pharm Bull. 1996;44:1174–1180. doi: 10.1248/cpb.44.1174. [DOI] [PubMed] [Google Scholar]; b) Lefevre PG, Marshall JK. J Biol Chem. 1959;234:3022–3026. [PubMed] [Google Scholar]
- 35.Nelson JA, Falk RE. Anticancer Res. 1993;13:2287–2292. [PubMed] [Google Scholar]
- 36.Kobori M, Shinmoto H, Tsushida T, Shinohara K. Cancer Lett. 1997;119:207–212. doi: 10.1016/s0304-3835(97)00271-1. [DOI] [PubMed] [Google Scholar]
- 37.Cao X, Fang XL, Gibbs S, Huang Y, Dai Z, Wen P, Zheng X, Sadee W, Sun D. Cancer Chemother Pharmacol. 2007;59:495–505. doi: 10.1007/s00280-006-0291-9. [DOI] [PubMed] [Google Scholar]
- 38.Zhan T, Digel M, Küch EM, Stremmel W, Füllekrug J. J Cell Biochem. 2011;112:849–859. doi: 10.1002/jcb.22984. [DOI] [PubMed] [Google Scholar]
- 39.Flaig TW, Gustafson DL, Su LJ, Zirrolli JA, Crighton F, Harrison GS, Pierson AS, Agarwal R, Glodé LM. Invest New Drugs. 2007;25:139–146. doi: 10.1007/s10637-006-9019-2. [DOI] [PubMed] [Google Scholar]
- 40.Singh RP, Dhanalakshmi S, Tyagi AK, Chan DCF, Agarwal C, Agarwal R. Cancer Res. 2002;62:3063–3069. [PubMed] [Google Scholar]
- 41.a) Devés R, Krupka RM. Biochim Biophys Acta. 1978;510:339–348. doi: 10.1016/0005-2736(78)90034-2. [DOI] [PubMed] [Google Scholar]; b) Klip A, Paquet MR. Diabetes Care. 1990;13:228. doi: 10.2337/diacare.13.3.228. [DOI] [PubMed] [Google Scholar]
- 42.Ulanovskaya OA, Cui J, Kron SJ, Kozmin SA. Chem Biol. 2011;18:222–230. doi: 10.1016/j.chembiol.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ramirez J, Yu LB, Li J, Braunschweiger PG, Wang PG. Bioorg Med Chem Lett. 1996;6:2575–2580. [Google Scholar]
- 44.Cantuaria G, Magalhaes A, Angioli R, Mendez L, Mirhashemi R, Wang J, Wang P, Penalver M, Averette H, Braunschweiger P. Cancer. 2000;88:381–388. [PubMed] [Google Scholar]
- 45.Briasoulis E, Judson I, Pavlidis N, Beale P, Wanders J, Groot Y, Veerman G, Schuessler M, Niebch G, Siamopoulos K, Tzamakou E, Rammou D, Wolf L, Walker R, Hanauske A. J Clin Oncol. 2000;18:3535–3544. doi: 10.1200/JCO.2000.18.20.3535. [DOI] [PubMed] [Google Scholar]
- 46.Halmos T, Santarromana M, Antonakis K, Scherman D. Eur J Pharmacol. 1996;318:477–484. doi: 10.1016/s0014-2999(96)00796-0. [DOI] [PubMed] [Google Scholar]
- 47.Schimmer AD, Thomas MP, Hurren R, Gronda M, Pellecchia M, Pond GR, Konopleva M, Gurfinke D, Mawji IA, Brown E, Reed JC. Cancer Res. 2006;66:2367–2375. doi: 10.1158/0008-5472.CAN-05-1061. [DOI] [PubMed] [Google Scholar]
- 48.Wood TE, Dalili S, Simpson CD, Hurren R, Mao X, Saiz FS, Gronda M, Eberhard Y, Minden MD, Bilan PJ, Klip A, Batey RA, Schimmer AD. Mol Cancer Ther. 2008;7:3546–3555. doi: 10.1158/1535-7163.MCT-08-0569. [DOI] [PubMed] [Google Scholar]
- 49.Chan DA, Sutphin PD, Nguyen P, Turcotte S, Lai EW, Banh A, Reynolds GE, Chi JT, Wu J, Solow-Cordero DE, Bonnet M, Flanagan JU, Bouley DM, Graves EE, Denny WA, Hay MP, Giaccia AJ. Sci Transl Med. 2011;3:94ra70. doi: 10.1126/scitranslmed.3002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kitagawa M, Misawa M, Ogawa S, Tashiro E, Imoto M. Biosci Biotechnol Biochem. 2011;75:367–369. doi: 10.1271/bbb.100693. [DOI] [PubMed] [Google Scholar]
- 51.Strigun A, Noor F, Pironti A, Niklas J, Yang TH, Heinzle E. J Biotechnol. 2011;155:299–307. doi: 10.1016/j.jbiotec.2011.07.028. [DOI] [PubMed] [Google Scholar]
- 52.Hellwig B, Joost HG. Mol Pharmacol. 1991;40:383–389. [PubMed] [Google Scholar]
- 53.Murata H, Hruz PW, Mueckler M. J Biol Chem. 2000;275:20251–20254. doi: 10.1074/jbc.C000228200. [DOI] [PubMed] [Google Scholar]
- 54.Wilson JE. J Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 55.Tsai HJ, Wilson JE. Arch Biochem Biophys. 1996;329:17–23. doi: 10.1006/abbi.1996.0186. [DOI] [PubMed] [Google Scholar]
- 56.Wolf A, Agnihotri S, Micallef J, Mukherjee J, Sabha N, Cairns R, Hawkins C, Guha A. J Exp Med. 2011;208:313–326. doi: 10.1084/jem.20101470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mathupala SP, Ko YH, Pedersen PL. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Robey RB, Hay N. Oncogene. 2006;25:4683–4696. doi: 10.1038/sj.onc.1209595. [DOI] [PubMed] [Google Scholar]
- 59.Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Biochim Biophys Acta Bioenerg. 2002;1555:14–20. doi: 10.1016/s0005-2728(02)00248-7. [DOI] [PubMed] [Google Scholar]
- 60.Marín-Hernández A, Rodríguez-Enríquez S, Vital-González PA, Flores-Rodríguez FL, Macías-Silva M, Sosa-Garrocho M, Moreno-Sánchez R. FEBS J. 2006;273:1975–1988. doi: 10.1111/j.1742-4658.2006.05214.x. [DOI] [PubMed] [Google Scholar]
- 61.Mathupala SP, Ko YH, Pedersen PL. Semin Cancer Biol. 2009;19:17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Penso J, Beitner R. Eur J Pharmacol. 1998;342:113–117. doi: 10.1016/s0014-2999(97)01507-0. [DOI] [PubMed] [Google Scholar]
- 63.Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, Rasola A. PLoS One. 2008;3:e1852. doi: 10.1371/journal.pone.0001852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.a) Penso J, Beitner R. Mol Genet Metab. 2002;76:181–188. doi: 10.1016/s1096-7192(02)00046-x. [DOI] [PubMed] [Google Scholar]; b) Penso J, Beitner R. Eur J Pharmacol. 2002;451:227–235. doi: 10.1016/s0014-2999(02)02103-9. [DOI] [PubMed] [Google Scholar]
- 65.Meira DD, Marinho-Carvalho MM, Teixeira CA, Veiga VF, Da Poian AT, Holandino C, de Freitas MS, Sola-Penna M. Mol Genet Metab. 2005;84:354–362. doi: 10.1016/j.ymgme.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 66.Khalid MH, Tokunaga Y, Caputy AJ, Walters E. J Neurosurg. 2005;103:79–86. doi: 10.3171/jns.2005.103.1.0079. [DOI] [PubMed] [Google Scholar]
- 67.Rotem R, Heyfets A, Fingrut O, Blickstein D, Shaklai M, Flescher E. Cancer Res. 2005;65:1984–1993. doi: 10.1158/0008-5472.CAN-04-3091. [DOI] [PubMed] [Google Scholar]
- 68.Goldin N, Arzoine L, Heyfets A, Israelson A, Zaslavsky Z, Bravman T, Bronner V, Notcovich A, Shoshan-Barmatz V, Flescher E. Oncogene. 2008;27:4636–4643. doi: 10.1038/onc.2008.108. [DOI] [PubMed] [Google Scholar]
- 69.Nakashima RA, Mangan PS, Colombini M, Pedersen PL. Biochemistry. 1986;25:1015–1021. doi: 10.1021/bi00353a010. [DOI] [PubMed] [Google Scholar]
- 70.Storm P, Aits S, Puthia MK, Urbano A, Northen T, Powers S, Bowen B, Chao Y, Reindl W, Lee DY, Sullivan NL, Zhang J, Trulsson M, Yang H, Watson JD, Svanborg C. Oncogene. 2011;30:4765–4779. doi: 10.1038/onc.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Floridi A, Paggi MG, Marcante ML, Silvestrini B, Caputo A, De Martino C. J Natl Cancer Inst. 1981;66:497–499. [PubMed] [Google Scholar]
- 72.Fanciulli M, Valentini A, Bruno T, Citro G, Zupi G, Floridi A. Oncol Res. 1996;8:111–120. [PubMed] [Google Scholar]
- 73.Floridi A, Bruno T, Miccadei S, Fanciulli M, Federico A, Paggi MG. Biochem Pharmacol. 1998;56:841–849. doi: 10.1016/s0006-2952(98)00054-9. [DOI] [PubMed] [Google Scholar]
- 74.De Lena M, Lorusso V, Latorre A, Fanizza G, Gargano G, Caporusso L, Guida M, Catino A, Crucitta E, Sambiasi D, Mazzei A. Eur J Cancer. 2001;37:364–368. doi: 10.1016/s0959-8049(00)00400-7. [DOI] [PubMed] [Google Scholar]
- 75.Di Cosimo S, Ferretti G, Papaldo P, Carlini P, Fabi A, Cognetti F. Drugs Today. 2003;39:157–174. doi: 10.1358/dot.2003.39.3.799451. [DOI] [PubMed] [Google Scholar]
- 76.Oudard S, Carpentier A, Banu E, Fauchon F, Celerier D, Poupon MF, Dutrillaux B, Andrieu JM, Delattre JY. J Neuro-Oncol. 2003;63:81–86. doi: 10.1023/a:1023756707900. [DOI] [PubMed] [Google Scholar]
- 77.Berruti A, Bitossi R, Gorzegno G, Bottini A, Alquati P, De Matteis A, Nuzzo F, Giardina G, Danese S, De Lena M, Lorusso V, Farris A, Sarobba MG, DeFabiani E, Bonazzi G, Castiglione F, Bumma C, Moro G, Bruzzi P, Dogliotti L. J Clin Oncol. 2002;20:4150–4159. doi: 10.1200/JCO.2002.08.012. [DOI] [PubMed] [Google Scholar]
- 78.Price GS, Page RL, Riviere JE, Cline JM, Thrall DE. Cancer Chemother Pharmacol. 1996;38:129–135. doi: 10.1007/s002800050460. [DOI] [PubMed] [Google Scholar]
- 79.Giorgioni G, Ruggieri S, Di Stefano A, Sozio P, Cinque B, Di Marzio L, Santoni G, Claudi F. Bioorg Med Chem Lett. 2008;18:2445–2450. doi: 10.1016/j.bmcl.2008.02.046. [DOI] [PubMed] [Google Scholar]
