

Abstract
One of the fundamental paradigms in the use of nanoparticles to treat disease is to evade or suppress the immune system in order to minimize systemic side effects and deliver sufficient nanoparticle quantities to the intended tissues. However, the immune system is the body's most important and effective defense against diseases. It protects the host by identifying and eliminating foreign pathogens as well as selfmalignancies. Here we report a nanoparticle engineered to work with the immune system, enhancing the intended activation of antigen presenting cells (APCs). We show that luminescent porous silicon nanoparticles (LPSiNPs), each containing multiple copies of an agonistic antibody (FGK45) to the APC receptor CD40, greatly enhance activation of B cells. The cellular response to the nanoparticle-based stimulators is equivalent to a 30–40 fold larger concentration of free FGK45. The intrinsic near-infrared photoluminescence of LPSiNPs is used to monitor degradation and track the nanoparticles inside APCs.
Keywords: nanoparticles, immunotherapy, CD40, vaccine, porous silicon
Nanomaterials of porous silicon have attracted intense interest for imaging and treatment of diseases including cancer due to their biocompatibility, large specific capacity for drug loading, non-toxic degradation products, and efficient photoluminescence.[1–9] One of the main barriers to any nanoparticle intended for in vivo use is the surveillance by the immune system.[10–12] For example, the mononuclear phagocyte system (MPS) recognizes and intercepts substantial quantities of systemically administered nanoparticles before they can reach the diseased tissues,[11–13] and this can lead to significant damage to major organs such as liver, spleen, etc., especially when the nanomaterials carry lethal anticancer drugs. In contrast, approaches to intentionally activate the body's own immune system to fight against diseases can be quite effective.[14–20] The goal of active immunotherapy is to elicit or amplify an immune response to harness the body's inherent defenses against foreign pathogens and self-malignancy. The experimental use of nanomaterials for such active immunotherapies has not been explored to a great extent in part due to the limited understanding of the interactions between the immune system and nanomaterials.[21] Nevertheless, recent studies have shown that some nanoparticle-based vaccines can be much more potent than soluble peptide or protein antigens,[22–30] and it has been proposed that nanovaccines are more adaptable and perhaps safer than viral vaccines.[31–33]
Most studies using nanomaterials in immunotherapies focus on antigen delivery, with little emphasis on the ability of nanomaterials to alter the potency of immunomodulators. In addition, the majority of nanovaccine systems are based on lipids or polymers such as poly(lactic-co-glycolic acid) (PLGA), or polystyrene,[23, 24, 34–39] many of which display some intrinsic immune stimulation that may limit their use for immunotherapies (due to unintended stimulation of APCs) or that may interfere with the function of loaded immunomodulators. For many immunotherapeutic or immunomodulation applications, a desirable criterion is that the nanomaterial itself shows low intrinsic immune stimulation.
CD40 is a co-stimulatory receptor as well as a member of the family of tumor necrosis factor (TNF) receptors found on APCs such as dendritic cells, B cells, and macrophages.[40–42] Agonistic monoclonal antibodies to CD40 (CD40 mAb) can activate APCs and improve immune responses when used in combination with antigens or vaccines.[18, 43, 44] In addition, CD40 mAb can produce substantial antitumor efficacy and can also potentially be used to treat chronic autoimmune inflammation.[45–48] However, the therapeutically effective dose of CD40 mAb is high and the high dose can result in severe side effects.[47, 49] We have developed nanoparticles based on luminescent porous silicon that display low systemic toxicity and degrade in vivo into renally cleared components.[1, 50, 51] The porous nanostructure and intrinsic near-infrared photoluminescence of porous silicon nanoparticles (LPSiNPs) enable the incorporation of drug payloads and the monitoring of distribution and degradation in vivo.[1] In this study, we show that when multiple copies of agonistic CD40 mAb (clone FGK45) are incorporated onto a LPSiNP, the activation potency on B cells is significantly amplified, equivalent to using ~ 30–40 fold larger concentration of free FGK45. LPSiNPs without FGK45 appear inert to B cells.
LPSiNPs were prepared by electrochemical etch of highly doped p-type single-crystal Si wafers in an electrolyte consisting of aqueous hydrofluoric acid and ethanol, lift-off of the porous layer, ultrasonic fracture, filtration of the resulting nanoparticles through a 0.22 μm filter membrane, and finally activation of luminescence by treatment in an aqueous solution following the published procedure.[1, 51] To incorporate FGK45 onto the nanoparticles, we first coated the LPSiNPs with avidin by physisorption (av-LPSiNPs). Biotinylated FGK45 was then conjugated to the nanoparticles through the strong biotinavidin binding interaction (FGK-LPSiNPs), Figure 1a. Approximately 0.058 mg of FGK45 was loaded per milligram of LPSiNPs, as measured by bicinchoninic acid (BCA) protein assay. The presence of FGK45 loaded on nanoparticle-FGK45 construct was also confirmed by gel electrophoresis and immunoblotting (Figure S1, Supporting Information). The FGK-LPSiNPs appeared similar to LPSiNPs in the transmission electron microscope (TEM) images (Figure 1b), but the mean hydrodynamic size measured by dynamic light scattering (DLS) increased from ~ 130 ± 10 nm to ~ 188 ± 15 nm after protein attachment (Figure S2, Supporting Information).
