

Recent developments in transition metal catalyzed processes with alkyne substrates have enabled the synthesis of a plethora of diverse products. Simple terminal alkynes are often used to prepare more elaborate disubstituted derivatives via metal promoted reactions. Early examples include the Glaser-Hay oxidative dimerization of alkynes[1] and Sonogashira coupling reaction.[2] Another milestone was Carreira's catalytic asymmetric aldehyde alkynylation to furnish highly enantioenriched propargylic alcohols.[3] Two very recent advances have broadened the classes of alkynes readily accessible via C-N and C-C bond-forming reactions.
Ynamides continue to receive increasing attention as synthetic intermediates.[4] Their synthesis typically entails halogenation of terminal alkynes followed by C–N cross-coupling.[5] A recent advance from the Stahl group described direct amidation of terminal alkynes via copper catalyzed C–H activation (Scheme 1).[6] Outside of the high efficiency, there are two noteworthy features of this breakthrough. First, like the Glaser-Hay oxidative dimerization of alkynes, dioxygen was used as the terminal oxidant. Second, a variety of nitrogen nucleophiles and alkynes participate to afford an array of ynamides (~70–90% yield, with up to 10 mmol scale).
Scheme 1.
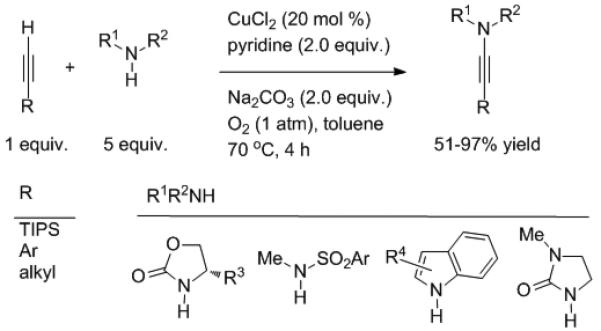
Oxidative amidation of alkynes.
Although catalyst loading can probably be lowered, it may prove difficult to reduce the excess equiv. of amine required in most cases. In the proposed mechanism (Scheme 2), the alkyne and nitrogen nucleophile compete for the copper alkynyl halide affording the C-C and C-N coupled products, respectively.
Scheme 2.
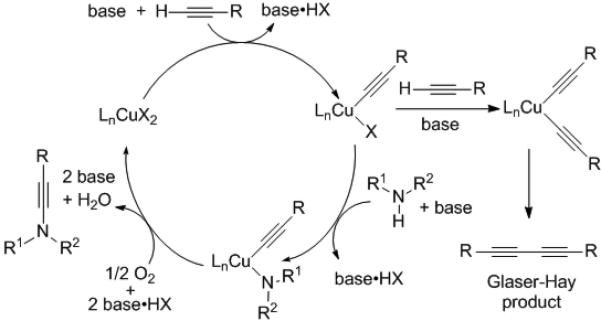
Mechanistic proposal for the C–N coupling reaction.
Another copper-catalyzed reaction involving activation of C–H bonds, this time adjacent to nitrogen, was recently introduced by Zhao and Li (Scheme 3).[7] The terminal oxidant was tert-butyl hydroperoxide (TBHP). After C–H activation coupling occurs with the alkyne. The analogous α-amino esters are unreactive under these conditions, setting the stage for chemoselective C–H functionalization (Scheme 4).
Scheme 3.
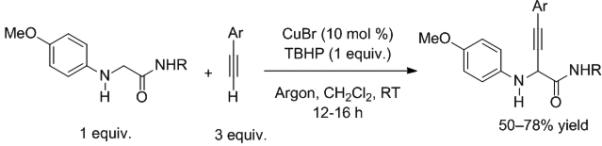
Cross-dehydrogenative coupling catalyzed by copper.
Scheme 4.
