

Dynamic DNA complexes are a new class of DNA technologies that can be engineered to function as programmable molecular machines,[1] detectors,[2] logic gates,[3] and chemical amplifiers.[4, 5] A unique feature of these devices is that, instead of purely classical hybridization mechanisms, they harness a process called strand displacement to facilitate the exchange of oligonucleotides between different thermodynamically-stable DNA complexes.[6, 7] As a result, adaptive and/or reconfigurable molecular devices can be created that operate through enzyme-free, isothermal chemical reactions between different oligonucleotide complexes. While improved understanding of strand displacement has opened new opportunities to engineer elaborate reaction networks for molecular computing,[8] a number of important biological applications for these devices have also emerged. Dynamic nucleic acid devices have been adapted for multiplexed in situ detection of proteins and mRNA,[9-11] and engineered to function as dynamic therapeutic devices[12] and molecular delivery vehicles.[13] Overall, such advances suggest dynamic oligonucleotide systems can function robustly within complex cellular environments and provide new molecular detection capabilities that are not available using existing nucleotide technologies.
Our group has been examining whether dynamic DNA complexes can function as erasable molecular imaging probes in order to increase the number of molecular pathway proteins that can be visualized within individual cells via fluorescence microscopy.[10,11] For this application, programmable, isothermal strand displacement reactions are employed to assemble and disassemble stable fluorescent reporting complexes that localize to their respective protein. These reactions therefore provide a minimally-perturbative route to image different sets of proteins via multiple rounds of fluorescence microscopy by allowing them to be labeled, imaged and erased sequentially.
The ability to visualize multiple sets of proteins within individual cells has become increasingly important, particularly considering that many contemporary biological studies now require more comprehensive, spatially-delineated analyses of protein pathways and networks within biological samples.[14] Such analyses are currently limited by the spectral overlap of the fluorophores used for immunostaining, and generic inabilities to remove fluorescent antibodies from a sample without employing harsh chemical reagents that perturb cell morphology and subsequent marker antigenicity. Hyperspectral imaging approaches can roughly double the number of markers that can be imaged simultaneously over conventional methods.[15] Yet, further increases have been minimal due to the increased noise[16] and decreased dynamic range that accompanies the integration of additional dye molecules into an immunofluorescence assay.[17]
Harnessing strand displacement reactions for multiplex imaging requires that dynamic DNA complexes can be interfaced with protein recognition reagents such as antibodies (Abs), and that their coupling and dispersion in a cell is efficient and uniform enough to generate images accurately reflecting protein intracellular distributions. Furthermore, the signal erasing steps must be sufficiently efficient to ensure residual signals do not compromise subsequent imaging and analyses. Prior kinetic studies outlined design principles that can be used to produce dynamic DNA complexes that possess most of these properties.[11] Yet, these analyses were performed using highly overexpressed autofluorescent proteins as model markers / internal protein standards that were outfitted with ssDNA using engineered protein polymers that were custom-tailored for DNA-protein labeling. Herein, we demonstrate that dynamic DNA complexes can react both selectively and efficiently with DNA-conjugated antibodies to facilitate multiplexed (multi-color) and reiterative (multiple sequential) in situ immunofluorescence analyses of endogenous proteins within individual cells.
The present protein labeling and erasing procedure is outlined in Scheme 1. The protein labeling reactions exploit ‘toehold domains’ within dynamic DNA probes to initiate strand-displacement reactions between a ssDNA targeting strand (TS) that is conjugated directly to antibodies, and a probe complex (PC) that contains a quenched fluorophore. These reactions result in the formation of a fluorescently active reporting complex (IR) containing a single DNA duplexed domain. Similarly, a toehold within the reporting complex is used to initiate a second displacement reaction between IR and an eraser complex (E). This reaction disassembles the IR complex and renders its fluorophore bearing strand inactive via the formation of a waste complex (W) that incorporates a quencher molecule. Consequently, the complete probe labeling / erasing cycle returns the Ab-conjugated TS oligonucleotide to its original ssDNA state.
Scheme 1.
