

Abstract
A versatile reaction cascade triggered by Rh(II)-catalyzed diazo decomposition followed by a vinylogous N-H insertion/Lewis acid catalyzed Mannich addition that produces highly substituted 1,2,3,6-tetrahydropyridazines in up to 97% ee with high yield and diastereocontrol has been developed.
Keywords: Asymmetric Catalysis, Tetrahydropyridazines, Enantioselective, Vinylogous N-H Insertion, Cycloaddition, Rhodium
Although rare, formal [3 + 3]-cycloaddition reactions are known to occur through sequential reactions of an activated substrate with the nucleophilic site of a dipole then with its electrophilic site to form the cycloaddition product.[1] Systems that follow this pathway include reactions of catalytically-generated substrates with nitrones[2,3] and azomethine imines;[2b,4] enantiocontrol, examples of which have only recently been published, occurs through the intervention of a chiral catalyst.[5] We envisioned that readily accessible hydrazones could be employed in place of azomethine imines to form tetrahydropyridazines[6] with enoldiazoacetates 1 (Scheme 1). Nucleophilic addition of hydrazone 3 to metal carbene intermediate 2 could occur either at the metal carbene centre (2 → TS I) or at the vinylogous position (2 → TS II) to give 6 or 7, respectively, after 1,2-hydrogen transfer between the two nitrogen atoms and Mannich addition (Path A and Path B). The 1,2-hydrogen transfer is formally a N-H insertion reaction but, in contrast to the direct process (TS I → 4),[7] the 1,4-hydrogen transfer (TS II → 5) from nitrogen to carbon is a vinylogous variant for which there has been no previous example of stereocontrol, although non-asymmetric vinylogous O-H insertion reactions and an asymmetric vinylogous C-H functionalization of several vinyldiazoacetates have recently been reported.[8] We now report a highly selective catalytic process involving enoldiazoacetates in combination with aldehyde-derived hydrazones that occur with high enantioselectivity and catalyst-controlled diastereoselectivity.
Scheme 1.

Competitive pathways for [3 + 3]-cycloaddition reactions of arylhydrazones.
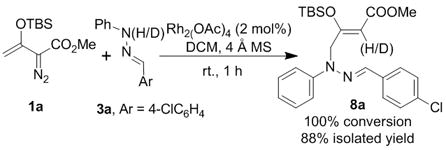
To determine the feasibility of the formal [3 + 3]-cycloaddition with hydrazones we treated 3-(tert-butyldimethylsiloxy)-2-diazo-3-butenoate 1a and the phenylhydrazone of 4-chlorobenzaldehyde (3a) with a catalytic amount of dirhodium tetraacetate at room temperature in dichloromethane. Complete conversion occurred within one hour but, instead of forming a tetrahydropyridazines, the product from vinylogous N-H insertion (8a) was formed exclusively as the terminal product (Eq 1). The (Z) geometry of the newly formed trisubstituted C=C bond in 8a was confirmed by single crystal X-ray diffraction.[9] That the reaction process was indeed a vinylogous N-H insertion was established by performing the reaction with deuterium-labeled 3a (exchange of the N-H proton) from which deuterium was found to reside exclusively on the vinyl carbon alpha to the carboxylate ester.[10] Instead of undergoing a 1,2-proton transfer, as envisioned in Scheme 1, the hydrogen on nitrogen replaced the dirhodium catalyst on the original carbene carbon. Recognizing the advantages of this methodology, we investigated this transformation for enantiocontrol in the vinylogous N-H insertion reaction and, since 8 is suitable for intramolecular iminium ion ring closure,[11] Lewis acid catalysis was probed for subsequent diastereoselective cyclization to tetrahydropyridazines.