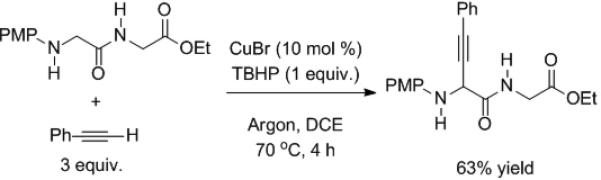
Chemoselective functionalization of the terminal glycine.
The selectivity observed in Scheme 4, and the minimal impact of radical inhibitors in related reactions, disfavour a radical pathway for the initial C–H cleavage. A proposed mechanism involves initial coordination of Cu(II) to the amino group followed by oxidation to generate an activated imine, which undergoes Cu-acetylide addition (Scheme 5). Based on this proposed mechanism, an asymmetric version seems plausible.
Scheme 5.
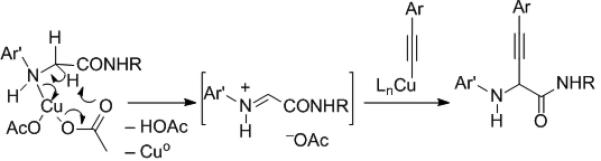
A mechanistic proposal for the amine-alkyne cross-dehydrogenative coupling reaction.
In many reactions the alkyne triple bond is not maintained in the product. Classic examples include Oppolzer's and Wipf's hydrometallation of terminal alkynes, transmetallation to zinc and asymmetric aldehyde vinylation.[8, 9] These reactions employ stoichiometric metal-based reagents. Krische's[10] advent of hydrogenative carbonyl vinylation employing alkynes, hydrogen, and catalytic metal complexes will revolutionize this chemistry.
The alkoxy-carbonylation of alkynes has received recent interest due to the biological activity of β-alkoxyacrylates. Although progress has been made in intramolecular alkoxy-carbonylations,[11] intermolecular version of this important transformation were lacking.[12] Recently Kato, Akita, and coworkers disclosed palladium-based catalysts for this process.[13] These authors rationalized that a metal center better able to activate the alkyne π-system would be more likely to accelerate the intermolecular addition of methanol to coordinated alkynes. A series of palladium sources and ligands were tested and it was found that rac-Ph2-Box/Pd(O2CCF3)2 was the most general (Scheme 6). The catalyst works well with R = alkyl, aryl, heteroaryl, and unprotected glycosides. Disubstituted alkynes gave regioisomeric mixtures in low yields (<50%).
Scheme 6.
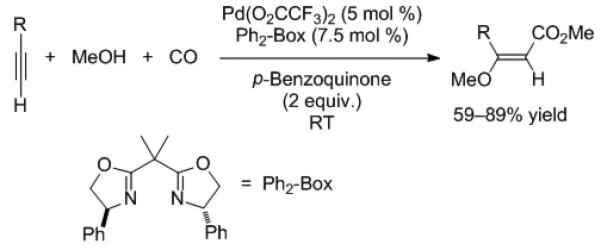
Catalytic intermolecular methoxy-carbonylation of alkynes.
The mechanism envisioned begins with alkyne coordination and addition of methanol to generate a vinyl palladium intermediate. Insertion of CO is likely followed by methanolysis to afford the product. This method for direct conversion of terminal alkynes into β-methoxyacrylates is likely to be synthetically very useful.
Mimicking Nature's approach to catalysis with small molecule catalysts often leads to increased reactivity, selectivity, and even new chemistry. Most enzymes contain a network of activating groups, such as metals and/or hydrogen bond donors and acceptors that cooperate to lower activation energies and stabilize intermediates. One of the leaders in small molecule bifunctional catalysts is Shibasaki with pioneering Li3(BINOLate)3Ln catalysts.[14] Recently Matsunaga, Shibasaki, and coworkers introduced a new class of dinuclear Schiff base catalysts (Figure 1) that promote a variety of highly enantioselective transformations,[15, 16] including additions to activated alkynes.[17]
Figure 1.