Figure 1. Preparation and characterization of FGK45 loaded luminescent porous silicon nanoparticles (FGK-LPSiNP).
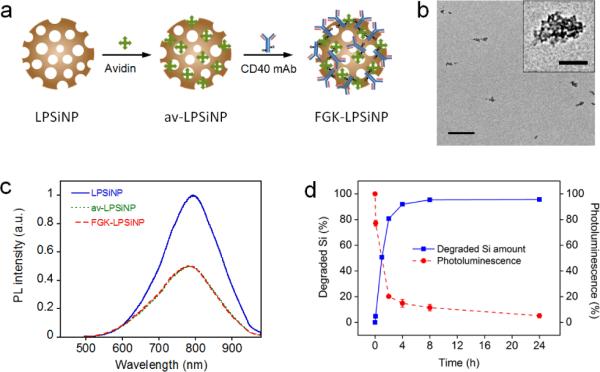
a) Schematic representation of the preparation of FGK-LPSiNP. LPSiNP was first coated with avidin by physisorption (av-LPSiNP). Biotinylated FGK45 was then conjugated to the nanoparticles through biotin-avidin binding to form FGK-LPSiNP. b) Transmission electron microscope image of FGKLPSiNP (inset shows the porous nanostructure of one of the nanoparticles). Scale bar is 1 μm (100 nm for the inset). c) Photoluminescence (PL) spectra of LPSiNP, av-LPSiNP and FGK-LPSiNP. PL was measured using UV excitation (λex= 370 nm). d) Appearance of dissolved silicon in solution (by ICP-OES) and decrease in photoluminescence intensity from a sample of FGK-LPSiNP (50 μg mL−1) incubated in PBS solution at 37°C as a function of time.
The intrinsic photoluminescence from the silicon nanostructures in FGK-LPSiNPs under ultraviolet excitation appeared in the near-infrared region of the spectrum (λmax = 790 nm), similar to the non-loaded LPSiNPs. However, the intensity of photoluminescence was somewhat lower from the protein-coated formulation (Figure 1c).[1] In a physiologically relevant aqueous solution of phosphate buffered saline (PBS) at pH 7.4 and 37 °C, the FGK-LPSiNP construct was observed to degrade by 95% within 24 h (Figure 1d). The degradation was tracked by monitoring disappearance of the photoluminescence signal, which decreased gradually upon dissolution of the quantum confined silicon nanostructure,[7, 52] and by appearance of free silicic acid in solution (by inductively coupled plasma-optical emission spectroscopy, ICP-OES), Figure 1d.
The FGK-LPSiNPs were more readily taken up by APCs compared to bare LPSiNPs. When cultured with mouse bone marrow-derived dendritic cells (BMDC), LPSiNPs showed limited (but still detectable) presence in the cells (Figure 2a); in contrast, BMDC incubated with FGK-LPSiNPs under the same conditions showed much higher uptake of nanoparticles (Figure 2b). This uptake was substantially blocked by pre-treatment of the BMDC with free FGK45, indicating that FGK45 binding is responsible for the increased internalization of FGK-LPSiNP (Figure 2c). It has been shown that the CD40 ligand and agonistic antibodies can induce endocytosis of CD40 upon binding,[53, 54] which may be responsible for cellular uptake of FGK-LPSiNP in the present case. However, because agonistic CD40 antibodies can also engage FcγRIIB on APCs,[55] interaction of cellular receptors for the Fc domain of the FGK45 antibody could also contribute to the uptake. We tested for inhibition of this interaction by pre-incubation of BMDC with anti-mouse FcγRII-III. The presence of FcγRII-III somewhat reduced, but did not eliminate, cellular uptake of FGK-LPSiNP (Figure S3, Supporting Information), which suggests that this is a relevant (but not exclusive) internalization element in the present system.
Figure 2. Dendritic cell uptake of FGK-LPSiNPs.
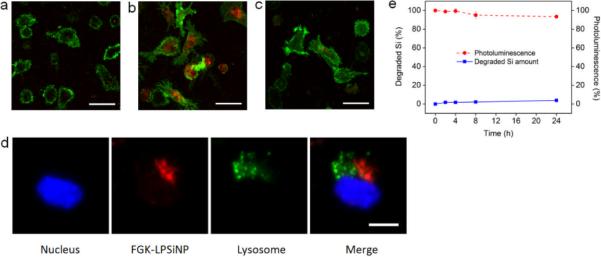
Fluorescence microscope images of mouse bone marrow-derived dendritic cells (BMDC) incubated with a) LPSiNPs or b) FGK-LPSiNPs for 1.5 h at 37 °C. c) free FGK45 inhibits uptake of FGK-LPSiNPs. BMDC were blocked with free FGK45 for 30 min and then incubated with FGK-LPSiNPs for 1.5 h at 37 °C. BMDC were detected by staining with Alexa Fluor 488 conjugated CD11c antibody (green). FGK-LPSiNPs were detected by their intrinsic visible/near-infrared photoluminescence (red, λex= 405 nm and λem= 700 ± 50 nm). The scale bars are 40 μm. d) FGK-LPSiNPs distribution in BMDC. BMDC were incubated with FGK-LPSiNP for 1.5 h at 37 °C. The lysosomes (green) of the cells were stained with LysoTracker (Invitrogen). Blue and red indicate the cell nucleus and FGK-LPSiNPs, respectively. The scale bar is 10 μm. e) Degradation of LPSiNPs (50 μg mL−1) in pH 4 buffer solution at 37 °C as a function of time.
