

Abstract
Chiral olefin metathesis catalysts enable chemists to access enantiomerically enriched small molecules with high efficiency; synthesis schemes involving such complexes can be substantially more concise than those that would involve enantiomerically pure substrates and achiral Mo alkylidenes or Ru-based carbenes. The scope of research towards design and development of chiral catalysts is not limited to discovery of complexes that are merely the chiral versions of the related achiral variants. A chiral olefin metathesis catalyst, in addition to furnishing products of high enantiomeric purity, can offer levels of efficiency, product selectivity and/or olefin stereoselectivity that are unavailable through the achiral variants. Such positive attributes of chiral catalysts (whether utilized in racemic or enantiomerically enriched form) should be considered as general, applicable to other classes of transformations.
Keywords: catalysis, catalytic olefin metathesis, enantioselective olefin metathesis, natural product synthesis, organometallic chemistry
“The beauty of images is situated in front of things, that of ideas behind them. So that the first sort of beauty ceases to astonish us as soon as we have reached the things themselves, but the second is something that we understand only when we have passed beyond them.”
Marcel Proust, In Search of Lost Time; Time Regained (Moncrief and Kilmartin translation)
1. Introduction
In this brief analysis, we provide evidence indicating that the availability of chiral catalysts for olefin metathesis[i] offers synthesis routes that afford natural products more efficiently than those involving the use of the corresponding achiral complexes and enantiomerically pure substrates. We demonstrate that research in enantioselective olefin metathesis has led to identification of catalysts that not only furnish products in high enantiomeric purity, but which deliver significantly higher efficiency as well.
As with any class of reactions, catalytic alkene metathesis has a vital symbiotic relationship with target-oriented synthesis.[ii] The utility of a new catalyst and the versatility of the related methods can be best illustrated in context of natural product synthesis.[ii] Perhaps more importantly, the molecular architectures provided by nature fuel the development of new catalysts that permit the introduction of increasingly efficient and selective C–C bond forming protocols. Efforts to access natural products expeditiously serves a crucial role in the advancement of catalytic olefin metathesis, a widely used set of reactions, but nonetheless one that, in spite of two decades of remarkable strides, remains in need of advances in catalyst design and development.[iii] Once more active, longer living, easily available and readily modifiable catalyst classes are discovered, an assortment of otherwise inaccessible molecules, from those that might exhibit unique biological activity to polymeric materials with special properties, can be prepared and myriad applications realized.
In designing a chiral catalyst, the structure – the steric as well as the electronic characteristics – of the molecule is altered to the extent that a new type of a promoter, one that likely has a different reactivity and/or selectivity profile, emerges. A chiral catalyst need not only be used in its enantiomerically pure form nor does it have to be utilized exclusively for the purpose of enantioselective synthesis. A chiral catalyst might also offer substantially improved product- and/or site-selectivity or allow for control of olefin stereoselectivity. The significance of research in enantioselective catalysis therefore extends beyond the development of metal-based complexes that are simply considered to be chiral versions of the initial achiral catalysts.
2. Examples of Enantioselective Natural Product Synthesis through the Use of Achiral Olefin Metathesis Catalysts
We begin with a brief discussion of a select number of total syntheses wherein an achiral olefin metathesis catalyst is employed together with an enantiomerically enriched substrate. The first example is selected to illustrate how catalytic olefin metathesis is often used in enantioselective synthesis; subsequent cases are chosen to demonstrate that use of a chiral catalyst could significantly enhance their efficiency. Such initial analysis is central to an understanding of the importance of chiral olefin metathesis catalysts. We will then present an overview of syntheses wherein a catalytic enantioselective olefin metathesis plays a pivotal role.
2.1. Enantioselective Total Synthesis of Fluvirucin B1
In the mid-nineties, we disclosed the Mo-catalyzed macrocyclization that delivers the fourteen-membered ring lactam 3 (Scheme 1) exclusively as a Z alkene.[iv] This instance of catalytic ring-closing metathesis (RCM) is one of the early examples illustrating that such transformations can be utilized to access macrocyclic structures during the late stages of a multistep total synthesis, and with substrates carrying (potentially catalyst deactivating) Lewis basic functionalities. The total synthesis of fluvirucin B1 served to underline the strategic significance of the reversible nature of catalytic olefin metathesis; unlike the macrocyclization process (generating a trisubstituted alkene), homodimerization of 1, involving reaction of its less sterically congested terminal alkene (affording a disubstituted olefin), is reversible.[v] Another notable aspect of the total synthesis is the highly stereoselective formation of the trisubstituted olefin (>98% Z),[vi] an attribute which led to an effective solution to the difficult problem of controlling the stereochemical identity of the remote Me-substituted stereogenic center (see Scheme 1).
Scheme 1.

Enantioselective synthesis of antifungal agent fluvirucin B1 through a late-stage macrocyclic Mo-catalyzed RCM.
Diene 1, used to synthesize macrocycle 3, is accessed through coupling of the corresponding carboxylic acid obtained in high enantiomeric purity by an enantioselective Zr-catalyzed carbometallation, and the requisite amine accessed through a sequence that includes a Ti-catalyzed enantioselective epoxidation.[iv] Thus, an achiral catalyst (Mo alkylidene 2) is used in a transformation involving an enantiomerically enriched intermediate. Such a strategy represents the manner in which different types of catalytic olefin metathesis (including enyne RCM, ring-opening/cross-metathesis (ROCM), and cross-metathesis) are commonly used in synthesis of enantiomerically enriched target molecules.[ii],[vii]
2.2. Synthesis of Targets through Diastereoselective Olefin Metathesis Reactions
One strategy used in enantioselective total synthesis involves catalytic diastereoselective olefin metathesis processes. In certain instances, a stereogenic element, temporarily installed to control stereoselectivity, is subsequently excised. The availability of an effective chiral olefin metathesis catalyst could obviate the need for installation of a stereocontrolling device and, therefore, elevate the overall efficiency of the synthesis route. Three illustrative examples are presented below.
2.2.1. Synthesis of enantiomerically enriched aspidospermine involving a catalytic diastereoselective RCM
One example of diastereoselective RCM of an enantiomerically pure substrate is in connection with a total synthesis of aspidospermine (Scheme 2).[viii] Catalytic cyclization of triene 4, obtained through a multi-step route that includes a Ti-catalyzed enantioselective epoxidation, is promoted by Ru carbene 5, furnishing diene 6 in 87:13 d.r. after silyl ether hydrolysis; a diastereomerically pure sample (>98:2 d.r.) of the chiral cyclohexenol product is obtained after silica gel chromatography. Subsequent to the catalytic RCM, the original stereogenic center, used to establish the all-carbon quaternary stereogenic center in 6, is oxidatively removed. Enantiomerically pure γ,γ-disubstituted cyclohexenone 7, which serves to complete the total synthesis of aspidospermine, is thus accessed. An enantioselective desymmetrization process promoted by a chiral olefin metathesis catalyst, involving the enone corresponding to triene 4, would directly afford the derived α,β-unsaturated carbonyl, rendering the total synthesis scheme more concise.
Scheme 2.
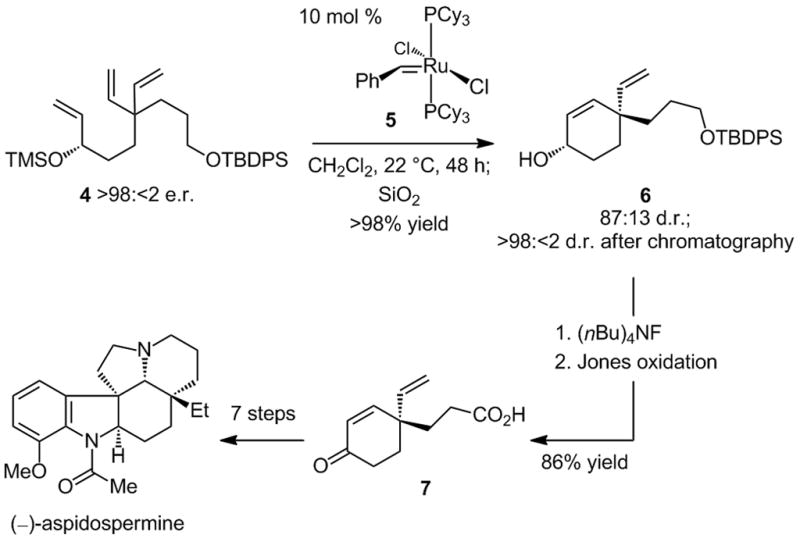
Total synthesis of aspidospermine through catalytic diastereoselective RCM.