 |
(1) |
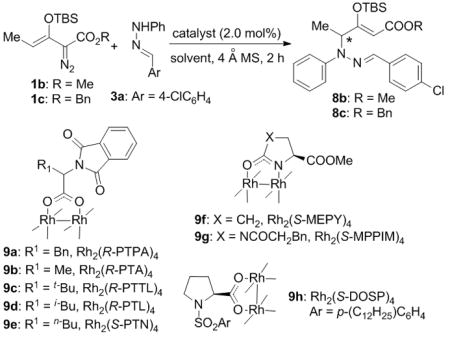
To address enantiocontrol in the vinylogous N-H insertion reaction, 3-(tert-butyldimethylsiloxy)-2-diazo-3-pentenoates 1b (R1 = Me, R2 = Me) and 1c (R1 = Me, R2 = Bn) were treated with hydrazone 3a in the presence of various chiral dirhodium catalysts, and the results from this investigation are summarized in Table 1. Except for the Rh2(R-PTTL)4 (entry 4), Hashimoto’s phthalimide-amino acid-based chiral dirhodium carboxylate catalysts[12] showed higher reactivity and enantioselectivity compared to Davies’ Rh2(S-DOSP)4 catalyst[13] (entries 1 and 2 vs entry 7), and dirhodium carboxamidate catalysts[14] were ineffective (entries 5 and 6). Adopting benzyl enoldiazoacetate 1c instead of the methyl ester 1b resulted in an improvement in enantioselectivity (entry 8 vs entry 3). Using Rh2(R-PTA)4 with reaction solvents at temperatures down to −40 °C provided additional improvements in the control of enantioselectivity, optimally giving 8c in up to 82% ee with high yield in toluene (entries 9–15). However, the highest yield and selectivity occurred with the Rh2(R-PTL)4 catalyst (R1 = iBu) which effected 100% conversion within 2 h to 8c which was isolated in 82% yield with 92% ee (entry 16). The (Z) geometry of the newly formed C=C bond in 8 was further confirmed by a 1D-nOe study.[9]
Table 1.
Optimization for the enantioselective vinylogous N-H insertion of a phenylhydrazone.[a]
| ||||||
|---|---|---|---|---|---|---|
| Entry | 1 | Catalyst (9) | T (°C) | Solvent | yield (%)[b] | ee (%)[c] |
| 1 | 1b | Rh2(S-PTPA)4 (9a) | 25 | DCM | 78 | 38 |
| 2 | 1b | Rh2(R-PTA)4 (9b) | 25 | DCM | 71 | 45 |
| 3 | 1b | Rh2(R-PTA)4 (9b) | 0 | DCM | 68 | 51 |
| 4 | 1b | Rh2(S-PTTL)4 (9c) | 25 | DCM | <5 | - |
| 5 | 1b | Rh2(S-MEPY)4 (9f) | 25 | DCM | <5 | - |
| 6 | 1b | Rh2(S-MPPIM)4 (9g) | 25 | DCM | <5 | - |
| 7 | 1b | Rh2(S-DOSP)4 (9h) | 0 | DCM | 65 | −35 |
| 8 | 1c | Rh2(R-PTA)4 (9b) | 0 | DCM | 91 | 56 |
| 9 | 1c | Rh2(R-PTA)4 (9b) | 0 | Toluene | 85 | 69 |
| 10 | 1c | Rh2(R-PTA)4 (9b) | −25 | Toluene | 78 | 74 |
| 11 | 1c | Rh2(R-PTA)4 (9b) | −25 | ClC6H5 | 72 | 69 |
| 12 | 1c | Rh2(R-PTA)4 (9b) | −25 | FC6H5 | 75 | 68 |
| 13 | 1c | Rh2(R-PTA)4 (9b) | −25 | CF3C6H5 | 71 | 70 |
| 14 | 1c | Rh2(R-PTA)4 (9b) | −40 | Toluene | 77 | 82 |
| 15 | 1c | Rh2(R-PTA)4 (9b) | −40 | TBME | 75 | 80 |
| 16 | 1c | Rh2(R-PTL)4 (9d) | −40 | Toluene | 82 | 92 |
| 17 | 1c | Rh2(S-PTN)4 (9e) | −40 | Toluene | 79 | −83 |
Reactions were carried out over 2 h on a 0.10 mmol scale: 1 (0.12 mmol), 3a (0.10 mmol), 4 Å MS (50 mg), in 1.0 mL solvent with 2.0 mol% catalyst at the stated temperature.