- 80.Dell’Antone P. Med Chem. 2009;5:491–496. doi: 10.2174/157340609790170551. [DOI] [PubMed] [Google Scholar]
- 81.Pelicano H, Martin DS, Xu RH, Huang P. Oncogene. 2006;25:4633–4646. doi: 10.1038/sj.onc.1209597. [DOI] [PubMed] [Google Scholar]
- 82.Ganapathy-Kanniappan S, Vali M, Kunjithapatham R, Buijs M, Syed LH, Rao PP, Ota S, Kwak BK, Loffroy R, Geschwind JF. Curr Pharm Biotechnol. 2010;11:510–517. doi: 10.2174/138920110791591427. [DOI] [PubMed] [Google Scholar]
- 83.Ganapathy-Kanniappan S, Geschwind JFH, Kunjithapatham R, Buijs M, Vossen JA, Tchernyshyov I, Cole RN, Syed LH, Rao PP, Ota S, Vali M. Anticancer Res. 2009;29:4909–4918. [PMC free article] [PubMed] [Google Scholar]
- 84.Ko YH, Pedersen PL, Geschwind JF. Cancer Lett. 2001;173:83–91. doi: 10.1016/s0304-3835(01)00667-x. [DOI] [PubMed] [Google Scholar]
- 85.Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, Torbenson MS, Hullihen J, Pedersen PL. Biochem Biophys Res Commun. 2004;324:269–275. doi: 10.1016/j.bbrc.2004.09.047. [DOI] [PubMed] [Google Scholar]
- 86.Xu RH, Pelicano H, Zhou Y, Carew JS, Feng L, Bhalla KN, Keating MJ, Huang P. Cancer Res. 2005;65:613–621. [PubMed] [Google Scholar]
- 87.Pereira Da Silva AP, El-Bacha T, Kyaw N, Dos Santos RS, Da-Silva WS, Almeida FC, Da Poian AT, Galina A. Biochem J. 2009;417:717–726. doi: 10.1042/BJ20080805. [DOI] [PubMed] [Google Scholar]
- 88.Kim JS, Ahn KJ, Kim JA, Kim HM, Lee JD, Lee JM, Kim SJ, Park JH. J Bioenerg Biomembr. 2008;40:607–618. doi: 10.1007/s10863-008-9188-0. [DOI] [PubMed] [Google Scholar]
- 89.Ihrlund LS, Hernlund E, Khan O, Shoshan MC. Mol Oncol. 2008;2:94–101. doi: 10.1016/j.molonc.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.El Sayed SM, Abou El-Magd RM, Shishido Y, Chung SP, Sakai T, Watanabe H. Cancer Gene Ther. 2012;19:1–18. doi: 10.1038/cgt.2011.59. [DOI] [PubMed] [Google Scholar]
- 91.Xu RH, Pelicano H, Zhang H, Giles FJ, Keating MJ, Huang P. Leukemia. 2005;19:2153–2158. doi: 10.1038/sj.leu.2403968. [DOI] [PubMed] [Google Scholar]
- 92.Cao X, Bloomston M, Zhang T, Frankel WL, Jia G, Wang B, Hall NC, Koch RM, Cheng H, Knopp MV, Sun D. Clin Cancer Res. 2008;14:1831–1839. doi: 10.1158/1078-0432.CCR-07-1607. [DOI] [PubMed] [Google Scholar]
- 93.Jae HJ, Chung JW, Park HS, Lee MJ, Lee KC, Kim HC, Yoon JH, Chung H, Park JH. Korean J Radiol. 2009;10:596–603. doi: 10.3348/kjr.2009.10.6.596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bachelard HS. Biochem J. 1972;127:83P. doi: 10.1042/bj1270083pa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Aft RL, Zhang FW, Gius D. Br J Cancer. 2002;87:805–812. doi: 10.1038/sj.bjc.6600547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang XD, Deslandes E, Villedieu M, Poulain L, Duval M, Gauduchon P, Schwartz L, Icard P. Anticancer Res. 2006;26:3561–3566. [PubMed] [Google Scholar]
- 97.Maher JC, Krishan A, Lampidis TJ. Cancer Chemother Pharmacol. 2004;53:116–122. doi: 10.1007/s00280-003-0724-7. [DOI] [PubMed] [Google Scholar]
- 98.Lynch RM, Fogarty KE, Fay FS. J Cell Biochem. 1991;112:385–395. doi: 10.1083/jcb.112.3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kurtoglu M, Gao N, Shang J, Maher JC, Lehrman MA, Wangpaichitr M, Savaraj N, Lane AN, Lampidis TJ. Mol Cancer Ther. 2007;6:3049–3058. doi: 10.1158/1535-7163.MCT-07-0310. [DOI] [PubMed] [Google Scholar]
- 100.Maschek G, Savaraj N, Priebe W, Braunschweiger P, Hamilton K, Tidmarsh GF, De Young LR, Lampidis TJ. Cancer Res. 2004;64:31–34. doi: 10.1158/0008-5472.can-03-3294. [DOI] [PubMed] [Google Scholar]
- 101.Tagg SLC, Foster PA, Leese MP, Potter BVL, Reed MJ, Purohit A, Newman SP. Br J Cancer. 2008;99:1842–1848. doi: 10.1038/sj.bjc.6604752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Egler V, Korur S, Failly M, Boulay JL, Imber R, Lino MM, Merlo A. Clin Cancer Res. 2008;14:3132–3140. doi: 10.1158/1078-0432.CCR-07-4182. [DOI] [PubMed] [Google Scholar]
- 103.Singh D, Banerji AK, Dwarakanath BS, Tripathi RP, Gupta JP, Mathew TL, Ravindranath T, Jain V. Strahlenther Onkol. 2005;181:507–514. doi: 10.1007/s00066-005-1320-z. [DOI] [PubMed] [Google Scholar]
- 104.Lin X, Zhang F, Bradbury CM, Kaushal A, Li L, Spitz DR, Aft RL, Gius D. Cancer Res. 2003;63:3413–3417. [PubMed] [Google Scholar]
- 105.Zhong D, Liu X, Schafer-Hales K, Marcus AI, Khuri FR, Sun SY, Zhou W. Mol Cancer Ther. 2008;74:809–817. doi: 10.1158/1535-7163.MCT-07-0559. [DOI] [PubMed] [Google Scholar]
- 106.Dearling JL, Qureshi U, Begent RH, Pedley RB. Clin Cancer Res. 2007;13:1903–1910. doi: 10.1158/1078-0432.CCR-06-2094. [DOI] [PubMed] [Google Scholar]
- 107.Mohanti BK, Rath GK, Anantha N, Kannan V, Das BS, Chandramouli BAR, Banerjee AK, Das S, Jena A, Ravichandran R, Sahi UP, Kumar R, Kapoor N, Kalia VK, Dwarakanath BS, Jain V. Int J Radiat Oncol Biol Phys. 1996;35:103–111. doi: 10.1016/s0360-3016(96)85017-6. [DOI] [PubMed] [Google Scholar]
- 108.Stein M, Lin H, Jeyamohan C, Dvorzhinski D, Gounder M, Bray K, Eddy S, Goodin S, White E, Dipaola RS. Prostate. 2010;70:1388–1394. doi: 10.1002/pros.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Y, Courtois P, Sener A, Malaisse WJ. Int J Mol Med. 2004;14:107–112. [PubMed] [Google Scholar]
- 110.Chen M, Whistler RL. Arch Biochem Biophys. 1975;169:392–396. doi: 10.1016/0003-9861(75)90180-0. [DOI] [PubMed] [Google Scholar]
- 111.Song CW, Sung JH, Clement JJ, Levitt SH. Br J Cancer. 1978;37:136–140. [PMC free article] [PubMed] [Google Scholar]
- 112.Fokt I, Szymanski S, Skora S, Cybulski M, Madden T, Priebe W. Carbohydr Res. 2009;344:1464–1473. doi: 10.1016/j.carres.2009.06.016. [DOI] [PubMed] [Google Scholar]
- 113.Lampidis TJ, Kurtoglu M, Maher JC, Liu H, Krishan A, Sheft V, Szymanski S, Fokt I, Rudnicki WR, Ginalski K, Lesyng B, Priebe W. Cancer Chemother Pharmacol. 2006;58:725–734. doi: 10.1007/s00280-006-0207-8. [DOI] [PubMed] [Google Scholar]
- 114.Kurtoglu M, Maher JC, Lampidis TJ. Antioxid Redox Signaling. 2007;9:1383–1390. doi: 10.1089/ars.2007.1714. [DOI] [PubMed] [Google Scholar]
- 115.Schibli R, Dumas C, Petrig J, Spadola L, Scapozza L, Garcia-Garayoa E, Schubiger PA. Bioconjugate Chem. 2005;16:105–112. doi: 10.1021/bc049774l. [DOI] [PubMed] [Google Scholar]
- 116.Hudock MP, Sanz-Rodrguez CE, Song Y, Chan JMW, Zhang Y, Odeh S, Kosztowski T, Leon-Rossell A, Concepcin JL, Yardley V, Croft SL, Urbina JA, Oldfield E. J Med Chem. 2006;49:215–223. doi: 10.1021/jm0582625. [DOI] [PubMed] [Google Scholar]
- 117.Sun AQ, Yüksel KU, Jacobson TM, Gracy RW. Arch Biochem Biophys. 1990;283:120–129. doi: 10.1016/0003-9861(90)90621-5. [DOI] [PubMed] [Google Scholar]
- 118.Baumann M, Kappl A, Lang T, Brand K, Siegfried W, Paterok E. Cancer Invest. 1990;8:351–356. doi: 10.3109/07357909009012053. [DOI] [PubMed] [Google Scholar]
- 119.Niinaka Y, Paku S, Haga A, Watanabe H, Raz A. Cancer Res. 1998;58:2667–2674. [PubMed] [Google Scholar]
- 120.Funasaka T, Haga A, Raz A, Nagase H. Biochem Biophys Res Commun. 2001;284:1116–1125. doi: 10.1006/bbrc.2001.4912. [DOI] [PubMed] [Google Scholar]
- 121.Yanagawa T, Funasaka T, Tsutsumi S, Watanabe H, Raz A. Endocr-Relat Cancer. 2004;11:749–759. doi: 10.1677/erc.1.00811. [DOI] [PubMed] [Google Scholar]
- 122.Tsutsumi S, Fukasawa T, Yamauchi H, Kato T, Kigure W, Morita H, Asao T, Kuwano H. Int J Oncol. 2009;35:1117–1121. doi: 10.3892/ijo_00000427. [DOI] [PubMed] [Google Scholar]
- 123.Funasaka T, Yanagawa T, Hogan V, Raz A. FASEB J. 2005;19:1422–1430. doi: 10.1096/fj.05-3699com. [DOI] [PubMed] [Google Scholar]
- 124.Funasaka T, Hu H, Hogan V, Raz A. J Biol Chem. 2007;282:36362–36369. doi: 10.1074/jbc.M706301200. [DOI] [PubMed] [Google Scholar]
- 125.Funasaka T, Hu H, Yanagawa T, Hogan V, Raz A. Cancer Res. 2007;67:4236–4243. doi: 10.1158/0008-5472.CAN-06-3935. [DOI] [PubMed] [Google Scholar]
- 126.Tanaka N, Haga A, Naba N, Shiraiwa K, Kusakabe Y, Hashimoto K, Funasaka T, Nagase H, Raz A, Nakamura KT. J Mol Biol. 2006;356:312–324. doi: 10.1016/j.jmb.2005.11.076. [DOI] [PubMed] [Google Scholar]