2.2.2. Synthesis of enantiomerically enriched longithorone A through two diastereoselective enyne RCM reactions
A total synthesis of longithorone,[ix] summarized in Scheme 3, is based on the fusion of macrocyclic polyenes 11 and 12. The two key intermediates are prepared, albeit inefficiently (50 mol % catalyst loading and <50% yield), through diastereoselective Ru-catalyzed enyne RCM. Whereas 11 is obtained in >95:5 d.r., generation of 12 is less selective (74:26 d.r.). Terminal alkynes 9 and 10, the requisite substrates for enyne RCM, are synthesized by enantioselective additions of the appropriate vinylzinc reagents to aldehyde 8 (Scheme 3) in the presence of stoichiometric amounts of N-methylephedrine.
Scheme 3.
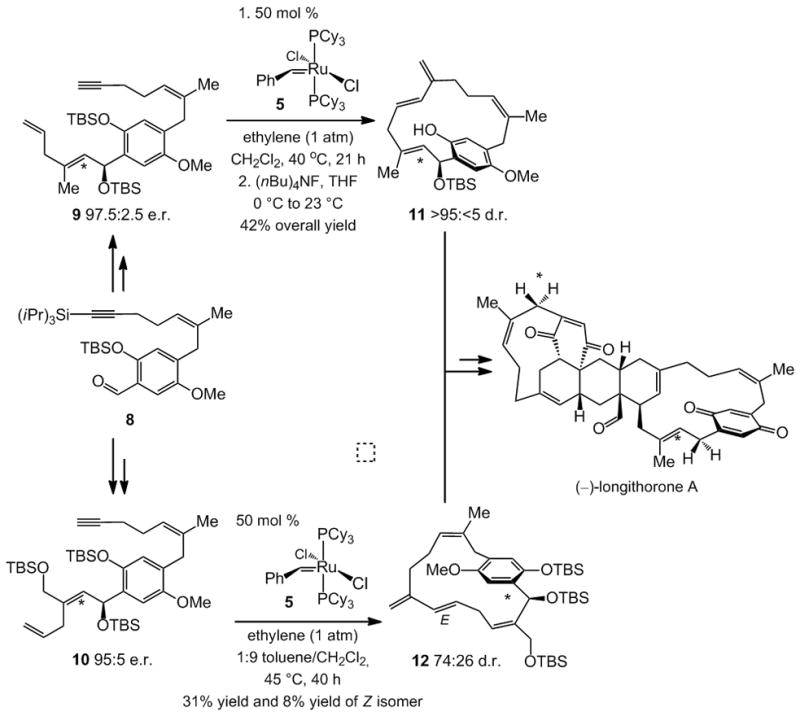
Total synthesis of (−)-longithorone A involving a diastereoselective enyne RCM. The silylether-bearing stereogenic carbon centers, installed for control of planar stereogenicity and subsequently removed, are highlighted.
The benzylic stereogenic centers in fragments 9 and 10 (Scheme 3) are initially installed as the means of controlling the stereochemical outcome of the enyne RCM, which establishes the stereogenic plane of the endo-diene macrocyclic products 11 and 12. The benzylic silyloxy groups must be deleted prior to completion of the total synthesis. A substantially more active (lower loading) and more efficient (higher yield) chiral catalyst for the enyne RCM[x] could allow for direct control of the planar stereogenicity,[xi] giving rise to a significantly more efficient synthesis route.
2.2.3. Synthesis of racemic coleophomones B and C. Control of atropisomerism through catalytic RCM reactions
Ru-catalyzed diastereoselective RCM has been employed towards controlling atropisomerism in total syntheses of rac-coleophomones B and C (Scheme 4).[xii] Treatment of tricarbonyl rac-13 with CH2N2 leads to the formation of a 3:2 mixture of 15 and 16, each of which was subjected to Ru carbene 14. It is likely that the metal complex reacts first with the sterically more accessible 1,1-disubstituted olefin in 15 and 16; RCM of the resulting carbene with one of the two diastereotopic prenyl groups establishes the identity of the quaternary carbon stereogenic center, while promoting the stereoselective formation of eleven-membered rings 17 and 18 (control of planar stereogenicity). It is noteworthy that ring-closure of 15 exclusively delivers the E-olefin isomer and ring-closure of 16 affords Z alkene 18; both processes are diastereoselective (>98%), with the prenyl unit syn to the adjacent methyl group participating in the RCM. A stereoselective desymmetrization of triene 13 promoted by an appropriate chiral RCM catalyst would directly furnish the desired target structures (17 and 18). This particular application raises the intriguing question of whether any alterations within the catalyst structure could allow one to access either the E (coleophomone B) or the Z (coleophomone C) trisubstituted alkene isomers.
Scheme 4.
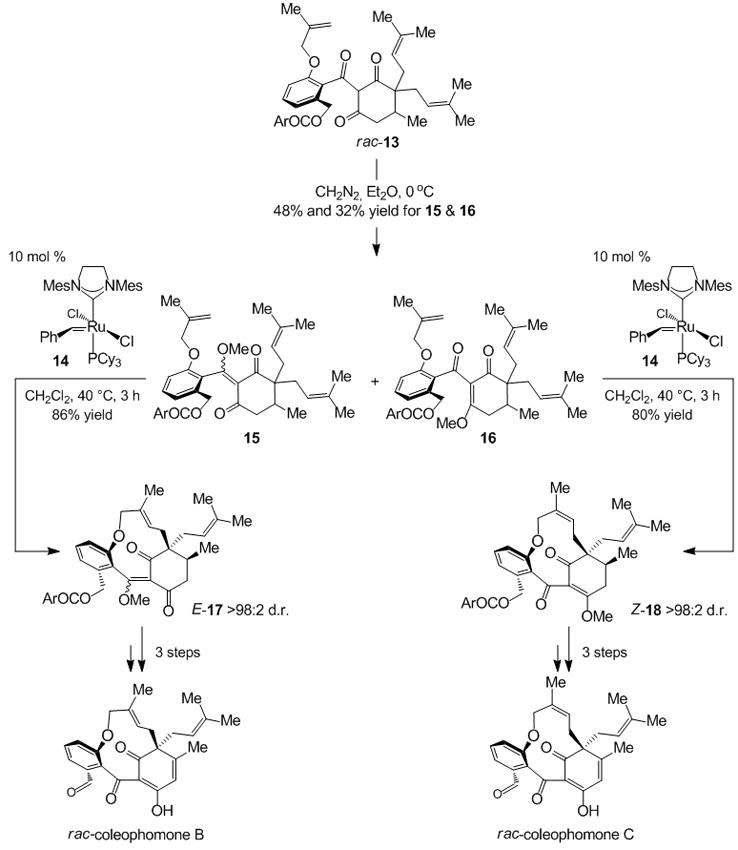
Control of atropisomerism in total syntheses of rac-coleophomones B and C. Ar = pBrC6H4; Mes = 2,4,6-Me3C6H2.
3. Synthesis of Natural Products with Enantiomerically Pure Chiral Olefin Metathesis Catalysts Bearing a C2-Symmetric Diolate
3.1. Synthesis of endo-brevicomin through Mo-catalyzed enantioselective RCM
In 1999, shortly after the disclosure of the first effective chiral catalyst for enantioselective RCM,[xiii] a total synthesis of endo-brevicomin was disclosed (Scheme 5).[xiv] Desymmetrization of meso acetal 19 is promoted by Mo-based diolate 20, affording the bicyclic acetal in 79.5:20.5 enantiomeric ratio (e.r.) and 90% yield; subsequent catalytic hydrogenation delivers the target molecule. It should be noted that the only effective chiral catalyst available at the time was complex 20; one of the chiral Mo alkylidenes developed[iia] after the study in Scheme 5 can likely furnish the desired bicyclic acetal with improved enantiomeric purity. All the Ru-catalyzed enantioselective RCM reactions reported thus far, however, proceed with high selectivity (≥95:5 e.r.) only when the substrate bears trisubstituted alkenes.[xv]
Scheme 5.