Isolated yield. Except for entries 4–6, reactions proceeded to 100% completion.
Determined by HPLC analysis with chiral columns.
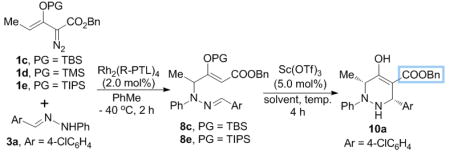
Having established the optimized conditions for the vinylogous N-H insertion, we investigated the potential of 8 to undergo Mannich addition to complete the tetrahydropyridazine synthesis. Thermal conditions (100 °C in toluene for 3 h) did not provide any evidence of ring-closing, and 8 was recovered intact. Various achiral Lewis acids were examined for cyclization of 8c,[15] and 5 mol% of Sc(OTf)3 was found to superior to other acid catalysts in smoothly promoting the formation of 1,2,3,6-tetrahydropyridazine 10a and in maintaining the high enantiomeric excess of the reactant. However, product diastereoselectivity was moderate (Table 2, entry 1, cis:trans = 72:28). In efforts to influence diastereocontrol by changing the size of OTBS attachment various organosilyl protective groups were used on the reactant enoldiazoacetates.[16] The labile TMS derivative 1d underwent vinylogous N-H insertion but gave the hydrolyzed product derived from 8 that, although proceeding to 10a,[10] did so with complete racemization (entry 2). With the TIPS derivative 1e, however, 10a was formed with 96% ee under same conditions (entry 3), although diastereoselectivity was only modestly improved to 76:24. Further investigation of the conditions for optimization found that solvent plays an important role in the Mannich addition process; the reaction performed in toluene was much slower than that which was run in dichloromethane and occurred with much lower diastereoselectivity, but when acetonitrile was used as reaction medium diastereoselectivity improved to 82:12 (entry 9). Surprisingly, the reaction performed in dichloromethane at 50 °C in a closed container reversed diastereoselectivity while retaining high enantioselectivity (entry 6). The major cis-diastereoisomer was confirmed by single-crystal X-ray diffraction analysis.[17]
Table 2.
Optimization of Lewis acid catalyzed Mannich addition for the synthesis of 1,2,3,6-tetrahydropyridazines.[a]
| ||||||
|---|---|---|---|---|---|---|
| Entry | 1 | Solvent | T (°C) | dr (10a)[b] cis:trans | conversion 8→10 (%)[c] | ee (%)[d] cis/trans-10a |
| 1 | 1c | DCM | 25 | 72:28 | >95 | 92/- |
| 2 | 1d | DCM | 25 | 70:30 | >95 | <5 |
| 3 | 1e | DCM | 25 | 76:24 | >95 | 96/- |
| 4[e] | 1e | Toluene | 25 | 60:40 | 65 | 95.5/- |
| 5 | 1e | DCM | 0 | 76:24 | >95 | 95.5/- |
| 6 | 1e | DCM | 50 | 23:77 | >95 | -/93 |
| 7 | 1e | Acetonitrile | 25 | 80:20 | >95 | 96/93 |
| 8 | 1e | Acetonitrile | 50 | 50:50 | >95 | 95/93 |
| 9[f] | 1e | Acetonitrile | 0 | 82:18 | >95 | 96/93 |
Reactions were carried out on 0.10 mol scale: 1 (0.12 mmol), 3a (0.10 mmol), and 4 Å MS (50 mg) in 1.0 mL toluene with 2.0 mol% Rh2(R-PTL)4 at −40 °C for 2 h. Then the reaction solution was passed through a short flash column chromatography (Φ0.5 cm X 10 mm), solvent was evaporated, and the solvent for the Mannich addition was added with 5.0 mol% Sc(OTf)3 at the indicated temperature.