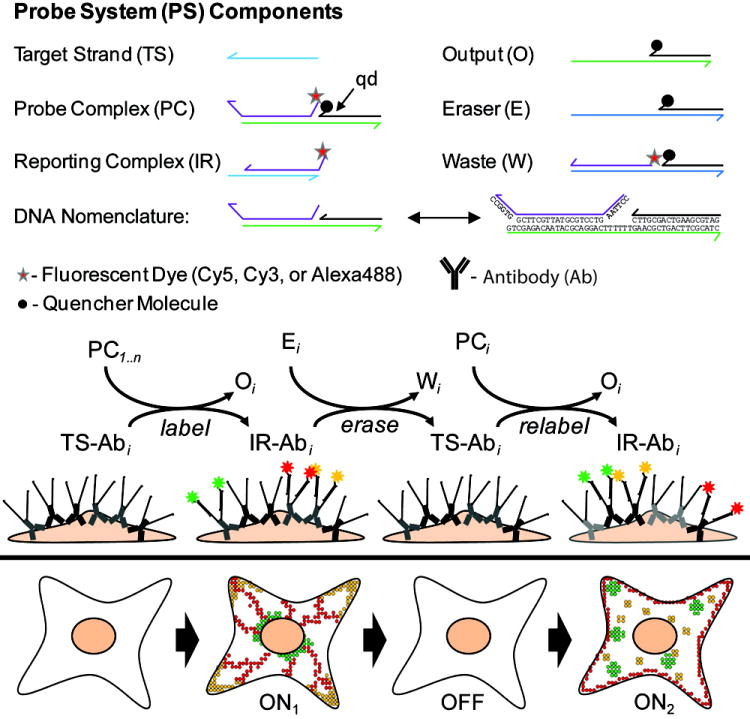
Multiplexed (multicolor) and reiterative (multiple sequential) in situ immunofluorescence labeling of proteins within fixed cells using dynamic DNA complexes.
The ability to selectively stain endogenous proteins using dynamic DNA probes was first tested by labeling native microtubule filaments within fixed HeLa cells using a primary Ab raised against α-tubulin and a secondary TS-Ab conjugate (Figure 1). The same reagents were also used to label microtubules that were counter-stained via the exogenous expression of mOrange-tubulin (Figure S1). In the later case, the signals generated by the DNA probes co-localize and linearly correlate with the mOrange signals, suggesting the probes react selectively and are dispersed evenly throughout the cells. Moreover, signal to background ratios were near-identical to those generated by standard dye-conjugated secondary antibodies, (varying between 10:1 and 20:1, Figure 1b; Figure S2). Importantly, these properties were also reproduced using multiple probe constructs / Ab-conjugates possessing different nucleotide sequences (Figure S3).
Figure 1.
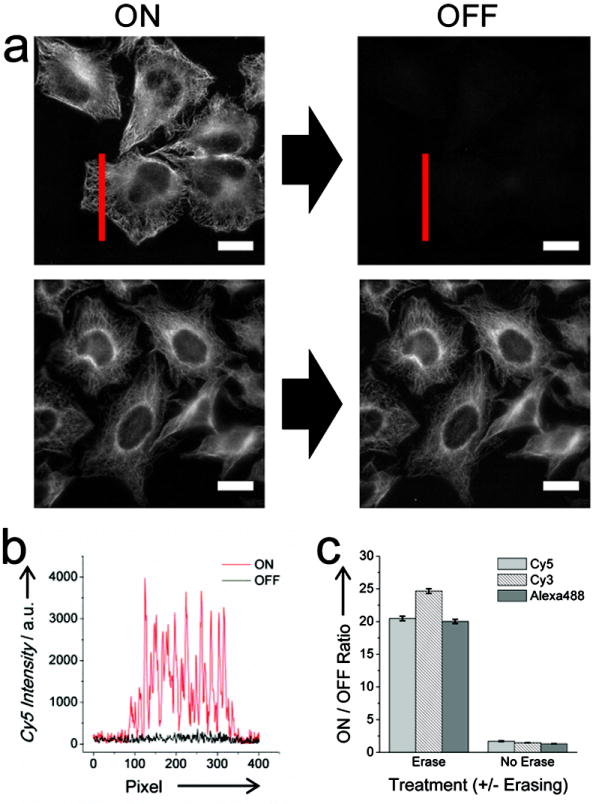
Labeling and erasing of DNA-conjugated Abs targeting microtubules using dynamic DNA probes. (a) Representative labeled (ON) and erased (OFF) images of microtubules imaged after cells were incubated with a probe complex incorporating a Cy5 molecule (ON) and subsequently with an eraser complex (OFF). Images where E was omitted from eraser reaction are also provided. (scale bars: 20 μM). (b) Line profile of microtubules before (ON) and after (OFF) erasing corresponding to the red line in (a). (c) Calculated ON/OFF intensity ratios for separate cell samples labeled with different probe complexes incorporating Alexa488, Cy3, or Cy5 dyes and incubated with or without E.