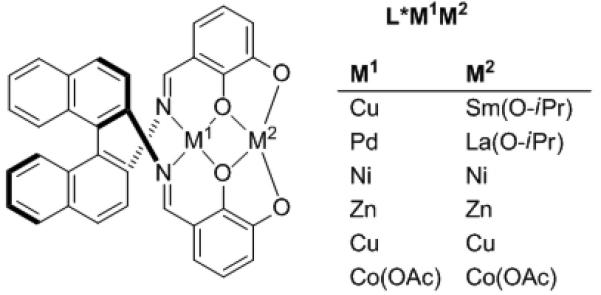
Dinuclear heterobimetallic catalysts.
Conjugate addition of β-keto esters to alkynones was targeted by Shibasaki and Matsunaga (Scheme 7).[18, 19] Initially, various metals were employed as M1 and M2 in Figure 1. Although the Ni2 catalyst gave excellent results with direct Mannich-type reactions,[15, 16] it was (CoOAc)2 that performed best in conjugate additions.[17] Enantioselectivities ranged from 91–99% with a variety of substituents R1–R3, including acyclic and cyclic β-keto esters (R2,R3 linked). Also noteworthy are the high E:Z ratios, which arise via the phosphine catalyzed isomerization. Highly enantioselective catalytic process conducted under solvent-free conditions, such as this, are rare.[20]
Scheme 7.
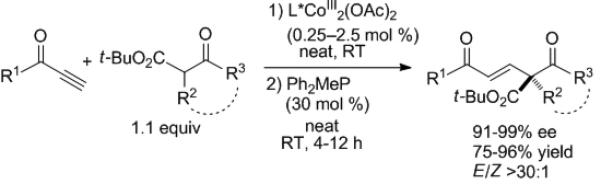
Conjugate additions of β-keto esters to alkynones.
To determine if both cobalts participate in the catalysis, the authors 1) used related monometallic (salen)CoOAc and 2) substituted either the inner or outer CoOAc in L*(CoOAc)2 with other metals. In each case, dramatic decreases in enantioselectivity were observed (maximum <40% ee). Although further studies are necessary to understand the mechanism, these experiments suggest that both cobalt centers are crucial to catalyst enantioselectivity.
The same features that increase the turn over frequency (TOF) and reactivity of bifunctional catalysts also complicate mechanistic studies. Detailed mechanistic insight on bifunctional catalysts, however, is invaluable for the development of new and improved catalysts. Recent advances mapping how bifunctional catalyst work their magic on alkynes has recently been reported from the Grotjahn laboratories.[21] These researchers made a remarkable find: that the acceleration of a ruthenium-based bifunctional catalyst for the anti-Markovnikov hydration of terminal alkynes was an enzyme-like 2.4 X 1011 faster than reaction in the absence of catalyst. Furthermore, the background reaction and acid catalyzed hydrations favor the ketone product. In contrast, the bifunctional catalyst provides exclusively aldehyde (Scheme 8).[22]
Scheme 8.
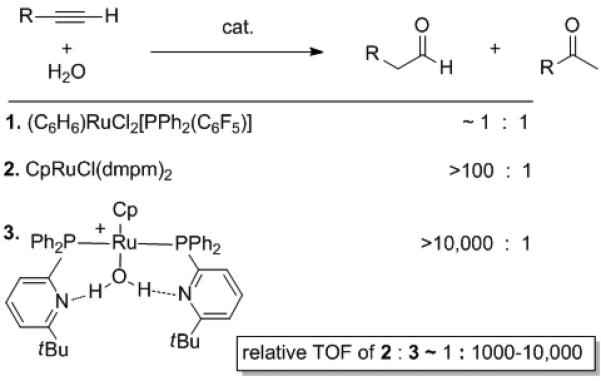
Alkyne hydration catalysts and relative TOFs.