- 127.Haga A, Tanaka N, Funasaka T, Hashimoto K, Nakamura KT, Watanabe H, Raz A, Nagase H. J Mol Biol. 2006;358:741–753. doi: 10.1016/j.jmb.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 128.Shih WL, Liao MH, Yu FL, Lin PY, Hsu HY, Chiu SJ. Cancer Lett. 2008;270:202–217. doi: 10.1016/j.canlet.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 129.Gibson DR, Gracy RW, Hartman FC. J Biol Chem. 1980;255:9369–9374. [PubMed] [Google Scholar]
- 130.Meng M, Chane TL, Sun YJ, Hsiao CD. Protein Sci. 1999;8:2438–2443. doi: 10.1110/ps.8.11.2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chou CC, Sun YJ, Meng M, Hsiao CD. J Biol Chem. 2000;275:23154–23160. doi: 10.1074/jbc.M002017200. [DOI] [PubMed] [Google Scholar]
- 132.Lee JH, Chang KZ, Patel V, Jeffery CJ. Biochemistry. 2001;40:7799–7805. doi: 10.1021/bi002916o. [DOI] [PubMed] [Google Scholar]
- 133.Arsenieva D, Jeffery CJ. J Mol Biol. 2002;323:77–84. doi: 10.1016/s0022-2836(02)00892-6. [DOI] [PubMed] [Google Scholar]
- 134.Hardré R, Bonnette C, Salmon L, Gaudemer A. Bioorg Med Chem Lett. 1998;8:3435–3438. doi: 10.1016/s0960-894x(98)00621-0. [DOI] [PubMed] [Google Scholar]
- 135.Arsenieva D, Hardre R, Salmon L, Jeffery CJ. Proc Natl Acad Sci USA. 2002;99:5872–5877. doi: 10.1073/pnas.052131799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jeffery CJ, Bahnson BJ, Chien W, Ringe D, Petsko GA. Biochemistry. 2000;39:955–964. doi: 10.1021/bi991604m. [DOI] [PubMed] [Google Scholar]
- 137.Jeffery CJ, Hardré R, Salmon L. Biochemistry. 2001;40:1560–1566. doi: 10.1021/bi0018483. [DOI] [PubMed] [Google Scholar]
- 138.Chirgwin JM, Parsons TF, Noltmann EA. J Biol Chem. 1975;250:7277–7279. [PubMed] [Google Scholar]
- 139.Watanabe H, Takehana K, Date M, Shinozaki T, Raz A. Cancer Res. 1996;56:2960–2963. [PubMed] [Google Scholar]
- 140.Graham Solomons JT, Zimmerly EM, Burns S, Krishnamurthy N, Swan MK, Krings S, Muirhead H, Chirgwin J, Davies C. J Mol Biol. 2004;342:847–860. doi: 10.1016/j.jmb.2004.07.085. [DOI] [PubMed] [Google Scholar]
- 141.Dunaway GA, Kasten TP, Sebo T, Trapp R. Biochem J. 1988;251:677–683. doi: 10.1042/bj2510677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rider MH, Bertrand L, Vertommen D, Michels PA, Rousseau GG, Hue L. Biochem J. 2004;381:561–579. doi: 10.1042/BJ20040752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chesney J, Mitchell R, Benigni F, Bacher M, Spiegel L, Al-Abed Y, Han JH, Metz C, Bucala R. Proc Natl Acad Sci USA. 1999;96:3047–3052. doi: 10.1073/pnas.96.6.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Minchenko O, Opentanova I, Caro J. FEBS Lett. 2003;554:264–270. doi: 10.1016/s0014-5793(03)01179-7. [DOI] [PubMed] [Google Scholar]
- 145.Minchenko A, Leshchinsky I, Opentanova I, Sang N, Srinivas V, Armstead V, Caro J. J Biol Chem. 2002;277:6183–6187. doi: 10.1074/jbc.M110978200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Atsumi T, Chesney J, Metz C, Leng L, Donnelly S, Makita Z, Mitchell R, Bucala R. Cancer Res. 2002;62:5881–5887. [PubMed] [Google Scholar]
- 147.Bando H, Atsumi T, Nishio T, Niwa H, Mishima S, Shimizu C, Yoshioka N, Bucala R, Koike T. Clin Cancer Res. 2005;11:5784–5792. doi: 10.1158/1078-0432.CCR-05-0149. [DOI] [PubMed] [Google Scholar]
- 148.Yalcin A, Telang S, Clem B, Chesney J. Exp Mol Pathol. 2009;86:174–179. doi: 10.1016/j.yexmp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 149.Jeon YK, Yoo DR, Jang YH, Jang SY, Nam MJ. Biochim Biophys Acta Proteins Proteomics. 2011;1814:1340–1348. doi: 10.1016/j.bbapap.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 150.Spitz GA, Furtado CM, Sola-Penna M, Zancan P. Biochem Pharmacol. 2009;77:46–53. doi: 10.1016/j.bcp.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 151.Riquelme PT, Wernette-Hammond ME, Kneer NM, Lardy HA. Proc Natl Acad Sci USA. 1983;80:4301–4305. doi: 10.1073/pnas.80.14.4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, Rasku MA, Arumugam S, Dean WL, Eaton J, Lane A, Trent JO, Chesney J. Mol Cancer Ther. 2008;7:110–120. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- 153.Seo M, Kim JD, Neau D, Sehgal I, Lee YH. PLoS One. 2011;6:e24179. doi: 10.1371/journal.pone.0024179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Zancan P, Rosas AO, Marcondes MC, Marinho-Carvalho MM, Sola-Penna M. Biochem Pharmacol. 2007;73:1520–1527. doi: 10.1016/j.bcp.2007.01.018. [DOI] [PubMed] [Google Scholar]
- 155.Gefflaut T, Blonski C, Perie J, Willson M. Prog Biophys Mol Biol. 1995;63:301–340. doi: 10.1016/0079-6107(95)00008-9. [DOI] [PubMed] [Google Scholar]
- 156.Pezza JA, Choi KH, Berardini TZ, Beernink PT, Allen KN, Tolan DR. J Biol Chem. 2003;278:17307–17313. doi: 10.1074/jbc.M209185200. [DOI] [PubMed] [Google Scholar]
- 157.Li C, Xiao Z, Chen Z, Zhang X, Li J, Wu X, Li X, Yi H, Li M, Zhu G, Liang S. Proteomics. 2006;6:547–558. doi: 10.1002/pmic.200500256. [DOI] [PubMed] [Google Scholar]
- 158.Khan KN, Nakata K, Nakao K, Kato Y, Eguchi K. Dig Dis Sci. 1999;44:1610–1618. doi: 10.1023/a:1026623312806. [DOI] [PubMed] [Google Scholar]
- 159.Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A. J Biol Chem. 1996;271:32529–32537. doi: 10.1074/jbc.271.51.32529. [DOI] [PubMed] [Google Scholar]
- 160.Blonski C, De Moissac D, Périé J, Sygusch J. Biochem J. 1997;323:71–77. doi: 10.1042/bj3230071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Dax C, Coinçon M, Sygusch J, Blonski C. Biochemistry. 2005;44:5430–5443. doi: 10.1021/bi0477992. [DOI] [PubMed] [Google Scholar]
- 162.Ginsburg A, Mehler AH. Biochemistry. 1966;5:2623–2634. doi: 10.1021/bi00872a021. [DOI] [PubMed] [Google Scholar]
- 163.Mabiala-Bassiloua CG, Zwolinska M, Therisod H, Sygusch J, Therisod M. Bioorg Med Chem Lett. 2008;18:1735–1737. doi: 10.1016/j.bmcl.2008.01.076. [DOI] [PubMed] [Google Scholar]
- 164.St-Jean M, Lafrance-Vanasse J, Liotard B, Sygusch J. J Biol Chem. 2005;280:27262–27270. doi: 10.1074/jbc.M502413200. [DOI] [PubMed] [Google Scholar]
- 165.Rale M, Schneider S, Sprenger GA, Samland AK, Fessner WD. Chem Eur J. 2011;17:2623–2632. doi: 10.1002/chem.201002942. [DOI] [PubMed] [Google Scholar]
- 166.Gess B, Hofbauer KH, Deutzmann R, Kurtz A. Pflugers Arch. 2004;448:175–180. doi: 10.1007/s00424-004-1241-1. [DOI] [PubMed] [Google Scholar]
- 167.Ationu A, Humphries A. Int J Mol Med. 1998;2:701–704. doi: 10.3892/ijmm.2.6.701. [DOI] [PubMed] [Google Scholar]
- 168.Opperdoes FR, Michels PAM. Int J Parasitol. 2001;31:482–490. doi: 10.1016/s0020-7519(01)00155-2. [DOI] [PubMed] [Google Scholar]
- 169.Sirover MA. J Cell Biochem. 1997;66:133–140. [PubMed] [Google Scholar]
- 170.Tokunaga K, Nakamura Y, Sakata K, Fujimori K, Ohkubo M, Sawada K, Sakiyama S. Cancer Res. 1987;47:5616–5619. [PubMed] [Google Scholar]
- 171.Schek N, Hall BL, Finn OJ. Cancer Res. 1988;48:6354–6359. [PubMed] [Google Scholar]
- 172.Desprez PY, Poujol D, Saez S. Cancer Lett. 1992;64:219–224. doi: 10.1016/0304-3835(92)90046-x. [DOI] [PubMed] [Google Scholar]
- 173.Epner DE, Partin AW, Schalken JA, Isaacs JT, Coffey DS. Cancer Res. 1993;53:1995–1997. [PubMed] [Google Scholar]
- 174.Bagui S, Ray M, Ray S. Eur J Biochem. 1999;262:386–395. doi: 10.1046/j.1432-1327.1999.00384.x. [DOI] [PubMed] [Google Scholar]
- 175.Song S, Finkel T. Nat Cell Biol. 2007;9:869–870. doi: 10.1038/ncb0807-869. [DOI] [PubMed] [Google Scholar]
- 176.Harris JL, Walters M. Glyceraldehyde-3-phosphate dehydrogenase. In: Boyer PD, editor. The Enzymes. Academic Press; New York: 1976. pp. 1–49. [Google Scholar]
- 177.Rodríguez-Enríquez S, Vital-González PA, Flores-Rodríguez FL, Marín-Hernández A, Ruiz-Azuara L, Moreno-Sánchez R. Toxicol Appl Pharmacol. 2006;215:208–217. doi: 10.1016/j.taap.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 178.Bhardwaj V, Rizvi N, Lai MB, Lai JC, Bhushan A. Anticancer Res. 2010;30:743–749. [PubMed] [Google Scholar]
- 179.Sakai K, Hasumi K, Endo A. Biochim Biophys Acta. 1991;1077:192–196. doi: 10.1016/0167-4838(91)90058-8. [DOI] [PubMed] [Google Scholar]
- 180.Endo A, Hasumi K, Sakai K, Kanbe T. J Antibiot. 1985;38:920–925. doi: 10.7164/antibiotics.38.920. [DOI] [PubMed] [Google Scholar]