Synthesis of (+)-endo-brevicomin through enantioselective RCM.
3.2. Total synthesis of coniine through enantioselective RCM with substrates bearing a tertiary amine
In 2005, the first examples of enantioselective olefin metathesis involving substrates that bear Lewis basic amines were reported.[xvi] The catalytic activity of a number of chiral Mo-based diolates (such as 20 in Scheme 5) were probed, illustrating that the corresponding enantioselective RCM reactions can afford azacyclic structures in up to 99:1 e.r. Ru-based catalysts can, at least in certain instances, be inhibited in the presence of the same class of amine substrates.[xvii]
The preparation of enantiomerically enriched coniine, the neurotoxin found in the hemlock plant, demonstrates the utility of the abovementioned class of catalytic transformations. As depicted in Scheme 6, Mo-catalyzed RCM of benzylamine 21 with 5 mol % of chiral Mo catalyst 22 gives rise to the formation of unsaturated piperidine 23 in 93.5:6.5 e.r. and 83% yield. A two-step sequence furnishes the poisonous alkaloid.[xvi]
Scheme 6.

Total synthesis of coniine through enantioselective RCM.
3.3. Enantioselective synthesis of africanol by a ring-opening/ring-closing metathesis reaction
An application of catalytic enantioselective ring-opening/ring-closing metathesis (RORCM) reaction was reported in 2004 in connection with a total synthesis of africanol.[xviii] Treatment of meso tertiary silyl ether 24, obtained as a single diastereomer through stereoselective alkylation of a norbornenone, with 3 mol % of chiral alkylidene 25 gives rise to the formation of 26 in 93.5:6.5 e.r. and 97% yield (Scheme 7). Thus, a bicyclic structure that bears nearly all the desired stereochemical attributes, including the quaternary carbon stereochemistry appropriate for the preparation of enantiomerically enriched africanol, is accessed in a single catalytic process.
Scheme 7.
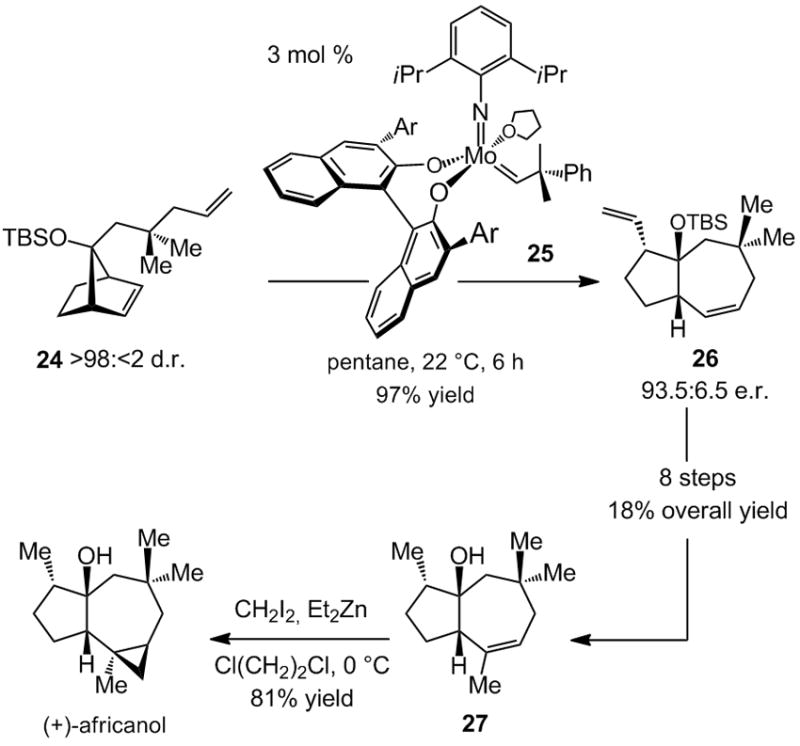
Synthesis of africanol through an enantioselective ring-opening/ring-closing metathesis (RORCM) reaction. Ar = 2,4,6-(iPr)3C6H2, TBS = t-butyldimethylsilyl
Conversion of 26 to 27 (Scheme 7) requires eight additional transformations; two nontrivial processes include site-selective reaction of the cyclic alkene to deliver a Me-bearing trisubstituted olefin and the transformation of the pendant vinyl to the requisite methyl unit. The somewhat lengthy sequence that connects 26 and 27 underlines a notable characteristic of catalytic desymmetrization processes promoted by enantioselective olefin metathesis reactions: such processes give rise to products bearing olefins that might not possess the desired substitution pattern and/or be readily differentiable. The issue of site-selective olefin functionalization is obviated in cases summarized in Schemes 5–6 due to catalytic hydrogenations of all alkenes in the product structure. An alternative strategy to synthesize africanol might have involved catalytic RORCM of the corresponding enantiomerically pure trisubstituted norbornyl alkene. Design of an enantioselective synthesis of such a chiral molecule, however, would not be trivial, again underlining the importance of enantioselective olefin metathesis.
Weaknesses of a total synthesis often point to intriguing problems that await effective new solutions; the route shown in Scheme 7 is a case in point. An efficient, site-selective, and preferably catalytic conversion of a disubstituted alkene to a trisubstituted olefin would strengthen the olefin metathesis-based approach developed for africanol. Such a transformation could involve alkylmetallation of the olefin followed by β-hydride elimination (Heck-type process).
3.4. Enantioselective synthesis of the lactone fragment of anti-HIV agent tipranavir
The ability of a chiral catalyst to rearrange a simple achiral molecule to a valuable chiral one in high enantiomeric purity finds an application in the context of a synthesis of tipranavir, a molecule active against the HIV-protease enzyme.[xix] An achiral substrate such as 28, through subjection to an appropriate chiral catalyst (5 mol % 25), is converted to a chiral product (29) that would otherwise be difficult to access in the non-racemic form (Scheme 8). Synthesis of 29 (95.5:4.5 e.r.) underlines the ability of catalytic enantioselective olefin metathesis to deliver molecules not easily prepared with achiral catalysts and enantiomerically enriched substrates.[xx] The latter approach would require an efficient method for enantioselective synthesis of the requisite O-substituted quaternary carbon stereogenic center. In spite of recent advances, efficient and facile protocols, particularly of the catalytic variety, to furnish tertiary alcohols in high enantiomeric purity remain largely unavailable.[xxi]
Scheme 8.
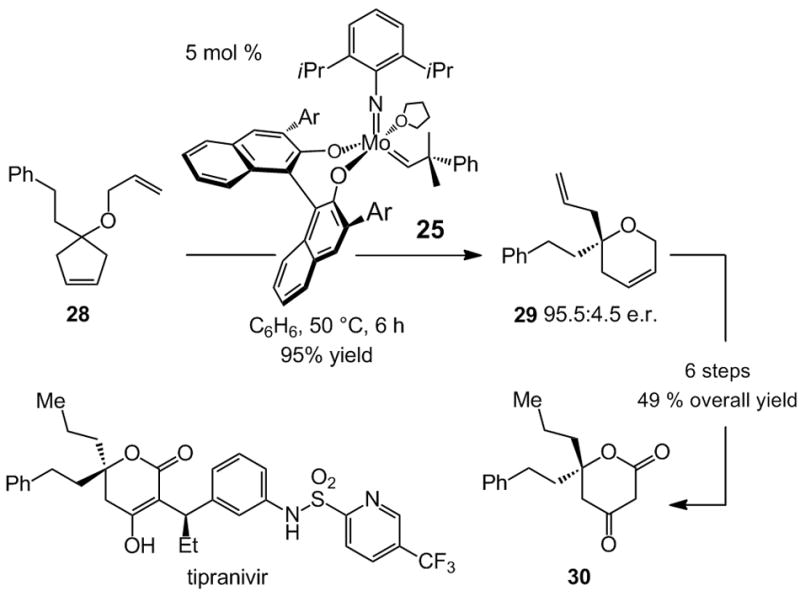
Enantioselective synthesis of the lactone fragment of HIV-protease inhibitor tipranivir through a Mo-catalyzed ring-opening/ring-closing metathesis (RORCM) reaction. Ar = 2,4,6-(iPr)3C6H2
Another noteworthy feature of the sequence shown in Scheme 8 relates to differentiation of the two olefins of 29. Site-selective allylic oxidation (PCC, CH2Cl2, 80 °C) of the cyclic allylic ether allows for a Rh-catalyzed hydrogenation [3 mol % ClRh(PPh3)3] to generate the desired lactone and the n-propyl side chain, respectively.