Subcellular localization of FGK-LPSiNPs was further examined by confocal fluorescence microscopy. By following the near-infrared photoluminescence spectrum of the nanoparticles, we observed FGK-LPSiNPs outside of the lysosomes of the dendritic cells (Figure 2d and Supporting Information, Video S1). The appearance of FGK-LPSiNPs outside of the lysosomes of the dendritic cells is consistent with previous reports that various types of silicon or silica based nanomaterials can escape from lysosomes and distribute inside the cytosol.[1, 56–58] In separate experiments aimed at simulating the low pH conditions inside the lysosome, only < 5% dissolution of LPSiNPs was observed in pH 4 buffer solution over a 24h period (Figure 2e and Supporting Information, Figure S4). Although LPSiNPs are expected to degrade within a few hours at pH 7.4 due to dissolution of the protective oxide coating,[1, 59] they are much more stable in acidic media such as the environment present in the interior of lysosomes. The observed ability of LPSiNPs to escape from lysosomes opens the possibility for delivery of antigens to the cytosol and inducing the MHC class I antigen presentation pathway.[18, 19]
We next investigated the activity of FGK-LPSiNPs added to B cells, by measuring the expression of cell surface molecules indicative of B cell activation. The nanoparticles in this experiment contained avidin labeled with fluorescein isothiocyanate (av-FITC). The resulting construct emits both in the green (from the FITC label) and in the near-infrared (from the silicon nanostructure) when excited with ultraviolet light (Figure 3a). B cells enriched from mouse splenocytes were incubated with FGK-LPSiNPs or av-LPSiNPs and then analyzed by flow cytometry. After 42 h of culture, the FITC signal was only detected from B cells that had been exposed to FGK-LPSiNPs (Figure 3b–e). B cells incubated with FGK-LPSiNPs also displayed increased expression of the cell surface receptors, CD86 and major histocompatibility complex class II (MHC II), the response typical of B cells that have received a signal through CD40 (Figure 3b–e).[45] Furthermore, the extent of activation induced by FGK-LPSiNPs was concentration dependent (Figure S5, Supporting Information). When exposed to a low concentration of FGK-LPSiNPs, not all of the B cells were activated, as indicated by the wide distribution of the fluorescence intensity from the cells in flow cytometry dot plots (Figure 3b, 3d). However, the population of B cells that displayed high FITC signals also expressed high levels of CD86 and MHC II, indicating that the cells bound with nanoparticles were the ones that were activated (Figure 3b, 3d). In contrast, B cells cultured with various concentrations of av-LPSiNP all showed low FITC signals and low activation marker levels (Figure S5, Supporting Information).
Figure 3. Interaction of FGK-LPSiNPs with B cells.
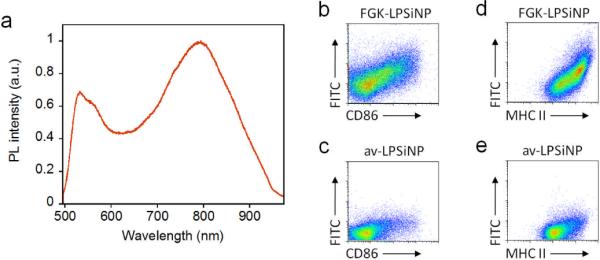
a) Photoluminescence spectrum of LPSiNPs coated with FITC-labeled avidin, showing the emission bands from both the FITC label (λmax ~ 520 nm) and porous silicon (λmax ~790 nm). b–e) Flow cytometry data quantifying the level of expression of the B cell activation markers CD86 (b, c) and MHC II (d, e) after incubation with 5 μg mL−1 of FGK-LPSiNPs (b, d) or av-LPSiNPs (c, e) for 42 h. The nanoparticles used in this experiment were coated with FITC-labeled avidin. The FITC signal from the cells is plotted against the expression level of CD86 (b, c) or MHC II (d, e) after stimulation. FGK-LPSiNPs used here contain 36 μg of FGK45 per milligram of nanoparticles. Note the quantity of FGK45 loaded is smaller when LPSiNPs are coated with FITC conjugated avidin compared with non-labeled native avidin.
Multivalency is one of the notable advantages of using nanomaterials for biomedical applications. For example, studies using nanoparticles as cancer diagnostic and therapeutic agents have shown that when multiple copies of tumor targeting ligand are displayed on an individual nanoparticle, its tumor targeting efficiency can be significantly enhanced.[60–63] This enhancement is generally ascribed to the multivalent effect which is also observed in many natural processes such as antibody interactions and clotting.[11] To determine if multivalency plays a strong role in the activation potency of the agonistic antibody to APCs, we cultured B cells with either FGK-LPSiNPs or an equivalent concentration of free FGK45 and analyzed the cells by flow cytometry. Both FGKLPSiNPs and free FGK45 activated B cells, and the activation level of the cells correlated with the concentration of FGK45 (Figure 4a). However, at a given total concentration of FGK45 antibody, FGK-LPSiNPs showed substantially higher activation potency than free FGK45. Activated B cells upregulated CD86 and MHC II to a detectable level when cultured with FGK-LPSiNPs containing as little as ~ 3.6 – 7.2 ng mL−1 of FGK45; whereas a similar level of B cell activation was only observed when the concentration of free FGK45 was ≥ 140 – 200 ng mL−1 (Figure 4a). Comparison of the titration curves of FGK-LPSiNPs and free FGK45 revealed that the B cell activation potency of FGK45 in the FGK-LPSiNP constructs is equivalent to using ~ 30–40 fold larger concentration of free FGK45 (Figure 4a).