To probe the mechanism, detailed isotopic labelling and computational studies were undertaken with 3 leading to the proposal in Scheme 9. The water adduct A is in equilibrium with κ2-PN B. Upon addition of alkyne, reaction is fast and alkyne coordination is not observed. It is, however, with closely related Ru-bifunctional catalysts and acetylene, which coordinates to Ru with both acetylenic C–H's hydrogen bonding to the pyridyl nitrogens of the ligands.[23] When alkyne is added to anhydrous B at –40 °C the first observed intermediate is vinylidene D. Reaction of this intermediate with water at 0 °C generated G, most likely through pyridyl assisted addition of water to the vinylidene (via E and F). When 15N labelled ligands were employed, the 15N NMR spectrum of G at –100 °C exhibited inequivalent pyridyl nitrogens, one of which was protonated giving rise to a doublet in the 1H NMR spectrum (JN-H=56.8 Hz) for the 15N-H. Strong evidence for an N–H•••Oacyl hydrogen bond was garnered when G was prepared from 13C-labeled acetylene in the presence of H2O. Once G formed, treatment with D2O caused a large isotopic perturbation of the 13C labelled acyl by 1.6 ppm. The remaining steps likely involve protonation of Ru by the pyridyl N–H followed by aldehyde reductive elimination or direct protonation of the acyl by the N–H.
Scheme 9.
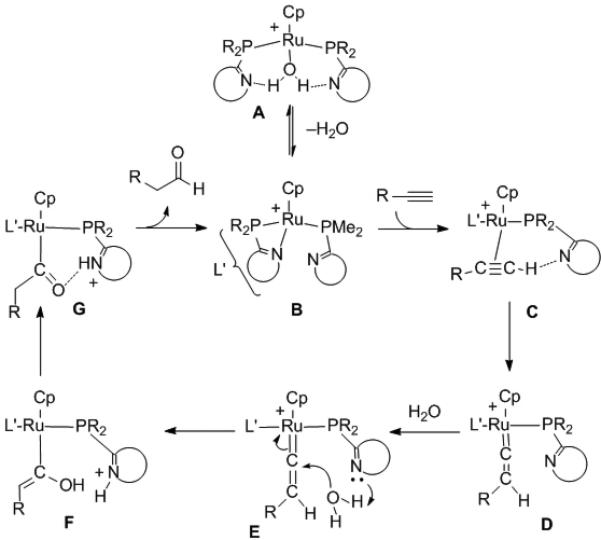
A mechanistic proposal for the hydration of alkynes.
These detailed mechanistic studies elucidated the roles of basic ligand sites, metal center, and proton transfers in this anti-Markovnikov alkyne hydration and help us understand the 1,000–10,000 fold rate increase over analogous catalysts without bifunctional ligands.[24] Likewise, Shibasaki's bifunctional heterobimetallic catalysts demonstrate that Lewis acidic sites acting in concert can lead to new reactivity and excellent enantioselectivity. In combination with direct C-C and C-N oxidative coupling and methoxy-carbonylation reactions, these works represent important developments at the forefront of synthetic chemistry.