4. Design, Synthesis and Application of Stereogenic-at-Mo Olefin Metathesis Catalysts. Enantioselective Synthesis of Quebrachamine through a Challenging RCM Reaction
Application of catalytic enantioselective olefin metathesis to natural product synthesis remains relatively uncommon, compared to myriad complex molecule syntheses achieved through utilization of the corresponding achiral catalysts.[ii] One reason for such paucity is that target molecules are often identified subsequent to development of a method. As a result, a protocol may not provide an ideal solution for a particular total synthesis application. It might be more expedient to secure first the most direct synthesis route, which would undoubtedly benefit from an effective catalytic enantioselective process. The effectiveness of the existing chiral complexes, if available, might then be evaluated and, if necessary, shortcomings can be addressed through new catalyst design.
A recent enantioselective synthesis of Aspidosperma alkaloid quebrachamine was accomplished through the above approach (Scheme 9).[xxii] That is, the total synthesis was principally conceived to challenge the current state-of-the-art olefin metathesis catalysts and serve as a springboard for catalyst development. As illustrated in Scheme 9, the late-stage RCM requires ring-closure onto one of two sterically hindered vinyl groups at a congested all-carbon quaternary center[xxiii] in the presence of a Lewis basic tertiary amine. Mo-based diolates, such as 20 (Scheme 5), fail to deliver the desired tetracycle 35 even under forcing conditions (up to 50 mol % catalyst, up to 80 °C, for as long as 48 h). With a chiral Ru catalyst, such as that shown in Scheme 10 (37, below), only rac-35 is obtained with little efficiency. Even when the more active achiral Mo complex 2 (Scheme 1) is used, >98% conversion (59% yield) is only achieved with 30 mol % catalyst loading.
Scheme 9.
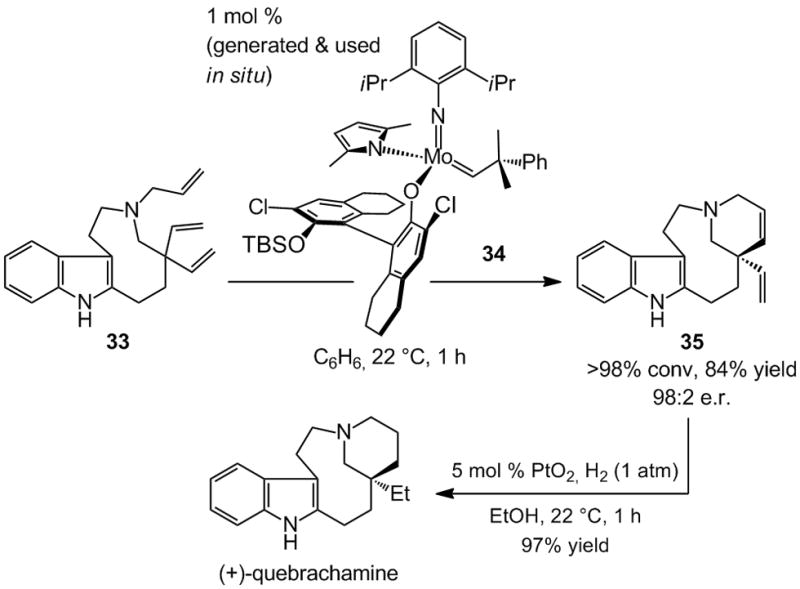
Catalyst development spurred by total synthesis: A stereogenic-at-Mo complex as a highly effective chiral catalyst developed for enantioselective RCM in total synthesis of quebrachamine.
Scheme 10.
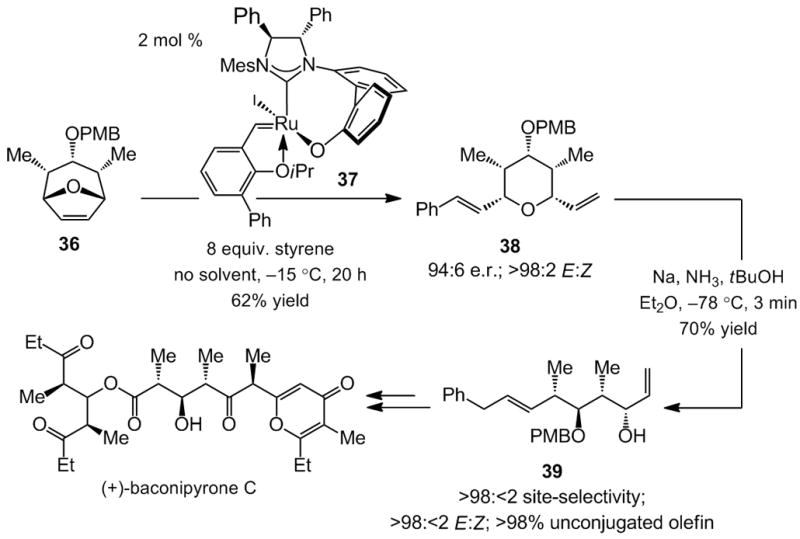
Total synthesis of baconipyrone C through an enantioselective Ru-catalyzed ring-opening/cross-metathesis (ROCM) reaction. PMB = pmethoxybenzyl; Mes = 2,4,6-Me3C6H2
To address the aforementioned shortcomings and identify an effective catalyst for efficient and enantioselective RCM of 33, a new class of stereogenic-at-Mo complexes, represented by monopyrrolide 34, was introduced.[xxiia] The design of the more active chiral Mo-based catalysts was largely based on mechanistic considerations. One such principle is that, in any olefin metathesis process the metal center is fluxional: it undergoes repeated inversion through intermediacy of trigonal bipyramidal or square pyramidal complexes. We argued that the absence of a rigid bidentate ligand reduces the energetic barriers that accompany such transformations within the catalytic cycle, enhancing catalyst activity. Furthermore, based on recent studies by Eisenstein and co-workers,[xxiv] the stereogenic-at-Mo complexes were outfitted with one relatively stronger electron-donating (e.g., pyrrolide in 34, Scheme 9) and a relatively weaker electron-donating ligand (e.g., aryloxide in 34, Scheme 9). Theoretical investigations suggested that the presence of an electron donor ligand (vs two identical aryloxides in chiral Mo diolates) could facilitate and control the stereochemical course of olefin coordination as well as increase the rate of metallacyclobutane cyclo-reversion. Experimental evidence strongly supported the validity of the above hypotheses: stereogenic-at-Mo complexes such as 34, generated in situ from the corresponding achiral Mo bispyrrolide and an equivalent of the chiral aryl alcohol (catalyst isolation not required), readily promote the difficult RCM (Scheme 9). The desired tetracyclic diene 35, containing an all-carbon quaternary stereogenic center, is thus obtained efficiently (84% yield) and with exceptional enantioselectivity (98:2 e.r.).[xxv]
5. Synthesis of Baconipyrone C by Ru-Catalyzed Enantioselective ROCM
Total synthesis of baconipyrone C, disclosed in 2007,[xxvi] represents the first and, thus far only, application of a Ru-catalyzed enantioselective olefin metathesis process to natural product synthesis. Treatment of oxabicycle 36 with styrene in the presence of 2 mol % Ru carbene 37[xxvii] delivers the fully substituted pyran 38 in 62% yield and 94:6 e.r. (Scheme 10). It should be noted that 37 is generated in situ by subjection of the corresponding Ag-based N-heterocyclic carbene (NHC) with the appropriate achiral Ru-PCy3 complex and NaI; there is no the need for isolation or purification of the catalyst. The chiral Ru catalyst does not only deliver high enantioselectivity: the ROCM with 37 is significantly more efficient than when achiral carbenes, which contain a non-stereogenic metal center and a monodentate NHC ligand (vs a stereogenic Ru center and a bidentate NHC in 37), are used.[xxviii]
It has already been mentioned that effective differentiation of the olefins within a product structure is a challenge typically associated with catalytic olefin metathesis reactions (particularly ring-opening processes; see Scheme 7). Alkene differentiation is achieved with 38 through a reductive cleavage (Scheme 10), which occurs with complete selectivity at the cinnamyl C–O (<2% conjugated alkene); a Birch reduction delivers only the E alkene as well. Acyclic diene 39 is used to access the polypropionate core (shown in red, Scheme 10).