Determined from the 1H NMR spectra of the reaction mixtures.
Determined from the 1H NMR spectra of the reaction mixtures based on limiting reagent 8.
Determined by HPLC analysis using chiral columns, see Supporting Information.
The reaction mixture from the first step was used for the second step without removal of solvent.
The reaction was run overnight at 0 °C.
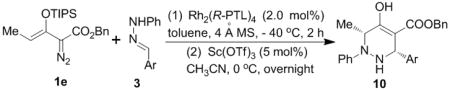
The generality of this enantioselective cascade reaction was further investigated using these optimum conditions, and the results of this investigation are given in Table 3. Product yields were high, and 1,2,3,6-tetrahydropyridazines 10 were the sole isolated reaction products. The position of the chloro substituent on the aryl group did not affect the efficiency of the reaction, and these substrates underwent the two-step process with high stereocontrol (entries 1–3). The electronic nature of the substituents had little influence on reactivity and selectivity (entries 4–10), except in cases where reactant solubility required that the vinylogous N-H insertion reaction be conducted at higher temperature (entries 5, 6, and 11–13). Reactions with sterically bulky mesityl and anthranyl substrates (entries 12 and 13) gave mixtures of the corresponding tetrahydropyridazines 10l or 10m along with hydrolyzed racemic ketone from the vinylogous N-H insertion reaction.[10] However, these sterically hindered substrates produced only one tetrahydropyridazine diastereoisomer with enantiomeric excesses up to 97%.
Table 3.
Enantioselective cascade sequences synthesis of 1,2,3,6-tetrahydropyridazines from 1e and hydrazones [a]
| |||||
|---|---|---|---|---|---|
| Entry | Ar (3) | 10 | dr[b] | yield (%)[c] | ee (%)[d] cis/trans |
| 1 | 4-ClC6H4 (3a) | 10a | 82:18 | 77 | 96/93 |
| 2 | 3-ClC6H4 (3b) | 10b | 84:16 | 82 | 95/93 |
| 3[e] | 2-ClC6H4 (3c) | 10c | 81:19 | 73 | 97/95 |
| 4 | 4-MeOC6H4 (3d) | 10d | 76:24 | 91 | 92/91 |
| 5[e,f] | 4-NO2C6H4 (3e) | 10e | 81:19 | 90 | 92/88 |
| 6[f] | 4-BrC6H4 (3f) | 10f | 95:5 | 72 | 90/78 |
| 7 | 4-FC6H4 (3g) | 10g | 86:14 | 80 | 91/91 |
| 8 | 4-MeC6H4 (3h) | 10h | 83:17 | 85 | 90/93 |
| 9 | 2-furyl (3i) | 10i | 79:21 | 77 | 78/77 |
| 10 | 4-PhC6H4 (3j) | 10j | >95:5 | 70 | 91/- |
| 11[f] | 2-CF3C6H4 (3k) | 10k | >95:5 | 67 | 87/- |
| 12[f] | 2,4,6-Me3C6H2 (3l) | 10l | >95:5 | 35 (57)[g] | 89/- |
| 13[e,f] | 9-anthryl (3m) | 10m | >95:5 | 29 (60)[g] | 97/- |
See experimental section.
Determined from the 1H NMR spectra of the reaction mixtures.
Isolated yield of 10 (cis + trans) based on limiting reagent 3.
Determined by HPLC analyses with chiral columns.
The second step was performed at room temperature.
The first step was performed at −20 °C.
The number in parenthesis is the yield of the hydrozed product from vinylogous N-H insertion.