Figure 4. Amplified activation potency of FGK-LPSiNPs compared to free FGK45.
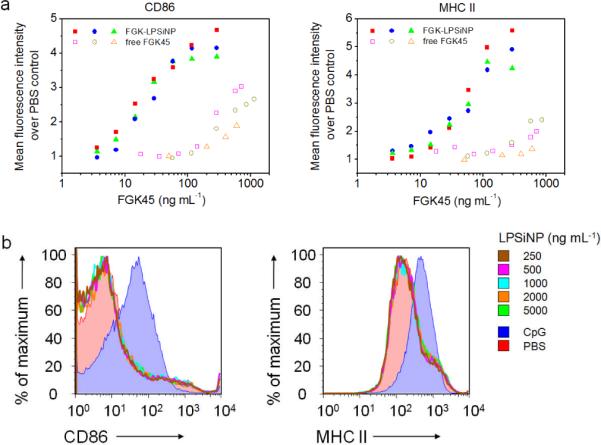
a) Flow cytometry analysis of the expression of B cell activation markers CD86 and MHC II, represented as the relative mean fluorescence intensity of the marker staining, after incubation with either FGK-LPSiNPs or free FGK45 for 42 h at 37 °C. The concentration of FGK45 on the FGK-LPSiNP constructs is reported based on the total loading of FGK45 on the nanoparticles (58 μg of FGK45 per mg of nanoparticles). Data are from independent experiments. b) Flow cytometry histograms of B cell activation markers CD86 and MHC II after incubation with various concentrations of LPSiNPs for 42 h at 37 °C. PBS (red shaded) and CpG (blue shaded) were used as negative and positive controls, respectively.
To test if the enhancement of APC activation is caused by the uncoated porous silicon nanomaterial itself, we cultured B cells with various concentrations of LPSiNPs as control experiments. No induction of CD86 or MHC II was observed at LPSiNP concentrations up to 5000 ng mL−1, equivalent to the highest concentration of FGK-LPSiNPs used in the stimulation study (Figure 4b and Supporting Information, Figure S6). This suggests that the amplification induced by the FGK-LPSiNP construct results from enhancement of the agonistic antibody's intrinsic function rather than an immune response to the nanomaterial itself. The very low stimulation of APCs by LPSiNPs that do not contain FGK45 is attributed to their primarily inorganic chemical composition; the chemical structure of the silicon-based nanoparticles (and their biodegradation products) possess little similarity to natural pathogens or other “danger signals” normally presented to the immune system.[8, 21, 64]
The present study represents the first example of amplification of the potency of an agonistic CD40 mAb to stimulate APCs using nanomaterials. Agonistic CD40 mAb has attracted considerable interest[48, 49, 55] as it has recently entered clinical trials as a therapeutic for solid tumors.[46] The amplification effect seen in the present study suggests that a nanoparticle-based CD40 antibody therapeutic may amplify the potency and decrease the dose required in the clinical application of CD40 mAb. The nanoparticle formulation may also provide a means of targeting specific types of immune cells by attaching additional targeting ligands on the surface of FGK-LPSiNPs. In addition to the enhancement effect, the inert inorganic composition and biodegradable property of LPSiNPs could overcome some of the disadvantages of lipid or polymer-based materials for immunotherapy applications. Their intrinsic photoluminescence can also be used to monitor the degradation of LPSiNPs and track their interaction with the immune system.
Experimental Section
Preparation of FGK45 loaded luminescent porous silicon nanoparticles (FGK-LPSiNPs)
LPSiNPs were first prepared using a previously described method.[1, 66] In brief, (100)-oriented p-type single-crystal Si wafers (0.8–1.2 mΩ cm, Siltronix) were electrochemically etched in an electrolyte containing aqueous 48% hydrofluoric acid and ethanol in a 3:1 ratio. The resulting porous Si films were lifted from the Si substrate, fractured by ultrasound and filtered through a 0.22 um membrane (Millipore, Inc). Finally, the photoluminescence of the nanoparticles were activated by soaking in deionized water for 14 d. To prepare FGK-LPSiNP, an avidin coating was first applied. An aliquot (1 mL) of an aqueous dispersion of LPSiNP (0.2 mg) was mixed with an aliquot (0.08 mL) of water containing 0.04 mg of avidin (Thermo Fisher Scientific, Inc.). The mixture was stirred for 1 h at room temperature, rinsed with water three times by centrifugation. The particles were resuspended in water to 0.2 mg mL−1 and were then mixed with an aliquot (0.045 mL) of water containing 0.022 mg of biotin-conjugated FGK45 (Enzo Life Sciences, Inc.; FGK45 is a monoclonal antibody to mouse CD40). The mixture was stirred for 1 h at room temperature, rinsed with water three times by centrifugation to remove any excess FGK45. The supernatant of each wash was combined and the quantity of excess FGK45 in the supernatant was measured by micro BCA (bicinchoninic acid) protein assay (Thermo Fisher Scientific, Inc.) to calculate the quantity of FGK45 loaded on LPSiNP.