Footnotes
PJW acknowledges the NSF and NIH, CAP thanks PRF and CONACYT
References
- 1.Siemsen P, Livingston RC, Diederich F. Angew. Chem. 2000;112:2740. [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2000;39:2632–2657. [PubMed] [Google Scholar]
- 2.Sonogashira K. Negishi E.-i., de Meijere A., editors. Palladium-catalyzed Alkynylations, Handbook of Organopalladium Chemistry for Organic Synthesis. 2002;1:493–529. 493. [Google Scholar]
- 3.Boyall D, Frantz DE, Carreira EM. Org. Lett. 2002;4:2605. doi: 10.1021/ol026282k. [DOI] [PubMed] [Google Scholar]
- 4.Mulder JA, Kurtz KCM, Hsung RP. Synlett. 2003:1379–1390. [Google Scholar]
- 5.Sagamanova IK, Kurtz KCM, Hsung RP. Org. Synth. 2007;84:359–367. [Google Scholar]
- 6.Hamada T, Ye X, Stahl SS. J. Am. Chem. Soc. 2008;130:833. doi: 10.1021/ja077406x. [DOI] [PubMed] [Google Scholar]
- 7.Zhao L, Li C-J. Angew. Chem. 2008;120:7183. [Google Scholar]; Angew. Chem., Int. Ed. 2008;47:7075–7078. doi: 10.1002/anie.200801367. [DOI] [PubMed] [Google Scholar]
- 8.Oppolzer W, Radinov RN. Helvetica Chimica Acta. 1992;75:170. [Google Scholar]
- 9.Wipf P, Ribe S. J. Org. Chem. 1998;63:6454. [Google Scholar]
- 10.Bower JF, Kim IS, Patman RL, Krische MJ. Angew. Chem., Int. Ed. 2009;121:36. doi: 10.1002/anie.200802938. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:34–46. [Google Scholar]
- 11.Zeni G, Larock RC. Chem. Rev. 2004;104:2285. doi: 10.1021/cr020085h. [DOI] [PubMed] [Google Scholar]
- 12.Tamaru Y, Hojo M, Yoshida Z. J. Org. Chem. 1991;56:1099. [Google Scholar]
- 13.Kato K, Motodate S, Mochida T, Kobayashi T, Akita H. Angew. Chem., Int. Ed. 2009;48:XXX. doi: 10.1002/anie.200806080. [DOI] [PubMed] [Google Scholar]
- 14.Shibasaki M, Kanai M, Funabashi K. J. Chem. Soc., Chem. Commun. 2002:1989. doi: 10.1039/b203495f. [DOI] [PubMed] [Google Scholar]
- 15.Handa S, Gnanadesikan V, Matsunaga S, Shibasaki M. J. Am. Chem. Soc. 2007;129:4900. doi: 10.1021/ja0701560. [DOI] [PubMed] [Google Scholar]
- 16.Chen Z, Morimoto H, Matsunaga S, Shibasaki M. J. Am. Chem. Soc. 2008;130:2170. doi: 10.1021/ja710398q. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z, Furutachi M, Kato Y, Matsunaga S, Shibasaki M. Angew. Chem. Int. Ed. 2009;48:xxx. doi: 10.1002/anie.200805967. [DOI] [PubMed] [Google Scholar]
- 18.Bella M, Jorgensen KA. J. Am. Chem. Soc. 2004;126:5672. doi: 10.1021/ja0493594. [DOI] [PubMed] [Google Scholar]
- 19.Wang X, Kitamura M, Maruoka K. J. Am. Chem. Soc. 2007;129:1038. doi: 10.1021/ja068119g. [DOI] [PubMed] [Google Scholar]
- 20.Walsh PJ, Li H, Anaya de Parrodi C. Chem. Rev. 2007;107:2503. doi: 10.1021/cr0509556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Grotjahn DB, Kragulj EJ, Zeinalipour-Yazdi CD, Miranda-Soto V, Lev DA, Cooksy AL. J. Am. Chem. Soc. 2008;130:10860. doi: 10.1021/ja803106z. [DOI] [PubMed] [Google Scholar]
- 22.Grotjahn DB, Lev DA. J. Am. Chem. Soc. 2004;126:12232. doi: 10.1021/ja046360u. [DOI] [PubMed] [Google Scholar]
- 23.Grotjahn DB, Miranda-Soto V, Kragulj EJ, Lev DA, Erdogan G, Zeng X, Cooksy AL. J. Am. Chem. Soc. 2008;130:20. doi: 10.1021/ja0774616. [DOI] [PubMed] [Google Scholar]
- 24.Tokunaga M, Suzuki T, Koga N, Fukushima T, Horiuchi A, Wakatsuki Y. J. Am. Chem. Soc. 2001;123:11917. doi: 10.1021/ja0119292. [DOI] [PubMed] [Google Scholar]