- 181.Sakai K, Hasumi K, Endo A. Biochim Biophys Acta Prot Struct Mol Enzymol. 1988;952:297–303. doi: 10.1016/0167-4838(88)90130-6. [DOI] [PubMed] [Google Scholar]
- 182.Kumagai S, Narasaki R, Hasumi K. Biochem Biophys Res Commun. 2008;365:362–368. doi: 10.1016/j.bbrc.2007.10.199. [DOI] [PubMed] [Google Scholar]
- 183.Brown-Woodman PDC, Mohri H, Mohri T, Suter D, White IG. Biochem J. 1978;170:23–37. doi: 10.1042/bj1700023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Ford WC, Harrison A. J Reprod Fertil. 1983;69:147–156. doi: 10.1530/jrf.0.0690147. [DOI] [PubMed] [Google Scholar]
- 185.Jones AR, Bubb WA, Murdoch SR, Stevenson DA. Aust J Biol Sci. 1986;39:395–406. doi: 10.1071/bi9860395. [DOI] [PubMed] [Google Scholar]
- 186.Stevenson D, Jones AR. J Reprod Fertil. 1985;74:157–165. doi: 10.1530/jrf.0.0740157. [DOI] [PubMed] [Google Scholar]
- 187.Skamarauskas J, Carter W, Fowler M, Madjd A, Lister T, Mavroudis G, Ray DE. Toxicology. 2007;232:268–276. doi: 10.1016/j.tox.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 188.Lopez BE, Rodriguez CE, Pribadi M, Cook NM, Shinyashiki M, Fukuto JM. Arch Biochem Biophys. 2005;442:140–148. doi: 10.1016/j.abb.2005.07.012. [DOI] [PubMed] [Google Scholar]
- 189.Lopez BE, Wink DA, Fukuto JM. Arch Biochem Biophys. 2007;465:430–436. doi: 10.1016/j.abb.2007.06.017. [DOI] [PubMed] [Google Scholar]
- 190.Norris AJ, Sartippour MR, Lu M, Park T, Rao JY, Jackson MI, Fukuto JM, Brooks MN. Int J Cancer. 2008;122:1905–1910. doi: 10.1002/ijc.23305. [DOI] [PubMed] [Google Scholar]
- 191.a) Aronov AM, Verlinde CL, Hol WG, Gelb MH. J Med Chem. 1998;41:4790–4799. doi: 10.1021/jm9802620. [DOI] [PubMed] [Google Scholar]; b) Aronov AM, Suresh S, Buckner FS, Van Voorhis WC, Verlinde CL, Opperdoes FR, Hol WG, Gelb MH. Proc Natl Acad Sci USA. 1999;96:4273–4278. doi: 10.1073/pnas.96.8.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bressi JC, Verlinde CL, Aronov AM, Shaw ML, Shin SS, Nguyen LN, Suresh S, Buckner FS, Van Voorhis WC, Kuntz ID, Hol WG, Gelb MH. J Med Chem. 2001;44:2080–2093. doi: 10.1021/jm000472o. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Suresh S, Bressi JC, Kennedy KJ, Verlinde CL, Gelb MH, Hol WG. J Mol Biol. 2001;309:423–435. doi: 10.1006/jmbi.2001.4588. [DOI] [PubMed] [Google Scholar]
- 192.Semenza GL, Roth PH, Fang HM, Wang GL. J Biol Chem. 1994;269:23757–23763. [PubMed] [Google Scholar]
- 193.Wang J, Ying G, Wang J, Jung Y, Lu J, Zhu J, Pienta KJ, Taichman RS. Cancer Res. 2010;70:471–480. doi: 10.1158/0008-5472.CAN-09-2863. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 194.Zieker D, Königsrainer I, Weinreich J, Beckert S, Glatzle J, Nieselt K, Bühler S, Löffler M, Gaedcke J, Northoff H, Mannheim JG, Wiehr S, Pichler BJ, von Weyhern C, Brücher BL, Königsrainer A. Cell Physiol Biochem. 2010;26:147–154. doi: 10.1159/000320545. [DOI] [PubMed] [Google Scholar]
- 195.Ai J, Huang H, Lv X, Tang Z, Chen M, Chen T, Duan W, Sun H, Li Q, Tan R, Liu Y, Duan J, Yang Y, Wei Y, Li Y, Zhou Q. Cell Physiol Biochem. 2011;27:207–216. doi: 10.1159/000327946. [DOI] [PubMed] [Google Scholar]
- 196.a) Lay AJ, Jiang XM, Kisker O, Flynn E, Underwood A, Condron R, Hogg PJ. Nature. 2000;408:869–873. doi: 10.1038/35048596. [DOI] [PubMed] [Google Scholar]; b) Daly EB, Wind T, Jiang XM, Sun L, Hogg PJ. Biochim Biophys Acta. 2004;1691:17–22. doi: 10.1016/j.bbamcr.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 197.Duan Z, Lamendola DE, Yusuf RZ, Penson RT, Preffer FI, Seiden MV. Anticancer Res. 2002;22:1933–1941. [PubMed] [Google Scholar]
- 198.Mcharg J, Littlechild JA. J Pharm Pharmacol. 1996;48:201–205. doi: 10.1111/j.2042-7158.1996.tb07123.x. [DOI] [PubMed] [Google Scholar]
- 199.Bressi JC, Choe J, Hough MT, Buckner FS, Van Voorhis WC, Verlinde CL, Hol WG, Gelb M. J Med Chem. 2000;43:4135–4150. doi: 10.1021/jm000287a. [DOI] [PubMed] [Google Scholar]
- 200.Vas M. Eur J Biochem. 1990;194:639–645. doi: 10.1111/j.1432-1033.1990.tb15663.x. [DOI] [PubMed] [Google Scholar]
- 201.Li YK, Byers LD. Biochim Biophys Acta. 1993;1164:17–21. doi: 10.1016/0167-4838(93)90106-2. [DOI] [PubMed] [Google Scholar]
- 202.Caplan NA, Pogson CI, Hayes DJ, Blackburn GM. Bioorg Med Chem Lett. 1998;8:515–20. doi: 10.1016/s0960-894x(98)00059-6. [DOI] [PubMed] [Google Scholar]
- 203.Jakeman DL, Ivory AJ, Williamson MP, Blackburn GM. J Med Chem. 1998;41:4439–4452. doi: 10.1021/jm970839y. [DOI] [PubMed] [Google Scholar]
- 204.Kotsikorou E, Sahota G, Oldfield E. J Med Chem. 2006;49:6692–6703. doi: 10.1021/jm0604833. [DOI] [PubMed] [Google Scholar]
- 205.Durany N, Carreras J. Comp Biochem Physiol Part B. 1996;114:217–223. doi: 10.1016/0305-0491(95)02135-3. [DOI] [PubMed] [Google Scholar]
- 206.a) Durany N, Joseph J, Campo E, Molina R, Carreras J. Br J Cancer. 1997;75:969–977. doi: 10.1038/bjc.1997.168. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Durany N, Joseph J, Cruz-Sánchez FF, Carreras J. Br J Cancer. 1997;76:1139–1149. doi: 10.1038/bjc.1997.525. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Durany N, Joseph J, Jimenez OM, Climent F, Fernández PL, Rivera F, Carreras J. Br J Cancer. 2000;82:20–27. doi: 10.1054/bjoc.1999.0871. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Usuba T, Ishibashi Y, Okawa Y, Hirakawa T, Takada K, Ohkawa K. Int J Cancer. 2001;94:662–668. doi: 10.1002/ijc.1524. [DOI] [PubMed] [Google Scholar]; e) Narayanan NK, Narayanan BA, Nixon DW. Cancer Detect Prev. 2004;28:443–452. doi: 10.1016/j.cdp.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 207.Takahashi Y, Takahashi S, Yoshimi T, Miura T. Eur J Biochem. 1998;254:497–504. doi: 10.1046/j.1432-1327.1998.2540497.x. [DOI] [PubMed] [Google Scholar]
- 208.Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, Martinez D, Carnero A, Beach D. Cancer Res. 2005;65:177–185. [PubMed] [Google Scholar]
- 209.Hallows WC, Yu W, Denu JM. J Biol Chem. 2012;287:3850–3858. doi: 10.1074/jbc.M111.317404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Rigden DJ, Walter RA, Phillips SE, Fothergill-Gilmore LA. J Mol Biol. 1999;289:691–699. doi: 10.1006/jmbi.1999.2848. [DOI] [PubMed] [Google Scholar]
- 211.Engel M, Mazurek S, Eigenbrodt E, Welter C. J Biol Chem. 2004;279:35803–35812. doi: 10.1074/jbc.M402768200. [DOI] [PubMed] [Google Scholar]
- 212.a) Evans MJ, Saghatelian A, Sorensen EJ, Cravatt BF. Nat Biotechnol. 2005;23:1303–1307. doi: 10.1038/nbt1149. [DOI] [PubMed] [Google Scholar]; b) Evans MJ, Morris GM, Wu J, Olson AJ, Sorensen EJ, Cravatt BF. Mol Biosyst. 2007;3:495–506. doi: 10.1039/b705113a. [DOI] [PubMed] [Google Scholar]
- 213.a) Larsen TM, Wedekind JE, Rayment I, Reed GH. Biochemistry. 1996;35:4349–4358. doi: 10.1021/bi952859c. [DOI] [PubMed] [Google Scholar]; b) Zhang E, Brewer JM, Minor W, Carreira LA, Lebioda L. Biochemistry. 1997;36:12526–12534. doi: 10.1021/bi9712450. [DOI] [PubMed] [Google Scholar]
- 214.a) He P, Naka T, Serada S, Fujimoto M, Tanaka T, Hashimoto S, Shima Y, Yamadori T, Suzuki H, Hirashima T, Matsui K, Shiono H, Okumura M, Nishida T, Tachibana I, Norioka N, Norioka S, Kawase I. Cancer Sci. 2007;98:1234–1240. doi: 10.1111/j.1349-7006.2007.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rütters H, Zürbig P, Halter R, Borlak J. Proteomics. 2006;6:3127–3137. doi: 10.1002/pmic.200500188. [DOI] [PubMed] [Google Scholar]
- 215.a) Tu SH, Chang CC, Chen CS, Tam KW, Wang YJ, Lee CH, Lin HW, Cheng TC, Huang CS, Chu JS, Shih NY, Chen LC, Leu SJ, Ho YS, Wu CH. Breast Cancer Res Treat. 2010;121:539–553. doi: 10.1007/s10549-009-0492-0. [DOI] [PubMed] [Google Scholar]; b) Tsai ST, Chien IH, Shen WH, Kuo YZ, Jin YT, Wong TY, Hsiao JR, Wang HP, Shih NY, Wu LW. Eur J Cancer. 2010;46:1712–1723. doi: 10.1016/j.ejca.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 216.Anderson VE, Weiss PM, Cleland WW. Biochemistry. 1984;23:2779–2786. doi: 10.1021/bi00307a038. [DOI] [PubMed] [Google Scholar]
- 217.Wedekind JE, Poyner RR, Reed GH, Rayment I. Biochemistry. 1994;33:9333–9342. doi: 10.1021/bi00197a038. [DOI] [PubMed] [Google Scholar]