Nanoparticle characterization
Transmission electron micrographs (TEM) were obtained with a FEI Tecnai G2 Sphera. Dynamic light scattering (Zetasizer Nano ZS90, Malvern Instruments) was used to determine the hydrodynamic size of the nanoparticles. The photoluminescence (PL, λex = 370 nm and 460 nm long pass emission filter) spectra of LPSiNP or FGK-LPSiNP were obtained using a Princeton Instruments/Acton spectrometer fitted with a liquid nitrogen-cooled silicon charge-coupled device (CCD) detector. Quantification of FGK45 loaded on the nanoparticles was determined by bicinchoninic acid protein assay (Micro BCA, Thermo Fisher Scientific, Inc.) using a subtraction method. In a typical experiment, av-LPSiNPs (0.2 mg mL–1) in 1 mL water were mixed with an aliquot (0.045 mL) of water containing 0.022 mg of biotin-conjugated FGK45. The mixture was stirred for 1 h at room temperature and rinsed with water three times by centrifugation. The supernatant of each rinse was collected and the excess FGK45 in the supernatant was quantified by Micro BCA assay following the manufacturer's instructions. The loading of FGK45 on the nanoparticles was calculated by subtracting the excess of FGK45 from the total quantity used for the loading experiment.
In vitro degradation of FGK-LPSiNP
A set of microtubes (typically 24 individual microtubes for a given experiment), each containing FGK-LPSiNP (0.05 mg mL−1) in 1 mL of PBS solution or pH 4.0 buffer solution were incubated at 37 °C. Three microtubes (triplicate measurements) were assayed for each time point. An aliquot (0.5 mL) of solution was removed from each microtube and filtered with a centrifugal filter (30,000 Da molecular weight cut-off, Millipore, Inc.) to remove undissolved LPSiNP. 0.4 mL of the filtered solution was then diluted with 5 mL HNO3 (2 %(v/v)) and subjected to analysis by inductively coupled plasma optical emission spectroscopy (ICP-OES, Perkin Elmer Optima 3000DV). In order to quantify percent degradation, we assumed the nanoparticles fully dissolved in 72 hours at 37 °C in PBS buffer (pH = 7.4), and used this timepoint to determine the 100% degradation level in the ICP-OES experiments. The decrease in PL of the above samples over time was also monitored, following a previously described method.[1]
Mice
C57BL/6 mice were maintained in specific pathogen-free facilities at the University of California, San Diego. Animal protocols were approved by the Institutional Animal Care and Use Committee.
Cell uptake of FGK-LPSiNP
Mouse bone marrow-derived dendritic cells (BMDC) were prepared as described [67] and harvested on day 8 for use in microscopy experiments. BMDC (40,000–60,000 cells per well) were seeded into 8-well chamber glass slides (Millipore, Inc.) and cultured overnight. The cells were washed with RPMI (Roswell Park Memorial Institute) medium once and incubated with 0.05 mg mL−1 LPSiNP or FGK-LPSiNP in RPMI medium for 1.5 hours at 37 °C. For the competitive binding experiment, BMDC were first incubated with free FGK45 (0.03 mg ml−1) for 30 min in RPMI medium, then incubated with FGK-LPSiNP (0.05 mg mL−1) as above. The cells were washed 3 times with RPMI medium and incubated with Alexa Fluor 488 conjugated CD11c antibody (clone N418, eBioscience—all antibodies are from eBioscience unless otherwise indicated; 1μg ml−1) in RPMI medium for 10 min to visualize the BMDC. The cells were then rinsed three times with PBS, fixed with 4% paraformaldehyde for 20 min and then observed with a Zeiss LSM 510 confocal fluorescence microscope. An excitation wavelength of 405 nm and an emission filter with a bandpass at 700 ± 50 nm were used to image the near-IR photoluminescence of the nanoparticles.
In vitro stimulation of B cells
Single-cell suspensions of C57BL/6 splenocytes were prepared and subjected to red blood cell lysis using ACK lysis buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM EDTA, pH 7.3). B cells were sorted out via CD43 (Miltenyi) magnetic bead depletion. Sorted cells were plated at 2×105 cells/well and incubated with LPSiNP, av-LPSiNP, FGK-LPSiNP, free agonistic anti-CD40 (clone FGK45), PBS, or CpG (Pfizer) for 42 h at 37 °C.
Flow cytometry
Approximately 1–2 million cells were resuspended in Hank's Balanced Salt Solution (Invitrogen) with 1% fetal calf serum (Omega Scientific) added (HBSS 1% FCS), incubated for 15 min at 4 °C with anti-mouse FcγRII-III (supernatant from hybridoma 2.4G2 cultures), and stained with fluorescently conjugated antibodies for 20 min at 4°C. For particles using avidin-FITC, cells were stained with MHC II biotin (M5/114.15.2), washed with HBSS 1% FCS, stained with streptavidin PerCP, CD86 Phycoerythrin (GL1; PE) and B220 Allophycocyanin (RA3-6B2) and analyzed by flow cytometry. For particles using non-labeled avidin, cells were stained with B220 FITC, CD86 PE and MHC II Allophycocyanin and analyzed by flow cytometry.