We next examined the staining equivalence of the dynamic DNA probes to standard secondary Ab labeling procedures using an array of primary Abs that recognize different proteins and localize to different cellular compartments (Figure S4). In each case, the subcellular localization and punctate staining patterns for each marker are very similar to those produced via standard immunofluorescence staining procedures. Overall, the two methods are primarily distinguished by their ability to resolve fine structures within cell nuclei since non-specific DNA-complex binding appears to influence the images within these regions of cells. Yet, these issues appear to simply require further optimization of DNA-antibody conjugation and cell passivation procedures to reduce non-specific DNA binding (Figure S5).
Image intensity measurements after the erasing reaction, a key step in our procedure, indicate that strand displacement reactions can also facilitate efficient removal of immunofluorescent signals from previously stained cells (Figure 1c). Here, the erasing reaction (IR + E → TS + W) yielded ON/OFF ratios between labeled and erased microtubules that ranged between 20.0/1 - 24.7/1. At this erasing level, residual signals are less than or equal to measured RMS fluctuations of background signals within the bare glass portions of the cell culture slides. In contrast, fluorescence intensities are largely unchanged in experiments where the cells were labeled using the same reaction conditions, but omitting the eraser complex (Figure S3). Signal ratios for sequential images of the same cells varied from 1.36/1 to 1.64/1 in this case (Figure 1c).
It should be noted that the present probe systems differ from prior designs since the present complexes incorporate a dedicated quencher domain (qd).[11] The nucleotide sequence of this domain was conserved in each PC and E complex so that the same quencher strand can be employed for all labeling and erasing reactions. This modification was introduced to reduce the costs associated with purchasing multiple modified oligonucleotides. While this domain does not participate in the displacement reaction, it precludes the use of erasing reactions between three-strand IR complexes and E based upon entropically driven circuit designs, which can proceed rapidly.[5,11] Furthermore, we found that including this domain influences abilities to erase fluorescent signals via a four-way branch migration mechanism depending on the type dye molecule employed (Figure S6). Our previous kinetic analyses showed this mechanism can yield increased in situ erasing reaction rates.[11] However, the qd domain appears to introduce steric constraints that limit rates the four-way branched migration reactions are initiated due to the use of internal toehold domains. Nevertheless, this issue was avoided by simply employing the two-strand E complexes depicted in Scheme 1, which exchange strands via a three-way branched migration reaction, and by allowing the erasing reactions to proceed overnight. Faster erasing kinetics could likely be achieved by removing the conserved domains from the probe complexes.
The low residual fluorescence signals remaining after erasing reactions suggests this procedure allows different proteins within the same cells to be visualized via subsequent staining rounds. To directly test this possibility, HeLa cell samples were incubated simultaneously with the rat α-tubulin Ab and a rabbit primary Ab that recognizes either (i) a light chains of kinesin (KLC4); or, (ii) a histone H3 complex that localizes to the cell nucleus (Figure 2a). Each marker/antibody was outfitted with a unique TS strand using a DNA-conjugated secondary Ab (goat anti-mouse, and goat anti-rabbit secondary Ab). The displacement reactions of two separate probe systems (PS1 and PS4, Table S1) were then used to couple Cy5 dye molecules to the microtubule networks, erase these signals, and then label the second marker sequentially (Figure 2a). Microtubule networks are clearly detected in each case, and are erased efficiently upon incubation with E (ON/OFF > 15:1). Moreover, residual signals were sufficiently low to facilitate a second round of immunofluorescence imaging. KLC4 and histone H3 intensity profiles within images obtained after the second labeling reaction are nearly indistinguishable if these images are processed by subtracting their corresponding erased microtubule images or if they are background corrected assuming a constant, spatially-invariant background intensity signal (Figure 2b). Considering these low residual signals will also be affected by lamp intensity fluctuations and focal shifts during imaging, we conclude multiple markers can be inspected using sequential displacement reactions with minimal crosstalk between the signals produced by each reaction.