- 218.Pietkiewicz J, Gamian A, Staniszewska M, Danielewicz R. J Enzyme Inhib Med Chem. 2009;24:356–364. doi: 10.1080/14756360802187679. [DOI] [PubMed] [Google Scholar]
- 219.Pietkiewicz J, Bronowicka-Szydelko A, Dzierzba K, Danielewicz R, Gamian A. Protein J. 2011;30:149–158. doi: 10.1007/s10930-011-9307-3. [DOI] [PubMed] [Google Scholar]
- 220.Qin J, Chai G, Brewer JM, Lovelace LL, Lebioda L. Biochemistry. 2006;45:793–800. doi: 10.1021/bi051558s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mazurek S. Int J Biochem Cell Biol. 2011;43:969–980. doi: 10.1016/j.biocel.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 222.Dombrauckas JD, Santarsiero BD, Mesecar AD. Biochemistry. 2005;44:9417–9429. doi: 10.1021/bi0474923. [DOI] [PubMed] [Google Scholar]
- 223.Luo W, Hu H, Chang R, Zhong J, Knabel M, O’Meally R, Cole RN, Pandey A, Semenza GL. Cell. 2011;145:732–744. doi: 10.1016/j.cell.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Mazurek S, Boschek CB, Hugo F, Eigenbrodt E. Semin Cancer Biol. 2005;15:300–308. doi: 10.1016/j.semcancer.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 225.Hitosugi T, Kang S, Vander Heiden MG, Chung TW, Elf S, Lythgoe K, Dong S, Lonial S, Wang X, Chen GZ, Xie J, Gu TL, Polakiewicz RD, Roesel JL, Boggon TJ, Khuri FR, Gilliland DG, Cantley LC, Kaufman J, Chen J. Sci Signaling. 2009;2:ra73. doi: 10.1126/scisignal.2000431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.a) Mazurek S, Grimm H, Boschek CB, Vaupel P, Eigenbrodt E. Br J Nutr. 2002;87:S23–S29. [PubMed] [Google Scholar]; b) Christofk HR, Vander Heiden MG, Wu N, Asara JM, Cantley LC. Nature. 2008;452:181–186. doi: 10.1038/nature06667. [DOI] [PubMed] [Google Scholar]
- 227.Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. Nature. 2008;452:230–233. doi: 10.1038/nature06734. [DOI] [PubMed] [Google Scholar]
- 228.Vander Heiden MG, Locasale JW, Swanson KD, Sharfi H, Heffron GJ, Amador-Noguez D, Christofk HR, Wagner G, Rabinowitz JD, Asara JM, Cantley LC. Science. 2010;329:1492–1499. doi: 10.1126/science.1188015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Steták A, Veress R, Ovádi J, Csermely P, Kéri G, Ullrich A. Cancer Res. 2007;67:1602–1608. doi: 10.1158/0008-5472.CAN-06-2870. [DOI] [PubMed] [Google Scholar]
- 230.Bluemlein K, Grüning NM, Feichtinger RG, Lehrach H, Kofler B, Ralser M. Oncotarget. 2011;2:393–400. doi: 10.18632/oncotarget.278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Chen J, Xie J, Jiang Z, Wang B, Wang Y, Hu X. Oncogene. 2011;30:4297–4306. doi: 10.1038/onc.2011.137. [DOI] [PubMed] [Google Scholar]
- 232.Papageorgiou VP, Assimopoulou AN, Couladouros EA, Hepworth D, Nicolaou KC. Angew Chem. 1999;111:280–311. doi: 10.1002/(SICI)1521-3773(19990201)38:3<270::AID-ANIE270>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 1999;38:270–301. doi: 10.1002/(SICI)1521-3773(19990201)38:3<270::AID-ANIE270>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 233.Chen J, Jiang Z, Wang B, Wang Y, Hu X. Cancer Lett. 2012;316:204–210. doi: 10.1016/j.canlet.2011.10.039. [DOI] [PubMed] [Google Scholar]
- 234.García-Alles LF, Erni B. Eur J Biochem. 2002;269:3226–3236. doi: 10.1046/j.1432-1033.2002.02995.x. [DOI] [PubMed] [Google Scholar]
- 235.Vander Heiden MG, Christofk HR, Schuman E, Subtelny AO, Sharfi H, Harlow EE, Xian J, Cantley LC. Biochem Pharmacol. 2010;79:1118–1124. doi: 10.1016/j.bcp.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Takada S, Nagato Y, Yamamura M. Biochem Mol Biol Int. 1997;43:9–17. doi: 10.1080/15216549700203761. [DOI] [PubMed] [Google Scholar]
- 237.a) Kéri G, Erchegyi J, Horváth A, Mezõ I, Idei M, Vántus T, Balogh A, Vadász Z, Bökönyi G, Seprödi J, Teplán I, Csuka O, Tejeda M, Gaál D, Szegedi Z, Szende B, Roze C, Kalthoff H, Ullrich A. Proc Natl Acad Sci USA. 1996;93:12513–12518. doi: 10.1073/pnas.93.22.12513. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Szende B, Kéri G. Anticancer Drugs. 2003;14:585–588. doi: 10.1097/00001813-200309000-00002. [DOI] [PubMed] [Google Scholar]
- 238.http://www.clinicaltrial.gov.
- 239.a) Spoden GA, Mazurek S, Morandell D, Bacher N, Ausserlechner MJ, Jansen-Dürr P, Eigenbrodt E, Zwerschke W. Int J Cancer. 2008;123:312–321. doi: 10.1002/ijc.23512. [DOI] [PubMed] [Google Scholar]; b) Spoden GA, Rostek U, Lechner S, Mitterberger M, Mazurek S, Zwerschke W. Exp Cell Res. 2009;315:2765–2774. doi: 10.1016/j.yexcr.2009.06.024. [DOI] [PubMed] [Google Scholar]
- 240.Duan HF, Hu XW, Chen JL, Gao LH, Xi YY, Lu Y, Li JF, Zhao SR, Xu JJ, Chen HP, Chen W, Wu CT. J Natl Cancer Inst. 2007;99:1551–1555. doi: 10.1093/jnci/djm132. [DOI] [PubMed] [Google Scholar]
- 241.a) Guo W, Zhang Y, Chen T, Wang Y, Xue J, Zhang Y, Xiao W, Mo X, Lu Y. J Cancer Res Clin Oncol. 2011;137:65–72. doi: 10.1007/s00432-010-0860-5. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Shi HS, Li D, Zhang J, Wang YS, Yang L, Zhang HL, Wang XH, Mu B, Wang W, Ma Y, Guo FC, Wei YQ. Cancer Sci. 2010;101:1447–1453. doi: 10.1111/j.1349-7006.2010.01562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Boxer MB, Jiang JK, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, Veith H, Leister W, Austin CP, Park HW, Inglese J, Cantley LC, Auld DS, Thomas CJ. J Med Chem. 2010;53:1048–1055. doi: 10.1021/jm901577g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Jiang JK, Boxer MB, Vander Heiden MG, Shen M, Skoumbourdis AP, Southall N, Veith H, Leister W, Austin CP, Park HW, Inglese J, Cantley LC, Auld DS, Thomas CJ. Bioorg Med Chem Lett. 2010;20:3387–3393. doi: 10.1016/j.bmcl.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Walsh MJ, Brimacombe KR, Veith H, Bougie JM, Daniel T, Leister W, Cantley LC, Israelsen WJ, Vander Heiden MG, Shen M, Auld DS, Thomas CJ, Boxer MB. Bioorg Med Chem Lett. 2011;21:6322–6327. doi: 10.1016/j.bmcl.2011.08.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.a) Koukourakis MI, Giatromanolaki A, Sivridis E, Bougioukas G, Didilis V, Gatter KC, Harris AL. Br J Cancer. 2003;89:877–885. doi: 10.1038/sj.bjc.6601205. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Koukourakis MI, Giatromanolaki A, Simopoulos C, Polychronidis A, Sivridis E. Clin Exp Metastasis. 2005;22:25–30. doi: 10.1007/s10585-005-2343-7. [DOI] [PubMed] [Google Scholar]; c) Giatromanolaki A, Sivridis E, Gatter KC, Turley H, Harris AL, Koukourakis MI. Gynecol Oncol. 2006;103:912–918. doi: 10.1016/j.ygyno.2006.05.043. [DOI] [PubMed] [Google Scholar]; d) Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Harris AL. J Clin Oncol. 2006;24:4301–4308. doi: 10.1200/JCO.2006.05.9501. [DOI] [PubMed] [Google Scholar]; e) Kolev Y, Uetake H, Takagi Y, Sugihara K. Ann Surg Oncol. 2008;15:2336–2344. doi: 10.1245/s10434-008-9955-5. [DOI] [PubMed] [Google Scholar]; f) Langhammer S, Najjar M, Hess-Stumpp H, Thierauch KH. Targeted Oncol. 2011;6:155–162. doi: 10.1007/s11523-011-0184-7. [DOI] [PubMed] [Google Scholar]; g) Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Trarbach T, Folprecht G, Shi MM, Lebwohl D, Jalava T, Laurent D, Meinhardt G, Harris AL. Clin Cancer Res. 2011;17:4892–4900. doi: 10.1158/1078-0432.CCR-10-2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Leiblich A, Cross SS, Catto JW, Phillips JT, Leung HY, Hamdy FC, Rehman I. Oncogene. 2006;25:2953–2960. doi: 10.1038/sj.onc.1209262. [DOI] [PubMed] [Google Scholar]
- 247.Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV. Proc Natl Acad Sci USA. 1997;94:6658–6663. doi: 10.1073/pnas.94.13.6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.a) Fantin VR, St-Pierre J, Leder P. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]; b) Wang ZY, Loo TY, Shen JG, Wang N, Wang DM, Yang DP, Mo SL, Guan XY, Chen JP. Breast Cancer Res Treat. 2012;131:791–800. doi: 10.1007/s10549-011-1466-6. [DOI] [PubMed] [Google Scholar]