The diketone fragment of baconipyrone C (shown in blue, Scheme 10) was synthesized through a tandem double-allylic alkylation, promoted by a chiral Cu complex that bears a bidentate NHC that is structurally related to that residing within Ru carbene 37. Development of chiral NHC ligands, initially designed for use in Ru-catalyzed alkene metathesis, have thus been successfully utilized in other important classes of C–C bond forming reactions.[xxix]
In the sequence summarized in Scheme 10, the transformation involving a chiral olefin metathesis catalyst (formation of 38 catalyzed by chiral Ru carbene 37) is performed at an early juncture. In contrast, total synthesis of quebrachamine, the final two transformations of which are depicted in Scheme 9, contains a penultimate enantioselective RCM. An application such as baconipyrone C illustrates that the catalytic enantioselective ROCM can be carried out in reasonable scale (multi-gram), so that sufficient material is made available for the completion of the total synthesis. The late-stage process involving the formation of 35 catalyzed by chiral Mo alkylidene 34 (Scheme 9) highlights a different noteworthy issue: a multi-step synthesis plan can be devised with a reasonable degree of reliability involving a set of transformations that includes a catalytic enantioselective olefin metathesis reaction.
6. A concept underlined by the development of new chiral Mo catalysts: The significance of chiral catalysts extends beyond enantioselectivity
In the course of our search for an efficient chiral complex that would promote the RCM of triene 33, en route to quebrachamine, we arrived at a class of chiral catalysts that readily initiates the requisite process enantioselectively and with significantly higher efficiency than other existing achiral or chiral Ru- or Mo-based catalysts.[xxii] A critical observation made in the above studies, as illustrated in Scheme 11, is that in the presence of only 1 mol % rac-40, RCM of 33 proceeds to >98% conversion within one hour, affording the desired rac-35 in 79% yield. As also shown in Scheme 11, reaction with achiral Mo alkylidene 2 is far less efficient. Various achiral Ru carbenes deliver 35% to >98% conversion at 5 mol % loading after 6 hours. Ru-catalyzed transformations generate a significant amount of byproducts; rac-35 can only be obtained in 36%–65% yield. The desired tetracyclic diene is isolated in 83% yield only when 7.5 mol % of a more efficient Ru complex is used.[xxiib] The above findings indicate that a chiral catalyst can offer reactivity patterns that are substantially different from those offered by the achiral variants. In other words, chiral catalysts can be relevant to cases other than those where the goal is achieving high enantioselectivity.
Scheme 11.

A stereogenic-at-Mo complex serving as a uniquely efficient RCM catalyst.
Design of a chiral Ru complex, utilized in the racemic form, to promote sequence-selective copolymerization processes,[xxx] constitutes a notable demonstration of how chiral catalysts can deliver unusual selectivity profiles.[xxxi] As illustrated in Scheme 12, the stereogenic-at-Ru complex 41 has a relatively more stable diastereomeric form (41 and 42), where the carbene resides beneath the sterically less demanding phenyl substituent of the phosphine ligand. This latter isomer reacts preferably with the more reactive (e.g., strained) alkene, leading to inversion at the Ru center and generation of a higher energy stereoisomer (43), where the carbene is forced to reside proximal to the sterically demanding t-butyl unit. The newly generated complex (43) reacts with all types of alkenes, as there is a significant driving force for re-inversion at the Ru center (formation of the lower energy metal carbene). Thus, by using an excess amount of the less reactive cross partner (versus the strained alkene partner), sequence-selective polymerization can be achieved when rac-41 is utilized.
Scheme 12.
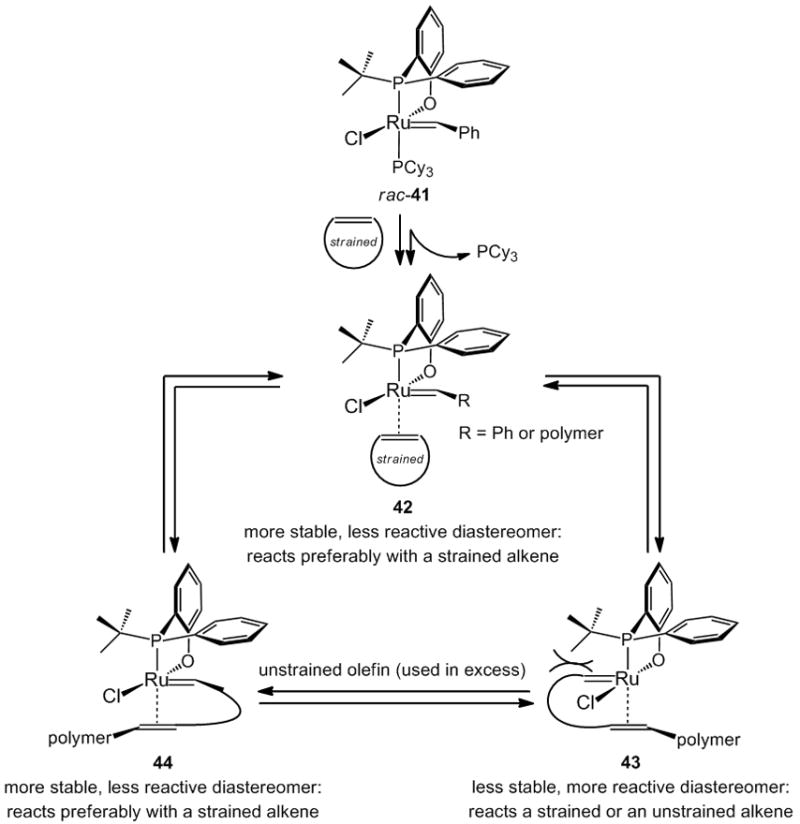
A racemic stereogenic-at-Ru catalyst used to promote sequence-selective polymerization; success of this strategy is due to energy differences between the two carbene diastereomers of the chiral complex.
Two more recent instances, where chiral olefin metathesis catalysts serve to address issues other than product enantiomeric purity, are summarized in Scheme 13. Chiral complex rac-46 readily promotes RCM of enyne 45 to afford diene 47 exclusively (Scheme 13);[xxxii] none of five-membered ring diene 48, generated predominantly by reactions with Ru carbene 49,[xxxiii] is obtained. In another example (Scheme 13), chiral Mo complex 51 promotes an exceptionally Z-selective (>98:<2 Z:E) ROCM of oxabicycle 50 and styrene.[xxxiv] Achiral alkylidene 2 (Scheme 1) delivers a 66:33 mixture of E:Z isomers, and chiral Mo-based diolates, such as 20 and 22 (Schemes 5 and 6) as well Ru complexes such as 14 (Scheme 4) and 37 (Scheme 10) afford only the corresponding E alkene (>98:2 E:Z).[17c,xxxv] In the most recent investigations, stereogenic-at-Mo complexes have been used to effect highly Z-selective polymerization reactions of norbornene-derived monomers.[xxxvi] The macromolecular systems obtained through the use of the new Mo alkylidenes were entirely inaccessible previously.
Scheme 13.

Chiral olefin metathesis catalysts are not only valuable for obtaining enantiomerically enriched products; they can also offer other types of selectivity. Mes = 2,4,6-Me3-C6H2.