Enoldiazoacetate 1 with R = Me at the vinylogous position is optimum for enantioselective vinoylogous reactions with hydrazones. However, when enoldiazoacetates with larger substituents at the vinylogous position (R = Et, Ph, Bn) were applied, enantioselectivity decreased with R = Et (Scheme 2) and vinylogous N-H insertion was effectively inhibited when R = Ph or Bn. Davies has reported that vinylogous carbenoid reactions from O-H insertion reactions using dirhodium(II) catalysts are highly restricted but can be overcome in selected cases with the use of more electron deficient silver(I), molybdenum, or diruthenium(I) catalysts.[8] The data of Scheme 2 suggests that steric effects are primarily responsible for the reactivity and selectivity that is observed for reactions with hydrazones.
Scheme 2.
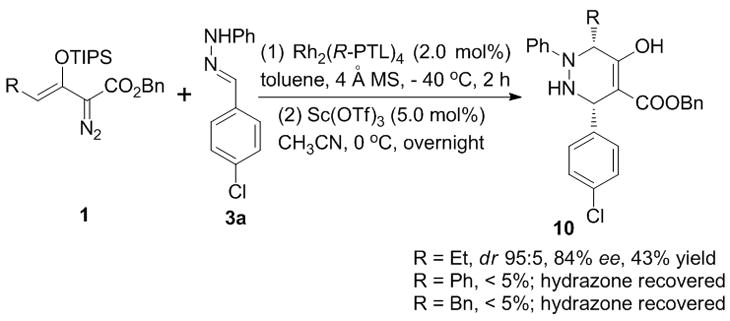
Effect on enantioselective cascade sequences with enoldiazoacetates having bulky substituents at the vinylogous position.
In summary, we have developed a cascade transformation that enables the efficient preparation of highly substituted 1,2,3,6-tetrahydropyridazines[18] starting from enoldiazoacetates and hydrazones in good overall yields, high diastereoselectivities, and excellent enantioselectivities that are controlled by catalysts and conditions. The sequence of reactions is triggered by Rh(II)-catalyzed dinitrogen extrusion followed by asymmetric vinylogous N-H insertion into hydrazones. Subsequent Lewis acid promoted Mannich addition of 8 smoothly produces 1,2,3,6-tetrahydropyridazines 10 (Scheme 3) with high diastereocontrol. To the best of our knowledge, this is the first example of highly enantioselective vinylogous N-H insertion. Further expansion of vinylogous reactivity with enoldiazoacetates applied broadly are being pursued.
Scheme 3.
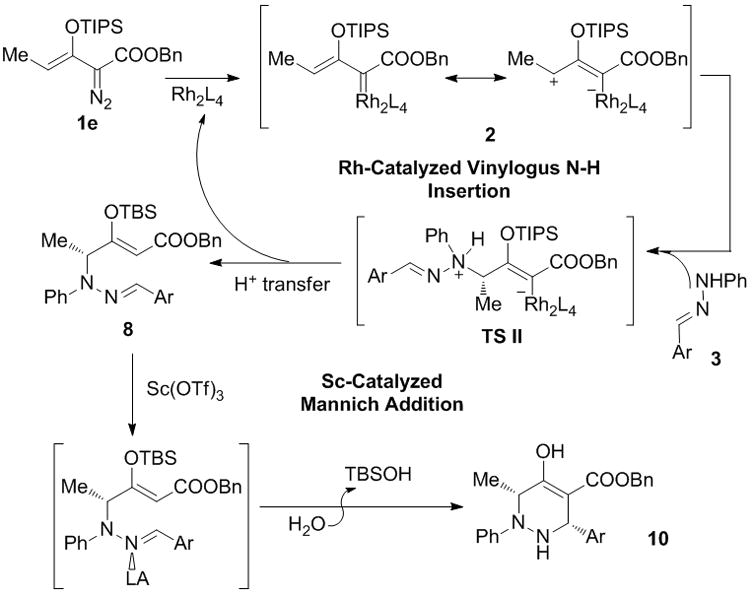
Tandem catalysis of the [3 + 3]-cycloaddition reaction from vinylogous N-H insertion/Mannich addition.