Supplementary Material
Acknowledgements
This work was supported by the National Cancer Institute of the National Institutes of Health through grant numbers 5-R01-CA124427 and R24EY022025-01, and by the National Science Foundation under Grant No. DMR-0806859. L.G. was partially supported by the UCSD HHMI/NIBIB Interfaces Graduate Training Program (HHMI grant HHMI56005681). L.E.R. was supported by University of California, San Diego Immunology training grant NIH T32AI060536-02. The authors thank the National Center for Microscopy and Imaging Research (NCRR Grant 5P41RR004050) for use of their facilities.
References
- [1].Park J-H, Gu L, Maltzahn G. v., Ruoslahti E, Bhatia SN, Sailor MJ. Nature Mater. 2009;8:331. doi: 10.1038/nmat2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tasciotti E, Liu XW, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MMC, Decuzzi P, Tour JM, Robertson F, Ferrari M. Nat. Nanotechnol. 2008;3:151. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- [3].Salonen J, Laitinen L, Kaukonen AM, Tuura J, Bjorkqvist M, Heikkila T, Vaha-Heikkila K, Hirvonen J, Lehto VP. J. Control. Release. 2005;108:362. doi: 10.1016/j.jconrel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- [4].Low SP, Voelcker NH, Canham LT, Willams KA. Biomaterials. 2009;30:2873. doi: 10.1016/j.biomaterials.2009.02.008. [DOI] [PubMed] [Google Scholar]
- [5].Xiao L, Gu L, Howell SB, Sailor MJ. ACS Nano. 2011;5:3651. doi: 10.1021/nn1035262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wolkin MV, Jorne J, Fauchet PM, Allan G, Delerue C. Phys. Rev. Lett. 1999;82:197. [Google Scholar]
- [7].Canham LT. Appl. Phys. Lett. 1990;57:1046. [Google Scholar]
- [8].Anderson SHC, Elliott H, Wallis DJ, Canham LT, Powell JJ. Phys. Status Solidi A-Appl. Res. 2003;197:331. [Google Scholar]
- [9].Erogbogbo F, Yong KT, Roy I, Xu GX, Prasad PN, Swihart MT. Acs Nano. 2008;2:873. doi: 10.1021/nn700319z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Peer D, Karp JM, Hong SP, FaroKhzad OC, Margalit R, Langer R. Nature Nanotech. 2007;2:751. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- [11].Ruoslahti E, Bhatia SN, Sailor MJ. J. Cell Biol. 2010;188:759. doi: 10.1083/jcb.200910104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moghimi SM, Hunter AC, Murray JC. Pharmacological Reviews. 2001;53:283. [PubMed] [Google Scholar]
- [13].Brigger I, Dubernet C, Couvreur P. Adv. Drug Deliv. Rev. 2002;54:631. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- [14].Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, Xu Y, Frohlich MW, Schellhammer PF, Ahmed T, Amin A, Arseneau J, Barth N, Bernstein G, Bracken B, Burch P, Caggiano V, Chin J, Chodak G, Chu F, Corman J, Curti B, Dawson N, Deeken JF, Dubernet T, Fishman M, Flanigan R, Gailani F, Garbo L, Gardner T, Gelmann E, George D, Godfrey T, Gomella L, Guerra M, Hall S, Hanson J, Israeli R, Jancis E, Jewett MAS, Kassabian V, Katz J, Klotz L, Koeneman K, Koh H, Kratzke R, Lance R, Lech J, Leichman L, Lemon R, Liang J, Libertino J, Lilly M, Malik I, Martin SE, McCaffrey J, McLeod D, McNeel D, Miles B, Murdock M, Nabhan C, Nemunaitis J, Notter D, Pantuck A, Perrotte P, Pessis D, Petrylak D, Polikoff J, Pommerville P, Ramanathan S, Rarick M, Richards J, Rifkin R, Rohatgi N, Rosenbluth R, Santucci R, Sayegh A, Seigne J, Shapira I, Shedhadeh N, Shepherd D, Sridhar S, Stephenson R, Teigland C, Thaker N, Vacirca J, Villa L, Jr., Vogelzang N, Wertheim M, Wolff JH, Wurzel R, Yang C, Young JR. N. Engl. J. Med. 2010;363:411. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- [15].Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJM, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbe C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. N. Engl. J. Med. 2010;363:711. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Porter DL, Levine BL, Kalos M, Bagg A, June CH. N. Engl. J. Med. 2011;365:725. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, Jungbluth A, Gnjatic S, Thompson JA, Yee C. N. Engl. J. Med. 2008;358:2698. doi: 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pardoll DM. Nature Rev. Immunol. 2002;2:227. doi: 10.1038/nri774. [DOI] [PubMed] [Google Scholar]