Application of chiral catalysts towards enhancing reaction efficiency or altering product selectivity trends does not pertain only to olefin metathesis. Two representative cases are provided in Scheme 14. Pd-catalyzed cross-coupling in the presence of achiral P(otolyl)3 is substantially less efficient in producing aniline 53 than when rac-55 is used (Scheme 14; 77% vs 93% yield, respectively).[xxxvii] A related Pd-catalyzed process involving ammonia is reported to proceed most efficiently with Josiphos, a chiral bisphosphine variant of 55.[xxxviii]
Scheme 14.
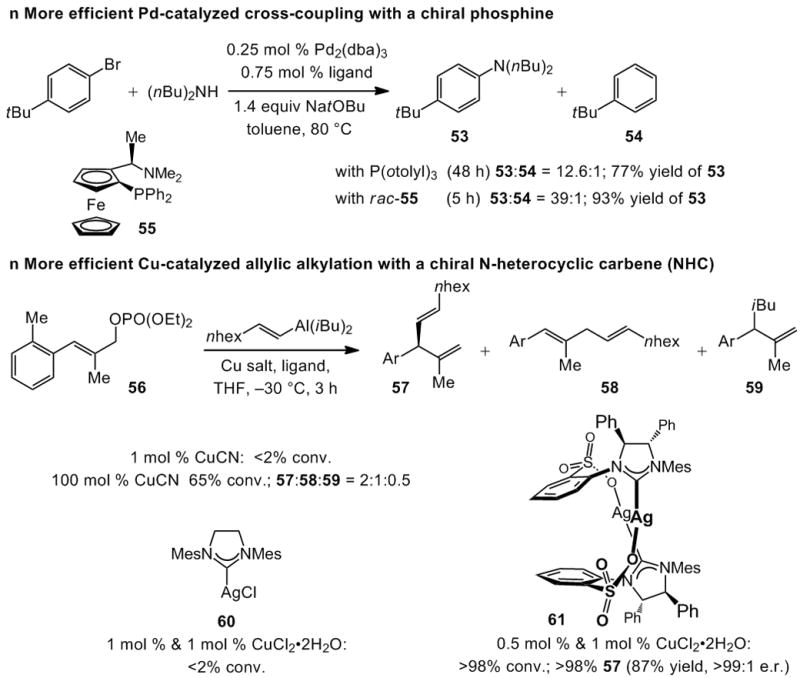
Chiral Pd- and Cu-based complexes generate product-selectivity, site-selectivity and efficiency levels that are superior to those afforded by the corresponding achiral catalysts.
Allylic alkylation of unsaturated phosphate 56, in the presence of various Cu salts, results either in complete recovery of the starting materials or, when stoichiometric amounts are used, a mixture of several products (57–59, Scheme 14). The NHC–Cu complex, formed in situ through reaction of achiral 60 and CuCl2·2H2O, is similarly ineffective in promoting alkylation. In stark contrast, the chiral NHC–Cu complex, derived from 61 and CuCl2·2H2O, not only delivers >98% conversion, promotes the generation of 57 exclusively (<2% 58 or 59) and as a single enantiomer (>99:1 e.r.).[xxxix]
The allylic alkylation shown in Scheme 14 represents a class of transformations where a chiral catalyst gives rise to the introduction of a new site-selective C–C bond forming protocol, one that might not necessarily have an effective non-enantioselective variant (as is typically the case). Herein lies a crucial lesson regarding research in catalyst and method design: examination of a process promoted by an achiral catalyst need not precede the development of an enantioselective version catalyzed by a chiral complex. Through the use of a chiral catalyst, we may be able to address – effectively and simultaneously – issues of reactivity and selectivity. A chiral catalyst may well deliver previously unattainable reactivity levels, while engendering an enantioselective variant. It is possible that a chiral catalyst delivers unprecendented reactivity levels without offering high stereoselectivity; for example, enantiomerically pure adamantylimido complex 51 (Scheme 13) promotes the RCM reaction that leads to quebrachamine (33→35, Scheme 9) far more readily than achiral 2 (Scheme 1), but with low enantioselectivity (70:30 e.r.).[xl]
7. Conclusions and Future Outlook
Catalytic olefin metathesis has made an enormous impact on chemical synthesis during the past two decades, but a number of its potential applications remain unrealized as a result of deficiencies in catalyst performance.[iii] The findings briefly touched upon in this article illustrate that target-oriented synthesis can serve as an invaluable indicator of the types of catalysts that remain to be discovered. In addition to the need for catalysts that are more efficient in promoting RCM and ROM processes that require relatively high loadings, most noteworthy, perhaps, are effective catalysts for stereoselective cross-metathesis reactions.[i] The example shown in Scheme 15, involving an inefficient cross-metathesis to obtain a trisubstituted olefin is illustrative.[viib] Examination of a variety of Ru- and Mo-based complexes revealed that optimal results are obtained with 35 mol % 49 in a transformation that affords the desired product with only 75:25 E:Z selectivity; E-60 is obtained in only 40% yield after purification. Use of phosphine-bearing Ru carbene 14 (Scheme 4) gives rise to some loss of enantiomeric purity,[xli] and with achiral Mo complex 2 (Scheme 1), still lower E:Z-selectivity is obtained (66:33).[xlii] High E:Z- or Z:E-selectivity in synthesis of di-, tri- or tetrasubstituted alkenes through catalytic cross-metathesis remains out of reach. Perhaps it will be a class of chiral catalysts that will provide effective solutions to the above problems; the high Z-selectivity provided by chiral Mo alkylidene 51 (Scheme 13) suggests that this might indeed be the case.
Scheme 15.
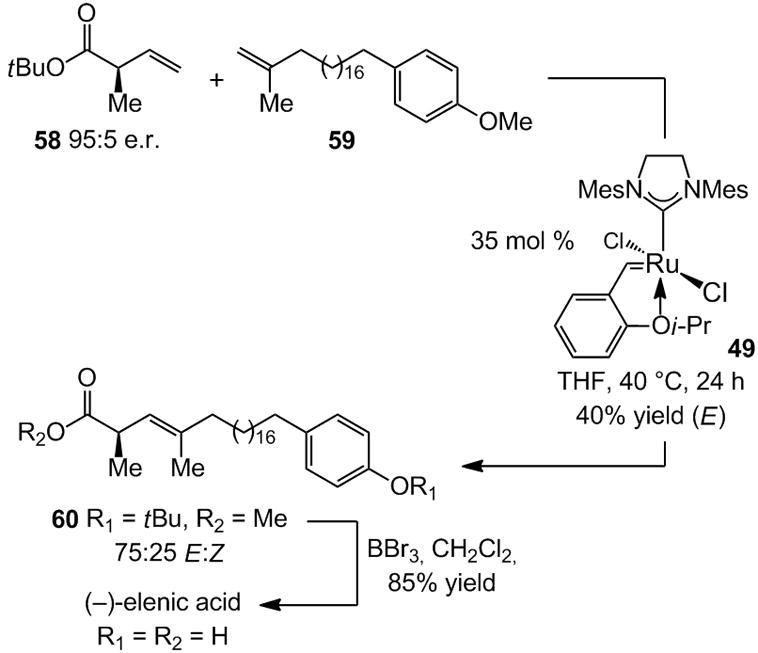
Natural product synthesis often underlines significant deficiencies in catalyst development, as indicated by an inefficient cross-metathesis reaction in an enantioselective synthesis of elenic acid.
A critical issue emerging through the above discussions is that a chiral catalyst should not be examined only in the context of an enantioselective process. A chiral catalyst might offer reactivity levels that are superior to those furnished by any achiral variant. If the goal of a study is the development of an enantioselective transformation, initial reactivity studies should not necessarily be focused only on achiral catalysts. One cannot help but wonder how many solutions to various problems in catalysis have been missed because only achiral catalysts have been probed?
Acknowledgments
We are grateful to the National Institutes of Health (GM-59426) and the National Science Foundation (CHE–0715138) for support of our investigations regarding development of new catalysts (Mo- and Ru-based, respectively) for olefin metathesis. We thank Professor Richard R. Schrock, Dr. Pamela J. Lombardi, Robert V. O’Brien III, Dr. Ismail Ibrahem, Miao Yu, and Dr. Yeon-Ju Lee for helpful suggestions and numerous discussions.