- 249.a) Zhou M, Zhao Y, Ding Y, Liu H, Liu Z, Fodstad O, Riker AI, Kamarajugadda S, Lu J, Owen LB, Ledoux SP, Tan M. Mol Cancer. 2010;9:33. doi: 10.1186/1476-4598-9-33. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao Y, Liu H, Liu Z, Ding Y, LeDoux SP, Wilson GL, Voellmy R, Lin Y, Lin W, Nahta R, Liu B, Fodstad O, Chen J, Wu Y, Price JE, Tan M. Cancer Res. 2011;71:4585–4597. doi: 10.1158/0008-5472.CAN-11-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Xie H, Valera VA, Merino MJ, Amato AM, Signoretti S, Linehan WM, Sukhatme VP, Seth P. Mol Cancer Ther. 2009;8:626–635. doi: 10.1158/1535-7163.MCT-08-1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.a) Kanno T, Sudo K, Maekawa M, Nishimura Y, Ukita M, Fukutake K. Clin Chim Acta. 1988;173:89–98. doi: 10.1016/0009-8981(88)90359-2. [DOI] [PubMed] [Google Scholar]; b) Miyajima H, Takahashi Y, Suzuki M, Shimizu T, Kaneko E. Neurology. 1993;43:1414–1419. doi: 10.1212/wnl.43.7.1414. [DOI] [PubMed] [Google Scholar]; c) Lee BJ, Zand L, Manek NJ, Hsiao LL, Babovic-Vuksanovic D, Wylam ME, Qian Q. Arthritis Care Res. 2011;63:1782–1786. doi: 10.1002/acr.20584. [DOI] [PubMed] [Google Scholar]
- 252.Granchi C, Bertini S, Macchia M, Minutolo F. Curr Med Chem. 2010;17:672–697. doi: 10.2174/092986710790416263. [DOI] [PubMed] [Google Scholar]
- 253.Thornburg JM, Nelson KK, Clem BF, Lane AN, Arumugam S, Simmons A, Eaton JW, Telang S, Chesney J. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.a) Fiume L, Manerba M, Vettraino M, Di Stefano G. Pharmacology. 2010;86:157–162. doi: 10.1159/000317519. [DOI] [PubMed] [Google Scholar]; b) Elwood JC. Cancer Res. 1968;28:2056–2060. [PubMed] [Google Scholar]; c) Papaconstantinou J, Colowick SP. J Biol Chem. 1961;236:278–284. [PubMed] [Google Scholar]; d) Papaconstantinou J, Colowick SP. J Biol Chem. 1961;236:285–288. [PubMed] [Google Scholar]; e). E. B. Goldberg, S. P. Colowick, J. Biol. Chem. 1965, 240, 2786–2790; f) Goldberg EB, Nitowsky HM, Colowick SP. J Biol Chem. 1965;240:2791–2796. [PubMed] [Google Scholar]
- 255.Shelley MD, Hartley L, Fish RG, Groundwater P, Morgan JJG, Mort D, Mason M, Evans A. Cancer Lett. 1999;135:171–180. doi: 10.1016/s0304-3835(98)00302-4. [DOI] [PubMed] [Google Scholar]
- 256.Gomez MS, Piper RC, Hunsaker LA, Royer RE, Deck LM, Makler MT, Vander Jagt DL. Mol Biochem Parasitol. 1997;90:235–246. doi: 10.1016/s0166-6851(97)00140-0. [DOI] [PubMed] [Google Scholar]
- 257.Jaroszewski JW, Kaplan O, Cohen JS. Cancer Res. 1990;50:6936–6943. [PubMed] [Google Scholar]
- 258.Tuszynski GP, Cossu G. Cancer Res. 1984;44:768–771. [PubMed] [Google Scholar]
- 259.a) Yu Y, Deck JA, Hunsaker LA, Deck LM, Royer RE, Goldberg E, Vander Jagt DL. Biochem Pharmacol. 2001;62:81–89. doi: 10.1016/s0006-2952(01)00636-0. [DOI] [PubMed] [Google Scholar]; b) Deck LM, Royer RE, Chamblee BB, Hernandez VM, Malone RR, Torres JE, Hunsaker LA, Piper RC, Makler MT, Vander Jagt DL. J Med Chem. 1998;41:3879–3887. doi: 10.1021/jm980334n. [DOI] [PubMed] [Google Scholar]
- 260.Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Proc Natl Acad Sci USA. 2010;107:2037–2042. doi: 10.1073/pnas.0914433107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Manerba M, Vettraino M, Fiume L, Di Stefano G, Sartini A, Giacomini E, Buonfiglio R, Roberti M, Recanatini M. ChemMedChem. 2012;7:311–317. doi: 10.1002/cmdc.201100471. [DOI] [PubMed] [Google Scholar]
- 262.Kotlyar AB, Randazzo A, Honbo N, Jin ZQ, Karliner JS, Cecchini G. FEBS Lett. 2010;584:159–165. doi: 10.1016/j.febslet.2009.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Moorhouse AD, Spiteri C, Sharma P, Zloh M, Moses JE. Chem Commun. 2011;47:230–232. doi: 10.1039/c0cc01166e. [DOI] [PubMed] [Google Scholar]
- 264.Minutolo F, Macchia M, Granchi C, Roy S, Giannaccini G, Lucacchini A. Università di Pisa; WO 2011054525, 2011. Chem Abstr. 2011;154:588710.
- 265.a) Granchi C, Roy S, Giacomelli C, Macchia M, Tuccinardi T, Martinelli A, Lanza M, Betti L, Giannaccini G, Lucacchini A, Funel N, León LG, Giovannetti E, Peters GJ, Palchaudhuri R, Calvaresi EC, Hergenrother PJ, Minutolo F. J Med Chem. 2011;54:1599–1612. doi: 10.1021/jm101007q. [DOI] [PubMed] [Google Scholar]; b) Granchi C, Roy S, De Simone A, Salvetti I, Tuccinardi T, Martinelli A, Macchia M, Lanza M, Betti L, Giannaccini G, Lucacchini A, Giovannetti E, Sciarrillo R, Peters GJ, Minutolo F. Eur J Med Chem. 2011;46:5398–5407. doi: 10.1016/j.ejmech.2011.08.046. [DOI] [PubMed] [Google Scholar]
- 266.Granchi C, Roy S, Del Fiandra C, Tuccinardi T, Lanza M, Betti L, Giannaccini G, Lucacchini A, Martinelli A, Macchia M, Minutolo F. Med Chem Commun. 2011;2:638–643. [Google Scholar]
- 267.Granchi C, Roy S, Mottinelli M, Nardini E, Campinoti F, Tuccinardi T, Lanza M, Betti L, Giannaccini G, Lucacchini A, Martinelli A, Macchia M, Minutolo F. Bioorg Med Chem Lett. 2011;21:7331–7336. doi: 10.1016/j.bmcl.2011.10.031. [DOI] [PubMed] [Google Scholar]
- 268.Ward RA, Brassington C, Breeze AL, Caputo A, Critchlow S, Davies G, Goodwin L, Hassall G, Greenwood R, Holdgate GA, Mrosek M, Norman RA, Pearson S, Tart J, Tucker JA, Vogtherr M, Whittaker D, Wingfield J, Winter J, Hudson K. J Med Chem. 2012 doi: 10.1021/jm201734r. in press. [DOI] [PubMed] [Google Scholar]
- 269.Chiche J, Brahimi-Horn MC, Pouysségur J. J Cell Mol Med. 2010;14:771–794. doi: 10.1111/j.1582-4934.2009.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Izumi H, Torigoe T, Ishiguchi H, Uramoto H, Yoshida Y, Tanabe M, Ise T, Murakami T, Yoshida T, Nomoto M, Kohno K. Cancer Treat Rev. 2003;29:541–549. doi: 10.1016/s0305-7372(03)00106-3. [DOI] [PubMed] [Google Scholar]
- 271.a) Merezhinskaya N, Fishbein WN. Histol Histopathol. 2009;24:243–264. doi: 10.14670/HH-24.243. [DOI] [PubMed] [Google Scholar]; b) Halestrap AP, Meredith D. Pflugers Arch. 2004;447:619–628. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]; c) Halestrap AP. IUBMB Life. 2012;64:1–9. doi: 10.1002/iub.573. [DOI] [PubMed] [Google Scholar]
- 272.Morris ME, Felmlee MA. AAPS J. 2008;10:311–321. doi: 10.1208/s12248-008-9035-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.a) Dimmer KS, Friedrich B, Lang F, Deitmer JW, Bröer S. Biochem J. 2000;350:219–227. [PMC free article] [PubMed] [Google Scholar]; b) Ullah MS, Davies AJ, Halestrap AP. J Biol Chem. 2006;281:9030–9037. doi: 10.1074/jbc.M511397200. [DOI] [PubMed] [Google Scholar]
- 274.Gallagher SM, Castorino JJ, Wang D, Philp NJ. Cancer Res. 2007;67:4182–4189. doi: 10.1158/0008-5472.CAN-06-3184. [DOI] [PubMed] [Google Scholar]; Kennedy KM, Dewhirst MW. Future Oncol. 2010;6:127–148. doi: 10.2217/fon.09.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.a) Pinheiro C, Longatto-Filho A, Scapulatempo C, Ferreira L, Martins S, Pellerin L, Rodrigues M, Alves VA, Schmitt F, Baltazar F. Virchows Arch. 2008;452:139–146. doi: 10.1007/s00428-007-0558-5. [DOI] [PubMed] [Google Scholar]; b) Pinheiro C, Albergaria A, Paredes J, Sousa B, Dufloth R, Vieira D, Schmitt F, Baltazar F. Histopathology. 2010;56:860–867. doi: 10.1111/j.1365-2559.2010.03560.x. [DOI] [PubMed] [Google Scholar]
- 276.Mathupala SP, Parajuli P, Sloan AE. Neurosurgery. 2004;55:1410–1419. doi: 10.1227/01.neu.0000143034.62913.59. [DOI] [PubMed] [Google Scholar]
- 277.Dhup S, Dadhich RK, Porporato PE, Sonveaux P. Curr Pharm Des. 2012;18:1319–1330. doi: 10.2174/138161212799504902. [DOI] [PubMed] [Google Scholar]
- 278.Gerlinger M, Santos CR, Spencer-Dene B, Martinez P, Endesfelder D, Burrell RA, Vetter M, Jiang M, Saunders RE, Kelly G, Rioux-Leclercq N, Stamp G, Patard JJ, Larkin J, Howell M, Swanton C. J Pathol. doi: 10.1002/path.4006. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Le Floch R, Chiche J, Marchiq I, Naïken T, Ilk K, Murray CM, Critchlow SE, Roux D, Simon MP, Pouysségur J. Proc Natl Acad Sci USA. 2011;108:16663–16668. doi: 10.1073/pnas.1106123108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.a) Halestrap AP, Denton RM. Biochem J. 1974;138:313–316. doi: 10.1042/bj1380313. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Halestrap AP, Denton RM. Biochem J. 1975;148:97–106. doi: 10.1042/bj1480097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Spencer TL, Lehninger AL. Biochem J. 1976;154:405–414. doi: 10.1042/bj1540405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Manning Fox JE, Meredith D, Halestrap AP. J Physiol. 2000;529:285–293. doi: 10.1111/j.1469-7793.2000.00285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.a) Wahl ML, Owen JA, Burd R, Herlands RA, Nogami SS, Rodeck U, Berd D, Leeper DB, Owen CS. Mol Cancer Ther. 2002;1:617–628. [PubMed] [Google Scholar]; b) Coss RA, Storck CW, Daskalakis C, Berd D, Wahl ML. Mol Cancer Ther. 2003;2:383–388. [PubMed] [Google Scholar]