- [19].Waldmann TA. Nature Med. 2003;9:269. doi: 10.1038/nm0303-269. [DOI] [PubMed] [Google Scholar]
- [20].Rosenberg SA, Yang JC, Restifo NP. Nature Med. 2004;10:909. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Dobrovolskaia MA, McNeil SE. Nature Nanotech. 2007;2:469. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- [22].Shen H, Ackerman AL, Cody V, Giodini A, Hinson ER, Cresswell P, Edelson RL, Saltzman WM, Hanlon DJ. Immunology. 2006;117:78. doi: 10.1111/j.1365-2567.2005.02268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Slutter B, Soema PC, Ding Z, Verheul R, Hennink W, Jiskoot W. J. Control Release. 2010;143:207. doi: 10.1016/j.jconrel.2010.01.007. [DOI] [PubMed] [Google Scholar]
- [24].Uto T, Wang X, Sato K, Haraguchi M, Akagi T, Akashi M, Baba M. J. Immunol. 2007;178:2979. doi: 10.4049/jimmunol.178.5.2979. [DOI] [PubMed] [Google Scholar]
- [25].Copland MJ, Rades T, Davies NM, Baird MA. Immunol. Cell Biol. 2005;83:97. doi: 10.1111/j.1440-1711.2005.01315.x. [DOI] [PubMed] [Google Scholar]
- [26].Elamanchili P, Diwan M, Cao M, Samuel J. Vaccine. 2004;22:2406. doi: 10.1016/j.vaccine.2003.12.032. [DOI] [PubMed] [Google Scholar]
- [27].Cho N-H, Cheong T-C, Min JH, Wu JH, Lee SJ, Kim D, Yang J-S, Kim S, Kim YK, Seong S-Y. Nature Nanotech. 2011;6:675. doi: 10.1038/nnano.2011.149. [DOI] [PubMed] [Google Scholar]
- [28].Li H, Li Y, Jiao J, Hu H-M. Nature Nanotech. 2011;6:645. doi: 10.1038/nnano.2011.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Makidon PE, Bielinska AU, Nigavekar SS, Janczak KW, Knowlton J, Scott AJ, Mank N, Cao ZY, Rathinavelu S, Beer MR, Wilkinson JE, Blanco LP, Landers JJ, Baker JR. PLoS One. 2008:3. doi: 10.1371/journal.pone.0002954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Makidon PE, Knowlton J, Groom JV, Blanco LP, LiPuma JJ, Bielinska AU, Baker JR. Med. Microbiol. Immunol. 2010;199:81. doi: 10.1007/s00430-009-0137-2. [DOI] [PubMed] [Google Scholar]
- [31].Peek LJ, Middaugh CR, Berkland C. Adv. Drug Deliv. Rev. 2008;60:915. doi: 10.1016/j.addr.2007.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Demento SL, Eisenbarth SC, Foellmer HG, Platt C, Caplan MJ, Saltzman WM, Mellman I, Ledizet M, Fikrig E, Flavell RA, Fahmy TM. Vaccine. 2009;27:3013. doi: 10.1016/j.vaccine.2009.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Moon JJ, Suh H, Bershteyn A, Stephan MT, Liu H, Huang B, Sohail M, Luo S, Um SH, Khant H, Goodwin JT, Ramos J, Chiu W, Irvine DJ. Nature Mater. 2011;10:243. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Diwan M, Elamanchili P, Lane H, Gainer A, Samuel J. J. Drug Targeting. 2003;11:495. doi: 10.1080/10611860410001670026. [DOI] [PubMed] [Google Scholar]
- [35].Petersen LK, Xue L, Wannemuehler MJ, Rajan K, Narasimhan B. Biomaterials. 2009;30:5131. doi: 10.1016/j.biomaterials.2009.05.069. [DOI] [PubMed] [Google Scholar]
- [36].Li A, Qin L, Zhu D, Zhu R, Sun J, Wang S. Biomaterials. 2010;31:748. doi: 10.1016/j.biomaterials.2009.09.095. [DOI] [PubMed] [Google Scholar]
- [37].Reddy ST, van der Vlies AJ, Simeoni E, Angeli V, Randolph GJ, O'Neill CP, Lee LK, Swartz MA, Hubbell JA. Nature Biotech. 2007;25:1159. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- [38].Klippstein R, Pozo D. Nanomed.-Nanotechnol. Biol. Med. 2010;6:523. doi: 10.1016/j.nano.2010.01.001. [DOI] [PubMed] [Google Scholar]
- [39].Cui Z, Han S-J, Vangasseri DP, Huang L. Molecular Pharmaceutics. 2005;2:22. doi: 10.1021/mp049907k. [DOI] [PubMed] [Google Scholar]
- [40].Banchereau J, Bazan F, Blanchard D, Briere F, Galizzi JP, Vankooten C, Liu YJ, Rousset F, Saeland S. Annu. Rev. Immunol. 1994;12:881. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- [41].Schoenberger SP, Toes REM, van der Voort EIH, Offringa R, Melief CJM. Nature. 1998;393:480. doi: 10.1038/31002. [DOI] [PubMed] [Google Scholar]
- [42].Bennett SRM, Carbone FR, Karamalis F, Flavell RA, Miller J, Heath WR. Nature. 1998;393:478. doi: 10.1038/30996. [DOI] [PubMed] [Google Scholar]