References
- i.Grubbs RH, editor. Handbook of Metathesis. Wiley–VCH; Weinheim: 2003. [Google Scholar]
- ii.a) Schrock RR, Hoveyda AH. Angew Chem Int Ed. 2003;42:4592–4633. doi: 10.1002/anie.200300576. [DOI] [PubMed] [Google Scholar]; b) Deiters A, Martin SF. Chem Rev. 2004;104:2199–2238. doi: 10.1021/cr0200872. [DOI] [PubMed] [Google Scholar]; c) Nicolaou KC, Bulger PG, Sarlah D. Angew Chem Int Ed. 2005;44:4490–4527. doi: 10.1002/anie.200500369. [DOI] [PubMed] [Google Scholar]
- iii.Hoveyda AH, Zhugralin AR. Nature. 2007;450:243–251. doi: 10.1038/nature06351. [DOI] [PubMed] [Google Scholar]
- iv.a) Houri AF, Xu Z, Cogan DA, Hoveyda AH. J Am Chem Soc. 1995;117:2943–2944. [Google Scholar]; b) Xu Z, Johannes CW, Salman SS, Hoveyda AH. J Am Chem Soc. 1996;118:10926–10927. [Google Scholar]; c) Xu Z, Johannes CW, Houri AF, La DS, Cogan DA, Hofilena GE, Hoveyda AH. J Am Chem Soc. 1997;119:10302–10316. [Google Scholar]
- v.For two noteworthy applications to natural product synthesis wherein the reversible nature of catalytic cross-metathesis is utilized, see: Smith AB, III, Adams CM, Kozmin SA, Paone DV. J Am Chem Soc. 2001;123:5925–5937. doi: 10.1021/ja0106164.Fürstner A, Thiel OR, Ackermann L. Org Lett. 2001;3:449–451. doi: 10.1021/ol0069554.
- vi.A recent study in connection with total syntheses fluvirucins B2–B5 indicates that RCM of this class of dienes with Ru-based carbenes leads to the formation of a mixture of E and Z isomers. Such observations suggest that, in contrast to the initial suppositions (ref. 4c), the high Z-selectivity obtained in the reaction illustrated in Scheme 1 is largely imposed by the Mo-based alkylidene (vs the conformational preferences of the substrate). See: Llàcer E, Urpì F, Vilarrasa J. Org Lett. 2009;11:3198–3201. doi: 10.1021/ol901030f.
- vii.For examples of these types of total syntheses reported from our laboratories, see: ring-opening/cross-metathesis: Johannes CW, Visser MS, Weatherhead GS, Hoveyda AH. J Am Chem Soc. 1998;120:8340–8347.cross-metathesis: Murphy KE, Hoveyda AH. J Am Chem Soc. 2003;125:4690–4691. doi: 10.1021/ja0300618.enyne ring-closing metathesis: Cesati RR, III, de Armas J, Hoveyda AH. J Am Chem Soc. 2004;126:96–101. doi: 10.1021/ja0305407.
- viii.Fukuda Y, Shindo M, Shishido K. Org Lett. 2003;5:749–751. doi: 10.1021/ol034020s. [DOI] [PubMed] [Google Scholar]
- ix.Layton ME, Morales CA, Shair MD. J Am Chem Soc. 2002;124:773–775. doi: 10.1021/ja016585u. [DOI] [PubMed] [Google Scholar]
- x.Formation of macrocyclic 11 is a rare example of an endo-selective processes catalyzed by Ru-based complex; Ru-based carbenes, unlike Mo alkylidenes, typically deliver the corresponding exo-dienes. For endo- and enantioselective Mo-catalyzed enyne RCM reactions, see: Singh R, Schrock RR, Müller P, Hoveyda AH. J Am Chem Soc. 2007;129:12654–12655. doi: 10.1021/ja075569f.Lee YJ, Schrock RR, Hoveyda AH. J, Am Chem Soc. 2009;131:10652–10661. doi: 10.1021/ja904098h.
- xi.There has been one report of control of planar stereogenicity through Mo-catalyzed enantioselective RCM. See: Ogasawara M, Watanabe S, Fan L, Nakajima K, Takahashi T. Organometallics. 2006;25:5201–5203.
- xii.a) Nicolaou KC, Vassilikogiannakis G, Montagnon T. Angew Chem Int Ed. 2002;41:3276–3281. doi: 10.1002/1521-3773(20020902)41:17<3276::AID-ANIE3276>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Montagnon T, Vassilikogiannakis G, Mathison CJN. J Am Chem Soc. 2005;127:8872–8888. doi: 10.1021/ja0509984. [DOI] [PubMed] [Google Scholar]
- xiii.a) Alexander JB, La DS, Cefalo DR, Hoveyda AH, Schrock RR. J Am Chem Soc. 1998;120:4041–4042. [Google Scholar]; b) La DS, Alexander JB, Cefalo DR, Graf DD, Hoveyda AH, Schrock RR. J Am Chem Soc. 1998;120:9720–9721. [Google Scholar]
- xiv.Burke SD, Müller N, Beaudry CM. Org Lett. 1999;1:1827–1829. doi: 10.1021/ol9910971. [DOI] [PubMed] [Google Scholar]
- xv.a) Seiders TJ, Ward DW, Grubbs RH. Org Lett. 2001;3:3225–3228. doi: 10.1021/ol0165692. [DOI] [PubMed] [Google Scholar]; b) Van Veldhuizen JJ, Gillingham DG, Garber SB, Kataoka O, Hoveyda AH. J Am Chem Soc. 2003;125:12502–12508. doi: 10.1021/ja0302228. [DOI] [PubMed] [Google Scholar]; c) Funk TW, Berlin JM, Grubbs RH. J Am Chem Soc. 2006;128:1840–1846. doi: 10.1021/ja055994d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xvi.Sattely ES, Cortez GA, Moebius DC, Schrock RR, Hoveyda AH. J Am Chem Soc. 2005;127:8526–8533. doi: 10.1021/ja051330s. [DOI] [PubMed] [Google Scholar]
- xvii.For representative examples where the presence of a Lewis basic amine leads to inhibition of Ru-catalyzed olefin metathesis, see: Lee KL, Goh JB, Martin SF. Tetrahedron Lett. 2001;42:1635–1638.Wipf P, Rector SR, Takahashi H. J Am Chem Soc. 2002;124:14848–14849. doi: 10.1021/ja028603t.Wipf P, Spencer SR. J Am Chem Soc. 2005;127:225–235. doi: 10.1021/ja044280k.Cortez GA, Schrock RR, Hoveyda AH. Angew Chem Int Ed. 2007;46:4534–4538. doi: 10.1002/anie.200605130.
- xviii.Weatherhead GS, Cortez GA, Schrock RR, Hoveyda AH. Proc Nat Acad Sci. 2004;101:5805–5809. doi: 10.1073/pnas.0307589101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xix.Fors KS, Gage JR, Heier RF, Kelly RC, Perrault WR, Wicnienski N. J Org Chem. 1998;63:7348–7356. doi: 10.1021/jo9809229. [DOI] [PubMed] [Google Scholar]
- xx.Cefalo DR, Kiely AF, Wuchrer M, Jamieson JY, Schrock RR, Hoveyda AH. J Am Chem Soc. 2001;123:3139–3140. [Google Scholar]
- xxi.For representative examples of catalytic enantioselective additions of C-based nucleophiles to ketones, see: Dosa PI, Fu GC. J Am Chem Soc. 1998;120:445–446.Tian SK, Deng L. J Am Chem Soc. 2001;123:6195–6196. doi: 10.1021/ja010690m.García GC, LaRochelle LK, Walsh PJ. J Am Chem Soc. 2002;124:10970–10971. doi: 10.1021/ja026568k.Wada R, Oisaki K, Kanai M, Shibasaki M. J Am Chem Soc. 2004;126:8910–8911. doi: 10.1021/ja047200l.Fuerst DE, Jacobsen EN. J Am Chem Soc. 2005;127:8964–8965. doi: 10.1021/ja052511x.Shintani R, Inoue M, Hayashi T. Angew Chem Int Ed. 2006;45:3353–3356. doi: 10.1002/anie.200600392.Zhao D, Oisaki K, Kanai M, Shibasaki M. J Am Chem Soc. 2006;128:14440–14441. doi: 10.1021/ja0652565.Komanduri V, Krische MJ. J Am Chem Soc. 2006;128:16448–16449. doi: 10.1021/ja0673027.Siewert J, Sandmann R, von Zezschwitz P. Angew Chem Int Ed. 2007;46:7122–7124. doi: 10.1002/anie.200701087.Friel DK, Snapper ML, Hoveyda AH. J Am Chem Soc. 2008;130:9942–9951. doi: 10.1021/ja802935w.