Figure 2.
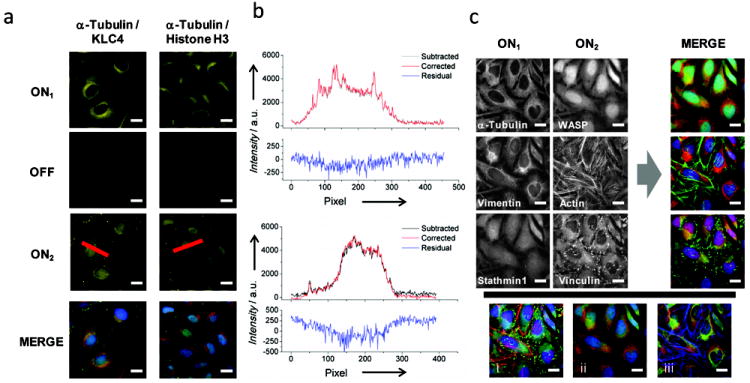
Multiplexed and reiterative immunofluorescence imaging of individual cells. (a) Different markers in the same cells were labeled using the same color dye molecule (Cy5). Microtubules were activated, imaged, and then erased to facilitate the detection of a second protein marker that either overlaps spatially with microtubules (KLC4; left) or that localizes to the nucleus (histone H3; right). (b) Line profiles from the images in (a) that were processed using different background subtraction procedures. The black lines indicate profiles obtained by subtracting the erased (OFF) signals from profiles measured after the cells were labeled a second time for KLC4 (top) and histone H3 (bottom). The red lines correspond to ON2 profiles measured after the KLC4 and histone H3 images where globally background subtracted by a spatially-invariant factor. Plots of the residual signals after subtraction of these profiles are also provided. (c) Abs against α-tubulin, vimentin, and stathmin 1 were detected with DNA-conjugated Abs that were labeled by three unique dynamic DNA probes incorporating Cy5, Cy3, or Alexa488, respectively (ON1). The signals were then erased to permit the detection of WASP and vinculin using dye-conjugated primary Abs (ON2). During this step, actin filaments were also imaged using Alexa532-phalloidin. Different permutations of merged images are also shown where markers are arranged in groups of actin-associated proteins (i), microtubule-associated proteins (ii), or cytoskeletal filaments (iii).
Finally, we examined the ability to implement multiplexed and reiterative imaging procedures to visualize multiple sets of markers within individual cells via sequential rounds of flourescence microscopy. Here, six different cytoskeletal-associated proteins (stathmin 1, vimentin, α-tubulin, Wiskott-Aldrich syndrome protein (WASP), F-actin, and vinculin) were imaged, three at a time, using the microscope‘s red, green and blue channels (Figure 2c). The first set of markers were detected using three different PC complexes to label DNA-conjugated Abs targeting stathmin 1, vimentin, and α-tubulin (Figure 2c; ON1). These signals were then erased simultaneously, allowing the second set of markers to be detected using either dye-conjugated primary antibodies (Alexa647 conjugated anti-WASP and FITC conjugated anti-vinculin), or with phalloidin-Alexa532 to stain actin filaments (Figure 2c, ON2). Cell nuclei were stained in each round using DAPI to register each set of images. Again, the resulting signals reflect the spatial distributions of their protein targets that are obtained using conventional immunofluorescence staining methods. Importantly, the ability to erase marker signals and stain cells a second time using conventional methods shows that strand displacement can not only be used to double the number of proteins that can be detected within a cell sample, but that the antigenicity of proteins targets within cells is also retained throughout these procedures. These results therefore illustrate the flexibility of this approach and suggest the novel detection modalities provided by dynamic DNA complexes can be integrated with various immuno-detection technologies.