- [43].Dougan M, Dranoff G. Annu. Rev. Immunol. 2009;27:83. doi: 10.1146/annurev.immunol.021908.132544. [DOI] [PubMed] [Google Scholar]
- [44].Palucka K, Banchereau J, Mellman I. Immunity. 2010;33:464. doi: 10.1016/j.immuni.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O'Dwyer PJ, Vonderheide RH. Science. 2011;331:1612. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Vonderheide RH, Flaherty KT, Khalil M, Stumacher MS, Bajor DL, Hutnick NA, Sullivan P, Mahany JJ, Gallagher M, Kramer A, Green SJ, O'Dwyer PJ, Running KL, Huhn RD, Antonia SJ. J. Clin. Oncol. 2007;25:876. doi: 10.1200/JCO.2006.08.3311. [DOI] [PubMed] [Google Scholar]
- [47].Jackaman C, Cornwall S, Graham PT, Nelson DJ. Immunol. Cell Biol. 2011;89:255. doi: 10.1038/icb.2010.88. [DOI] [PubMed] [Google Scholar]
- [48].Mauri C, Mars LT, Londei M. Nature Med. 2000;6:673. doi: 10.1038/76251. [DOI] [PubMed] [Google Scholar]
- [49].Barr TA, McCormick AL, Carlring J, Heath AW. Immunology. 2003;109:87. doi: 10.1046/j.1365-2567.2003.01634.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Heinrich JL, Curtis CL, Credo GM, Kavanagh KL, Sailor MJ. Science. 1992;255:66. doi: 10.1126/science.255.5040.66. [DOI] [PubMed] [Google Scholar]
- [51].Gu L, Park J-H, Duong KH, Ruoslahti E, Sailor MJ. Small. 2010;6:2546. doi: 10.1002/smll.201000841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lehmann V, Gosele U. Appl. Phys. Lett. 1991;58:856. [Google Scholar]
- [53].Chen J, Chen L, Wang G, Tang H. Arterioscler. Thromb. Vasc. Biol. 2007;27:2005. doi: 10.1161/ATVBAHA.107.145961. [DOI] [PubMed] [Google Scholar]
- [54].Chen Y, Chen J, Xiong Y, Da Q, Xu Y, Jiang X, Tang H. Biochem. Biophys. Res. Comm. 2006;345:106. doi: 10.1016/j.bbrc.2006.04.034. [DOI] [PubMed] [Google Scholar]
- [55].Li F, Ravetch JV. Science. 2011;333:1030. doi: 10.1126/science.1206954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Slowing I, Trewyn BG, Lin VSY. J. Am. Chem. Soc. 2006;128:14792. doi: 10.1021/ja0645943. [DOI] [PubMed] [Google Scholar]
- [57].Serda RE, Go J, Bhavane RC, Liu X, Chiappini C, Decuzzi P, Ferrari M. Biomaterials. 2009;30:2440. doi: 10.1016/j.biomaterials.2009.01.019. [DOI] [PubMed] [Google Scholar]
- [58].Ashley CE, Carnes EC, Phillips GK, Padilla D, Durfee PN, Brown PA, Hanna TN, Liu J, Phillips B, Carter MB, Carroll NJ, Jiang X, Dunphy DR, Willman CL, Petsev DN, Evans DG, Parikh AN, Chackerian B, Wharton W, Peabody DS, Brinker CJ. Nature Mater. 2011;10:389. doi: 10.1038/nmat2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wu EC, Park J-H, Park J, Segal E, Cunin F, Sailor MJ. ACS Nano. 2008;2:2401. doi: 10.1021/nn800592q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hong S, Leroueil PR, Majoros IJ, Orr BG, Baker JR, Jr., Banaszak Holl MM. Chem Biol. 2007:14. doi: 10.1016/j.chembiol.2006.11.015. [DOI] [PubMed] [Google Scholar]
- [61].Park J-H, Maltzahn G. v., Zhang L, Derfus AM, Simberg D, Harris TJ, Bhatia SN, Ruoslahti E, Sailor MJ. Small. 2009;5:694. doi: 10.1002/smll.200801789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liu Z, Cai WB, He LN, Nakayama N, Chen K, Sun XM, Chen XY, Dai HJ. Nat. Nanotech. 2007;2:47. doi: 10.1038/nnano.2006.170. [DOI] [PubMed] [Google Scholar]
- [63].Montet X, Funovics M, Montet-Abou K, Weissleder R, Josephson L. J. Med. Chem. 2006;49:6087. doi: 10.1021/jm060515m. [DOI] [PubMed] [Google Scholar]
- [64].Van Dyck K, Robberecht H, Van Cauwenbergh R, Van Vlaslaer V, Deelstra H. Biol. Trace Elem. Res. 2000;77:25. doi: 10.1385/BTER:77:1:25. [DOI] [PubMed] [Google Scholar]
- [65].Kwong B, Liu H, Irvine DJ. Biomaterials. 2011;32:5134. doi: 10.1016/j.biomaterials.2011.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sailor MJ. Porous Silicon in Practice: Preparation, Characterization, and Applications. Wiley-VCH; Weinheim, Germany: 2012. [Google Scholar]
- [67].Dejean AS, Beisner DR, Ch'en IL, Kerdiles YM, Babour A, Arden KC, Castrillon DH, DePinho RA, Hedrick SM. Nature Immunol. 2009;10:504. doi: 10.1038/ni.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.