- xxii.a) Malcolmson SJ, Meek SJ, Sattely ES, Schrock RR, Hoveyda AH. Nature. 2008;456:933–937. doi: 10.1038/nature07594. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sattely ES, Meek SJ, Malcolmson SJ, Schrock RR, Hoveyda AH. J Am Chem Soc. 2009;131:943–953. doi: 10.1021/ja8084934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxiii.a) Christophers J, Baro A, editors. Quaternary Stereocenters: Challenges and Solutions for Organic Synthesis. Wiley-VCH; Weinheim: 2006. [Google Scholar]; b) Denissova I, Barriault L. Tetrahedron. 2003;59:10105–10146. [Google Scholar]
- xxiv.a) Solans-Monfort X, Clot E, Copéret C, Eisenstein O. J Am Chem Soc. 2005;127:14015–14025. doi: 10.1021/ja053528i. [DOI] [PubMed] [Google Scholar]; b) Poater A, Solans-Monfort X, Clot E, Copéret C, Eisenstein O. J Am Chem Soc. 2007;129:8207–8216. doi: 10.1021/ja070625y. [DOI] [PubMed] [Google Scholar]
- xxv.Formation of an all-carbon quaternary stereogenic center through enantioselective olefin metathesis is relatively rare. For the first example of such a transformation, see: Lee AL, Malcolmson SJ, Puglisi A, Schrock RR, Hoveyda AH. J Am Chem Soc. 2006;128:5153–5157. doi: 10.1021/ja058428r.
- xxvi.Gillingham DG, Hoveyda AH. Angew Chem Int Ed. 2007;46:3860–3864. doi: 10.1002/anie.200700501. [DOI] [PubMed] [Google Scholar]
- xxvii.Van Veldhuizen JJ, Campbell JE, Giudici RE, Hoveyda AH. J Am Chem Soc. 2005;127:6877–6882. doi: 10.1021/ja050179j. [DOI] [PubMed] [Google Scholar]
- xxviii.In contrast to reactions with stereogenic-at-Ru catalysts with bidentate chiral ligands, transformations of oxabicyclic alkenes in the presence of achiral Ru-carbenes bearing monodentate NHCs lead to formation of substantial amounts of oligomerization; the desired ROCM products are isolated in only 12–30% yield (unpublished results of A. R. Zhugralin, M. Yu, A. H. Hoveyda). The mechanistic details for the difference in efficiencies of these two classes of Ru-based carbenes in promoting ROCM reactions will be disclosed shortly in a separate account.
- xxix.Chiral bidentate NHCs originally designed for Ru-catalyzed olefin metathesis have been used to promote other classes of transformations. See: (Cu-catalyzed enantioselective conjugate additions) Lee K-s, Brown MK, Hoveyda AH. J Am Chem Soc. 2006;128:7182–7184. doi: 10.1021/ja062061o.Brown MK, May TL, Hoveyda AH. Angew Chem Int Ed. 2007;46:1097–1100. doi: 10.1002/anie.200604511.May TL, Brown MK, Baxter CA, Hoveyda AH. Angew Chem Int Ed. 2008;47:7358–7362. doi: 10.1002/anie.200802910.(Cu-catalyzed enantioselective allylic alkylations): Kacprzynski MA, May TL, Kazane SA, Hoveyda AH. Angew Chem Int Ed. 2007;46:4554–4558. doi: 10.1002/anie.200700841.(Cu-catalyzed hydroboration reactions): Lee Y, Hoveyda AH. J Am Chem Soc. 2009;131:3160–3161. doi: 10.1021/ja809382c.
- xxx.Bornand M, Chen P. Angew Chem Int Ed. 2005;44:7909–7911. doi: 10.1002/anie.200502606. [DOI] [PubMed] [Google Scholar]
- xxxi.For other chiral, stereogenic-at-Ru, catalysts that have been used in olefin metathesis but only in the racemic form, see: Fürstner A, Liebl M, Lehmann CW, Picquet M, Kunz R, Bruneau C, Touchard D, Dixneuf PH. Chem Eur J. 2000;6:1847–1857. doi: 10.1002/(sici)1521-3765(20000515)6:10<1847::aid-chem1847>3.0.co;2-1.Conrad JC, Parnas HH, Snelgrove JL, Fogg DE. J Am Chem Soc. 2005;127:11882–11883. doi: 10.1021/ja042736s.
- xxxii.Lee YJ, Schrock RR, Hoveyda AH. J Am Chem Soc. 2009;131:10652–10661. doi: 10.1021/ja904098h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxiii.For Ru-catalyzed enyne RCM reactions, see: Mori M, Sakakibara N, Kinoshita A. J Org Chem. 1998;63:6082–6083. doi: 10.1021/jo980896e.Dieltiens N, Moonen K, Stevens CV. Chem Eur J. 2007;13:203–214. doi: 10.1002/chem.200600789.Diver ST, Giessert AJ. Chem Rev. 2004;104:1317–1382. doi: 10.1021/cr020009e.Lloyd-Jones GC, Margue RG, de Vries JG. Angew Chem Int Ed. 2005;44:7442–7447. doi: 10.1002/anie.200502243.
- xxxiv.Ibrahem I, Yu M, Schrock RR, Hoveyda AH. J Am Chem Soc. 2009;131:3844–3845. doi: 10.1021/ja900097n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxv.a) Gillingham DG, Kataoka O, Garber SB, Hoveyda AH. J Am Chem Soc. 2004;126:12288–12290. doi: 10.1021/ja0458672. [DOI] [PubMed] [Google Scholar]; b) Cortez GA, Baxter CA, Schrock RR, Hoveyda AH. Org Lett. 2007;9:2871–2874. doi: 10.1021/ol071008h. [DOI] [PubMed] [Google Scholar]
- xxxvi.Flook MM, Jiang AJ, Schrock RR, Müller P, Hoveyda AH. J Am Chem Soc. 2009;131:7962–7963. doi: 10.1021/ja902738u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxvii.Wolfe JP, Wagaw S, Marcoux JF, Buchwald SL. Acc Chem Res. 1998;31:805–818. [Google Scholar]
- xxxviii.Shen Q, Hartwig JF. J Am Chem Soc. 2006;128:10028–10030. doi: 10.1021/ja064005t. [DOI] [PubMed] [Google Scholar]; b) Shen Q, Ogata T, Hartwig JF. J Am Chem Soc. 2008;130:6586–6596. doi: 10.1021/ja077074w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xxxix.Lee Y, Akiyama K, Gillingham DG, Brown MK, Hoveyda AH. J Am Chem Soc. 2008;130:446–447. doi: 10.1021/ja0782192. [DOI] [PubMed] [Google Scholar]
- xl.Meek SJ, Hoveyda AH. unpublished results [Google Scholar]
- xli.For another notable example of complications arising from undesirable epimerization promoted by P-containing Ru-based catalysts (vs 45), see: Yee NK, Farina V, Houpis IN, Haddad N, Frutos RP, Gallou F, Wang X-j, Wei X, Simpson RD, Feng X, Fuchs V, Xu Y, Tan J, Zhang L, Xu J, Smith-Keenan LL, Vitous J, Ridges MD, Spinelli EM, Johnson M, Donsbach K, Nicola T, Brenner M, Winter E, Kreye P, Samstag W. J Org Chem. 2006;71:7133–7145. doi: 10.1021/jo060285j.
- xlii.Murphy KE, Hoveyda AH. unpublished results [Google Scholar]