In summary, we have demonstrated that dynamic DNA complexes can be employed to selectively activate and erase immunofluorescence signals within fixed cell samples. Provided steric and kinetic constraints affecting their reactions are addressed, these probe technologies can be used to at least double the number of markers that can be detected within individual cells through sequential rounds of fluorescent microscopy. This benefit could be further leveraged using hyperspectral imaging techniques by allowing additional proteins to be stained simultaneously in each imaging round. Furthermore, the displacement reactions incorporated into the present DNA probe systems constitute elementary components of various programmable chemical networks that have been designed to perform more complex detection functions.[7, 18] Our analyses therefore suggest the chemical logic gates and amplifiers of these systems can be integrated with immuno-targeting procedures to facilitate even more detailed, sophisticated and sensitive spatially-dependent analyses of protein pathways within individual cells.
Supplementary Material
Footnotes
This work was supported in whole or in part by grants from the NIH (1R21CA147912) and the Welch Foundation (C-1625). R.M.S. is supported by the Nanobiology Interdisciplinary Graduate Training Program of the W. M. Keck Center for Interdisciplinary Bioscience Training of the Gulf Coast Consortia (NIH grant no. T32 EB009379).
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Dr. Ryan M. Schweller, Department of Bioengineering and Department of Chemistry, Rice University, 6100 Main Street, Houston, Texas 77005 (USA)
Jan Zimak, Department of Bioengineering and Department of Chemistry, Rice University, 6100 Main Street, Houston, Texas 77005 (USA).
Dr. Dzifa Y. Duose, Department of Bioengineering and Department of Chemistry, Rice University, 6100 Main Street, Houston, Texas 77005 (USA)
Dr. Amina A. Qutub, Department of Bioengineering and Department of Chemistry, Rice University, 6100 Main Street, Houston, Texas 77005 (USA)
Prof. Walter N. Hittelman, Department of Experimental Therapeutics, M. D. Anderson Cancer Center, 1515 Holcombe Blvd, Houston, Texas 77030 (USA)
Dr. Michael R. Diehl, Email: diehl@rice.edu, Department of Bioengineering and Department of Chemistry, Rice University, 6100 Main Street, Houston, Texas 77005 (USA).
References
- 1.a) Elbaz J, Wang ZG, Wang F, Willner I. Angew Chem. 2012;124:2399–2403. [Google Scholar]; Angew Chem Int Ed. 2012;51:2349–2353. doi: 10.1002/anie.201107591. [DOI] [PubMed] [Google Scholar]; b) Lubrich D, Lin J, Yan J. Angew Chem. 2008;120:7134–7136. [Google Scholar]; Angew Chem Int Ed. 2008;47:7026–7028. doi: 10.1002/anie.200800476. [DOI] [PubMed] [Google Scholar]; c) Turberfield AJ, Mitchell JC, Yurke B, Mills AP, Jr, Blakey MI, Simmel FC. Phys Rev Lett. 2003;90:118102. doi: 10.1103/PhysRevLett.90.118102. [DOI] [PubMed] [Google Scholar]; d) Yurke B, Turberfield AJ, Mills AP, Simmel FC, Neumann JL. Nature. 2000;406:605–608. doi: 10.1038/35020524. [DOI] [PubMed] [Google Scholar]