- 284.Colen CB, Seraji-Bozorgzad N, Marples B, Galloway MP, Sloan AE, Mathupala SP. Neurosurgery. 2006;59:1313–1323. doi: 10.1227/01.NEU.0000249218.65332.BF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Colen CB, Shen Y, Ghoddoussi F, Yu P, Francis TB, Koch BJ, Monterey MD, Galloway MP, Sloan AE, Mathupala SP. Neoplasia. 2011;13:620–632. doi: 10.1593/neo.11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Poole RC, Halestrap AP. Biochem J. 1991;275:307–312. doi: 10.1042/bj2750307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Jackson VN, Halestrap AP. J Biol Chem. 1996;271:861–868. doi: 10.1074/jbc.271.2.861. [DOI] [PubMed] [Google Scholar]
- 288.Wilson MC, Meredith D, Manning Fox JE, Manoharan C, Davies AJ, Halestrap AP. J Biol Chem. 2005;280:27213–27221. doi: 10.1074/jbc.M411950200. [DOI] [PubMed] [Google Scholar]
- 289.Johnson JH, Belt JA, Dubinsky WP, Zimniak A, Racker E. Biochemistry. 1980;19:3836–3840. doi: 10.1021/bi00557a029. [DOI] [PubMed] [Google Scholar]
- 290.Donovan JA, Jennings ML. Biochemistry. 1985;24:561–564. doi: 10.1021/bi00324a003. [DOI] [PubMed] [Google Scholar]
- 291.Volk C, Kempski B, Kempski OS. Neurosci Lett. 1997;223:121–124. doi: 10.1016/s0304-3940(97)13420-6. [DOI] [PubMed] [Google Scholar]
- 292.Ben-Horin H, Tassini M, Vivi A, Navon G, Kaplan O. Cancer Res. 1995;55:2814–2821. [PubMed] [Google Scholar]
- 293.Fang J, Quinones QJ, Holman TL, Morowitz MJ, Wang Q, Zhao H, Sivo F, Maris JM, Wahl ML. Mol Pharmacol. 2006;70:2108–2115. doi: 10.1124/mol.106.026245. [DOI] [PubMed] [Google Scholar]
- 294.Murray CM, Hutchinson R, Bantick JR, Belfield GP, Benjamin AD, Brazma D, Bundick RV, Cook ID, Craggs RI, Edwards S, Evans LR, Harrison R, Holness E, Jackson AP, Jackson CG, Kingston LP, Perry MW, Ross AR, Rugman PA, Sidhu SS, Sullivan M, Taylor-Fishwick DA, Walker PC, Whitehead YM, Wilkinson DJ, Wright A, Donald DK. Nat Chem Biol. 2005;1:371–376. doi: 10.1038/nchembio744. [DOI] [PubMed] [Google Scholar]
- 295.Guile SD, Bantick JR, Cheshire DR, Cooper ME, Davis AM, Donald DK, Evans R, Eyssade C, Ferguson DD, Hill S, Hutchinson R, Ingall AH, Kingston LP, Martin I, Martin BP, Mohammed RT, Murray C, Perry MW, Reynolds RH, Thorne PV, Wilkinson DJ, Withnall J. Bioorg Med Chem Lett. 2006;16:2260–2265. doi: 10.1016/j.bmcl.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 296.a) Bueno V, Binet I, Steger U, Bundick R, Ferguson D, Murray C, Donald D, Wood K. Transplantation. 2007;84:1204–1207. doi: 10.1097/01.tp.0000287543.91765.41. [DOI] [PubMed] [Google Scholar]; b) Ekberg H, Qi Z, Pahlman C, Veress B, Bundick RV, Craggs RI, Holness E, Edwards S, Murray CM, Ferguson D, Kerry PJ, Wilson E, Donald DK. Transplantation. 2007;84:1191–1199. doi: 10.1097/01.tp.0000287541.53389.be. [DOI] [PubMed] [Google Scholar]; c) Påhlman C, Malm H, Qi Z, Veress B, Ferguson D, Bundick R, Murray C, Donald D, Ekberg H. Transplantation. 2008;86:1135–1138. doi: 10.1097/TP.0b013e318186b978. [DOI] [PubMed] [Google Scholar]
- 297.a) Ovens MJ, Davies AJ, Wilson MC, Murray CM, Halestrap AP. Biochem J. 2010;425:523–530. doi: 10.1042/BJ20091515. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ovens MJ, Manoharan C, Wilson MC, Murray CM, Halestrap AP. Biochem J. 2010;431:217–225. doi: 10.1042/BJ20100890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.http://science.cancerresearchuk.org.
- 299.Kobayashi M, Otsuka Y, Itagaki S, Hirano T, Iseki K. Int J Pharm. 2006;317:19–25. doi: 10.1016/j.ijpharm.2006.02.043. [DOI] [PubMed] [Google Scholar]
- 300.Hodel C. Toxicol Lett. 2002;128:159–168. doi: 10.1016/s0378-4274(02)00010-3. [DOI] [PubMed] [Google Scholar]
- 301.Izumi H, Takahashi M, Uramoto H, Nakayama Y, Oyama T, Wang KY, Sasaguri Y, Nishizawa S, Kohno K. Cancer Sci. 2011;102:1007–1013. doi: 10.1111/j.1349-7006.2011.01908.x. [DOI] [PubMed] [Google Scholar]
- 302.Harris RA, Bowker-Kinley MM, Huang B, Wu P. Adv Enzyme Regul. 2002;42:249–259. doi: 10.1016/s0065-2571(01)00061-9. [DOI] [PubMed] [Google Scholar]
- 303.a) Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Biochem J. 1998;329:191–196. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Roche TE, Hiromasa Y. Cell Mol Life Sci. 2007;64:830–849. doi: 10.1007/s00018-007-6380-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.a) Korotchkina LG, Patel MS. J Biol Chem. 2001;276:37223–37229. doi: 10.1074/jbc.M103069200. [DOI] [PubMed] [Google Scholar]; b) Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Biochem J. 2001;358:69–77. doi: 10.1042/0264-6021:3580069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.a) Kim JW, Tchernyshyov I, Semenza GL, Dang CV. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]; b) Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. Cell Metab. 2006;3:187–197. doi: 10.1016/j.cmet.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 306.Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL. Br J Cancer. 2008;98:1975–1984. doi: 10.1038/sj.bjc.6604356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, Jeoung NH, Harris RA, Verma A. J Biol Chem. 2008;283:22700–22708. doi: 10.1074/jbc.M801765200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Mann WR, Dragland CJ, Vinluan CC, Vedananda TR, Bell PA, Aicher TD. Biochim Biophys Acta. 2000;1480:283–292. doi: 10.1016/s0167-4838(00)00079-0. [DOI] [PubMed] [Google Scholar]
- 309.a) Stacpoole PW, Lorenz AC, Thomas RG, Harman EM. Ann Intern Med. 1988;108:58–63. doi: 10.7326/0003-4819-108-1-58. [DOI] [PubMed] [Google Scholar]; b) Stacpoole PW, Kurtz TL, Han Z, Langaee T. Adv Drug Delivery Rev. 2008;60:1478–1487. doi: 10.1016/j.addr.2008.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Whitehouse S, Randle PJ. Biochem J. 1973;134:651–653. doi: 10.1042/bj1340651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.a) Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]; b) Michelakis ED, Webster L, Mackey JR. Br J Cancer. 2008;99:989–994. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Papandreou I, Goliasova T, Denko NC. Int J Cancer. 2011;128:1001–1008. doi: 10.1002/ijc.25728. [DOI] [PubMed] [Google Scholar]
- 313.a) Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Sci Transl Med. 2010;2:31ra34. doi: 10.1126/scitranslmed.3000677. [DOI] [PubMed] [Google Scholar]; b) Pearson H. Nature. 2007;446:474–475. doi: 10.1038/446474a. [DOI] [PubMed] [Google Scholar]
- 314.Khan A. J Palliat Med. 2011;14:973–977. doi: 10.1089/jpm.2010.0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, Stacpoole PW, DiMauro S, De Vivo DC. Neurology. 2006;66:324–330. doi: 10.1212/01.wnl.0000196641.05913.27. [DOI] [PubMed] [Google Scholar]
- 316.Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, Kasten SA, Roche TE, Brown DG. Biochemistry. 2006;45:402–415. doi: 10.1021/bi051402s. [DOI] [PubMed] [Google Scholar]
- 317.Cairns RA, Papandreou I, Sutphin PD, Denko NC. Proc Natl Acad Sci USA. 2007;104:9445–9450. doi: 10.1073/pnas.0611662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Breast Cancer Res Treat. 2010;120:253–260. doi: 10.1007/s10549-009-0435-9. [DOI] [PubMed] [Google Scholar]
- 319.Xie J, Wang BS, Yu DH, Lu Q, Ma J, Qi H, Fang C, Chen HZ. Int J Oncol. 2011;38:409–417. doi: 10.3892/ijo.2010.851. [DOI] [PubMed] [Google Scholar]
- 320.Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Gynecol Oncol. 2008;109:394–402. doi: 10.1016/j.ygyno.2008.01.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Prostate. 2008;68:1223–1231. doi: 10.1002/pros.20788. [DOI] [PubMed] [Google Scholar]