- 2.a) Li B, Ellington AD, Chen X. Nucleic Acids Res. 2011;39:e110. doi: 10.1093/nar/gkr504. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Modi S, M GS, Goswami D, Gupta GD, Mayor S, Krishnan Y. Nat Nanotechnol. 2009;4:325–330. doi: 10.1038/nnano.2009.83. [DOI] [PubMed] [Google Scholar]; c) Niemeyer CM, Adler M. Angew Chem. 2002;114:3933–3937. [Google Scholar]; Angew Chem Int Ed. 2002;41:3779–3783. doi: 10.1002/1521-3773(20021018)41:20<3779::AID-ANIE3779>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 3.a) Genot AJ, Bath J, Turberfield AJ. J Am Chem Soc. 2011;133:20080–20083. doi: 10.1021/ja208497p. [DOI] [PubMed] [Google Scholar]; b) Macdonald J, Li Y, Sutovic M, Lederman H, Pendri K, Lu W, Andrews BL, Stefanovic D, Stojanovic MN. Nano Lett. 2006;6:2598–2603. doi: 10.1021/nl0620684. [DOI] [PubMed] [Google Scholar]; c) Seelig G, Soloveichik D, Zhang DY, Winfree E. Science. 2006;314:1585–1588. doi: 10.1126/science.1132493. [DOI] [PubMed] [Google Scholar]
- 4.a) Dirks RM, Pierce NA. Proc Natl Acad Sci U S A. 2004;101:15275–15278. doi: 10.1073/pnas.0407024101. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yin P, Choi HMT, Calvert CR, Pierce NA. Nature. 2008;451:318–322. doi: 10.1038/nature06451. [DOI] [PubMed] [Google Scholar]
- 5.Zhang DY, Turberfield AJ, Yurke B, Winfree E. Science. 2007;318:1121–1125. doi: 10.1126/science.1148532. [DOI] [PubMed] [Google Scholar]
- 6.Zhang DY, Winfree E. J Am Chem Soc. 2009;131:17303–17314. doi: 10.1021/ja906987s. [DOI] [PubMed] [Google Scholar]
- 7.Krishnan Y, Simmel FC. Angew Chem. 2011;123:3180–3215. doi: 10.1002/anie.200907223. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:3124–3156. doi: 10.1002/anie.200907223. [DOI] [PubMed] [Google Scholar]; b) Feldkamp U, Niemeyer CM. Angew Chem. 2008;120:3933–3935. [Google Scholar]; Angew Chem Int Ed. 2008;47:3871–3873. doi: 10.1002/anie.200800675. [DOI] [PubMed] [Google Scholar]
- 8.a) Hagiya M, Yaegashi S, Takahashi K. In: Nanotechnology: Science and Computation. Chen J, Jonoska N, Rozenberg G, editors. Springer; 2006. pp. 293–308. [Google Scholar]; b) Qian L, Winfree E. Science. 2011;332:1196–1201. doi: 10.1126/science.1200520. [DOI] [PubMed] [Google Scholar]; c) Qian L, Winfree E, Bruck J. Nature. 2011;475:368–372. doi: 10.1038/nature10262. [DOI] [PubMed] [Google Scholar]
- 9.Choi HMT, Chang JY, Trinh LA, Padilla JE, Fraser SE, Pierce NA. Nat Biotechnol. 2010;28:1208–1212. doi: 10.1038/nbt.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duose DY, Schweller RM, Hittelman WN, Diehl MR. Bioconjugate Chem. 2010;21:2327–2331. doi: 10.1021/bc100348q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duose DY, Schweller RM, Zimak J, Rogers AR, Hittelman WN, Diehl MR. Nucleic Acids Res. 2012;40:3289–3298. doi: 10.1093/nar/gkr1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Venkataraman S, Dirks RM, Ueda CT, Pierce NA. Proc Natl Acad Sci U S A. 2010;107:16777–16782. doi: 10.1073/pnas.1006377107. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13.Douglas SM, Bachelet I, Church GM. Science. 2012;335:831–834. doi: 10.1126/science.1214081. [DOI] [PubMed] [Google Scholar]
- 14.Collins FS, Green ED, Guttmacher AE, Guyer MS. Nature. 2003;422:835–847. doi: 10.1038/nature01626. [DOI] [PubMed] [Google Scholar]
- 15.Schieker M, Pautke C, Haasters F, Schieker J, Docheva D, Bocker W, Guelkan H, Neth P, Jochum M, Mutschler W. J Anat. 2007;210:592–599. doi: 10.1111/j.1469-7580.2007.00716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zimmermann T. Adv Biochem Eng Biotechnol. 2005;95:245–265. doi: 10.1007/b102216. [DOI] [PubMed] [Google Scholar]
- 17.Constantinou P, Dacosta RS, Wilson BC. J Microsc. 2009;234:137–146. doi: 10.1111/j.1365-2818.2009.03155.x. [DOI] [PubMed] [Google Scholar]
- 18.Zhang DY, Seelig G. Nat Chem. 2011;3:103–113. doi: 10.1038/nchem.957. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.