- 322.Dhar S, Lippard SJ. Proc Natl Acad Sci USA. 2009;106:22199–22204. doi: 10.1073/pnas.0912276106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323.Xue X, You S, Zhang Q, Wu Y, Zou GZ, Wang PC, Zhao YL, Xu Y, Jia L, Zhang X, Liang XJ. Mol Pharm. 2012;9:634–644. doi: 10.1021/mp200571k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Whitehouse S, Cooper RH, Randle PJ. Biochem J. 1974;141:761–774. doi: 10.1042/bj1410761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Espinal J, Leesnitzer T, Hassman A, Beggs M, Cobb J. Drug Dev Res. 1995;35:130–136. [Google Scholar]
- 326.Tuganova A, Yoder MD, Popov KM. J Biol Chem. 2001;276:17994–17999. doi: 10.1074/jbc.M009327200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 328.a) Aicher TD, Anderson RC, Bebernitz GR, Coppola GM, Jewell CF, Knorr DC, Liu C, Sperbeck DM, Brand LJ, Strohschein RJ, Gao J, Vinluan CC, Shetty SS, Dragland C, Kaplan EL, DelGrande D, Islam A, Liu X, Lozito RJ, Maniara WM, Walter RE, Mann WR. J Med Chem. 1999;42:2741–2746. doi: 10.1021/jm9902584. [DOI] [PubMed] [Google Scholar]; b) Aicher TD, Anderson RC, Gao J, Shetty SS, Coppola GM, Stanton JL, Knorr DC, Sperbeck DM, Brand LJ, Vinluan CC, Kaplan EL, Dragland CJ, Tomaselli HC, Islam A, Lozito RJ, Liu X, Maniara WM, Fillers WS, DelGrande D, Walter RE, Mann WR. J Med Chem. 2000;43:236–249. doi: 10.1021/jm990358+. [DOI] [PubMed] [Google Scholar]; c) Hiromasa Y, Yan X, Roche TE. Biochemistry. 2008;47:2312–2324. doi: 10.1021/bi7014772. [DOI] [PubMed] [Google Scholar]
- 329.Bebernitz GR, Aicher TD, Stanton JL, Gao J, Shetty SS, Knorr DC, Strohschein RJ, Tan J, Brand LJ, Liu C, Wang WH, Vinluan CC, Kaplan EL, Dragland CJ, DelGrande D, Islam A, Lozito RJ, Liu X, Maniara WM, Mann WR. J Med Chem. 2000;43:2248–2257. doi: 10.1021/jm0000923. [DOI] [PubMed] [Google Scholar]
- 330.Kato M, Li J, Chuang JL, Chuang DT. Structure. 2007;15:992–1004. doi: 10.1016/j.str.2007.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.a) Mayers RM, Butlin RJ, Kilgour E, Leighton B, Martin D, Myatt J, Orme JP, Holloway BR. Biochem Soc Trans. 2003;31:1165–1167. doi: 10.1042/bst0311165. [DOI] [PubMed] [Google Scholar]; b) Morrell JA, Orme J, Butlin RJ, Roche TE, Mayers RM, Kilgour E. Biochem Soc Trans. 2003;31:11668–11670. doi: 10.1042/bst0311168. [DOI] [PubMed] [Google Scholar]; c) Mayers RM, Leighton B, Kilgour E. Biochem Soc Trans. 2005;33:367–370. doi: 10.1042/BST0330367. [DOI] [PubMed] [Google Scholar]
- 332.Aicher TD, Damon RE, Koletar J, Vinluan CC, Brand LJ, Gao J, Shetty SS, Kaplan EL, Mann WR. Bioorg Med Chem Lett. 1999;9:2223–2228. doi: 10.1016/s0960-894x(99)00380-7. [DOI] [PubMed] [Google Scholar]
- 333.Kukimoto-Niino M, Tokmakov A, Terada T, Ohbayashi N, Fujimoto T, Gomi S, Shiromizu I, Kawamoto M, Matsusue T, Shirouzu M, Yokoyama S. Acta Crystallogr D. 2011;67:763–773. doi: 10.1107/S090744491102405X. [DOI] [PubMed] [Google Scholar]
- 334.Ayyasamy V, Owens KM, Desouki MM, Liang P, Bakin A, Thangaraj K, Buchsbaum DJ, LoBuglio AF, Singh KK. PLoS One. 2011;6:e24792. doi: 10.1371/journal.pone.0024792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.a) Yen KE, Bittinger MA, Su SM, Fantin VR. Oncogene. 2010;29:6409–6417. doi: 10.1038/onc.2010.444. [DOI] [PubMed] [Google Scholar]; b) Prensner JR, Chinnaiyan AM. Nat Med. 2011;17:291–293. doi: 10.1038/nm0311-291. [DOI] [PubMed] [Google Scholar]
- 336.Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, Thompson CB. Nature. 2012;483:474–478. doi: 10.1038/nature10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.a) Riggins GJ, Seltzer MJ, Tsukamoto T, Dang CV, Emadi A. The Johns Hopkins University; WO 2011143160, 2011. Chem Abstr. 2011;155:1475730.; b) Fantin V, Su S. Agios Pharmaceuticals, Inc; WO 2011123618, 2011. Chem Abstr. 2011;155:1270655.; c) Salituro FG, Saunders JO. Agios Pharmaceuticals, Inc; WO 2011072174, 2011. Chem Abstr. 2011;155:750870.; c) Su S, Dang L, Gross S, Jin S, Fantin V. Agios Pharmaceuticals, Inc; WO 2011050210, 2011. Chem Abstr. 2011;154:530647.; d) Dang L, Fantin V, Gross S, Jang HG, Jin S, Salituro F, Saunders JO, Su S, Yen K. Agios Pharmaceuticals, Inc; WO 2010105243, 2010. Chem Abstr. 2010;153:1157605.
- 338.a) Bucay AH. Med Hypotheses. 2007;69:826–828. doi: 10.1016/j.mehy.2007.02.002. [DOI] [PubMed] [Google Scholar]; b) Bucay AH. Med Hypotheses. 2009;73:271–271. doi: 10.1016/j.mehy.2009.03.018. [DOI] [PubMed] [Google Scholar]; c) Bucay AH. Clin Res Hepatol Gastroenterol. 2011;35:241–241. doi: 10.1016/j.clinre.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 339.Lu Y, Zhang X, Zhang H, Lan J, Huang G, Varin E, Lincet H, Poulain L, Icard P. Anticancer Res. 2011;31:797–805. [PubMed] [Google Scholar]
- 340.Goldberg JS. Perspect Med Chem. 2011;5:1–10. doi: 10.4137/PMC.S6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.González A, Murcia M, Benhamú B, Campillo M, López-Rodríguez ML, Pardo L. Med Chem Commun. 2011;2:160–164. [Google Scholar]
- 342.Yaromina A, Meyer S, Fabian C, Zaleska K, Sattler UGA, Kunz-Schughart LA, Mueller-Klieser W, Zips D, Baumann M. Strahlenther Onkol. 2012 doi: 10.1007/s00066-011-0054-3. in press. [DOI] [PubMed] [Google Scholar]
- 343.Martin M. J Natl Cancer Inst. 2009;101:220–222. doi: 10.1093/jnci/djp013. [DOI] [PubMed] [Google Scholar]
- 344.Kareva I. PLoS One. 2011;6:e28576. doi: 10.1371/journal.pone.0028576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.a) Taubes G. Science. 2012;335:28–32. doi: 10.1126/science.335.6064.28. [DOI] [PubMed] [Google Scholar]; b) Pollak M. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 346.a) Ben Sahra I, Le Marchand-Brustel Y, Tanti JF, Bost F. Mol Cancer Ther. 2010;9:1092–1099. doi: 10.1158/1535-7163.MCT-09-1186. [DOI] [PubMed] [Google Scholar]; b) Algire C, Zakikhani M, Blouin MJ, Shuai JH, Pollak M. Endocr-Relat Cancer. 2008;15:833–839. doi: 10.1677/ERC-08-0038. [DOI] [PubMed] [Google Scholar]; c) Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- 347.a) Maurer GD, Brucker DP, Bähr O, Harter PN, Hattingen E, Walenta S, Mueller-Klieser W, Steinbach JP, Rieger J. BMC Cancer. 2011;11:315. doi: 10.1186/1471-2407-11-315. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ho VW, Leung K, Hsu A, Luk B, Lai J, Shen SY, Minchinton AI, Waterhouse D, Bally MB, Lin W, Nelson BH, Sly LM, Krystal G. Cancer Res. 2011;71:4484–4493. doi: 10.1158/0008-5472.CAN-10-3973. [DOI] [PubMed] [Google Scholar]; c) Lee C, Longo VD. Oncogene. 2011;30:3305–3316. doi: 10.1038/onc.2011.91. [DOI] [PubMed] [Google Scholar]
- 348.a) Wolin KY, Colditz GA. Br J Cancer. 2008;99:995–999. doi: 10.1038/sj.bjc.6604623. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hu FB, Willett WC, Li T, Stampfer MJ, Colditz GA, Manson JE. N Engl J Med. 2004;351:2694–2703. doi: 10.1056/NEJMoa042135. [DOI] [PubMed] [Google Scholar]
- 349.a) Blagosklonny MV. Int J Oncol. 2001;19:257–267. doi: 10.3892/ijo.19.2.257. [DOI] [PubMed] [Google Scholar]; b) Rapisarda A, Melillo G. Drug Resist Update. 2009;12:74–80. doi: 10.1016/j.drup.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Keunen O, Johansson M, Oudin A, Sanzey M, Abdul Rahim SA, Fack F, Thorsen F, Taxt T, Bartos M, Jirik R, Miletic H, Wang J, Stieber D, Stuhr L, Moen I, Brekke Rygh C, Bjerkvig R, Niclou SP. Proc Natl Acad Sci USA. 2011;108:3749–3754. doi: 10.1073/pnas.1014480108. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS. Proc Natl Acad Sci USA. 2012;109:2784–2789. doi: 10.1073/pnas.1018866109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.a) Ishii H, Doki Y, Mori M. Yonsei Med J. 2010;51:617–621. doi: 10.3349/ymj.2010.51.5.617. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Prigione A, Fauler B, Lurz R, Lehrach H, Adjaye J. Stem Cells. 2010;28:721–733. doi: 10.1002/stem.404. [DOI] [PubMed] [Google Scholar]; c) Shyh-Chang N, Zheng Y, Locasale JW, Cantley LC. EMBO J. 2011;30:4851–4852. doi: 10.1038/emboj.2011.436. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Scatena R. Adv Exp Med Biol. 2012;942:287–308. doi: 10.1007/978-94-007-2869-1_13. [DOI] [PubMed] [Google Scholar]