Experimental Section
To an oven-dried flask containing a magnetic stirring bar, hydrazone 3 (0.1 mmol), 4 Å molecular sieves (50 mg), and Rh2(R-PTL)4 (2.0 mol%) in toluene (0.5 mL), was added enoldiazoacetate 1e (0.12 mmol) in toluene (0.5 mL) over 1 h period via a syringe pump at the indicated temperature (either −40 or −20 °C). The reaction mixture was stirred for another hour under these conditions, then passed through a short flash column of silica gel (Φ0.5 cm X 10 cm) and, after removal of the solvent under reduced pressure, acetonitrile (2.0 mL) was added. This solution was transferred to a reaction tube containing a magnetic stirring bar, and the temperature of the solution was decreased to 0 °C (or to room temperature, as indicated), followed by adding Sc(OTf)3 (5.0 mol%). The reaction mixture was stirred for another 4 h, and then subjected to 1H NMR spectroscopic analysis after solvent removal to determine product diastereoselectivity. The crude reaction mixture was purified by column chromatography on silica gel (eluent hexanes:EtOAc = 100:0 to 90:10) to give the pure tetrahydropyridazines 10 in moderate to high yield with high to excellent enantioselectivity.
Supplementary Material
Acknowledgments
Support for this research to MPD from the National Institutes of Health (GM 46503) is gratefully acknowledged.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For reviews of formal [3 + 3]-cycloaddition reactions, see: Hsung RP, Wei LL, Sklenicka HM, Shen HC, McLaughlin MJ, Zehnder LO. Org Biomol Chem. 2005;3:1349.Buchanan GS, Feltenberger JB, Hsung RP. Curr Org Synth. 2010;7:363. doi: 10.2174/157017910791414490.For enantioselective organocatalytic formal [3+3]-cycloadditions see: Sklenicka HM, Hsung RP, McLaughlin MJ, Wie LL, Gerasyuto AI, Brennessel WB. J Am Chem Soc. 2002;124:10435. doi: 10.1021/ja020698b.Hong BC, Wu MF, Tseng HC, Liao JH. Org Lett. 2006;8:2217. doi: 10.1021/ol060486+.Chan A, Scheidt KA. J Am Chem Soc. 2007;129:5334. doi: 10.1021/ja0709167.
- 2.a) Liu F, Qian D, Li L, Zhao X, Zhang J. Angew Chem. 2010;122:6819. doi: 10.1002/anie.201003136. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:6669. [Google Scholar]; b) Shintani R, Hayashi T. J Am Chem Soc. 2006;128:6330. doi: 10.1021/ja061662c. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Xu X, Zavalij PY, Doyle MP. J Am Chem Soc. 2011;133:16402. doi: 10.1021/ja207664r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Shapiro ND, Shi Y, Toste FD. J Am Chem Soc. 2009;131:11654. doi: 10.1021/ja903863b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Perreault C, Goudreau SR, Ee Zimmer L, Charette AB. Org Lett. 2008;10:689. doi: 10.1021/ol702414e. [DOI] [PubMed] [Google Scholar]; c) Chan A, Scheidt KA. J Am Chem Soc. 2007;129:5334. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Hayashi Y, Gotoh H, Masui R, Ishikawa H. Angew Chem. 2008;120:4076. doi: 10.1002/anie.200800662. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2008;47:4012. [Google Scholar]; b) Kanao K, Miyake Y, Nishibayashi Y. Organometal. 2010;29:2126. [Google Scholar]; c) Al-Harrasi A, Reiβig H-U. Angew Chem. 2005;117:6383. [Google Scholar]; Angew Chem Int Ed. 2005;44:6227. [Google Scholar]; d) Helms M, Schade W, Pulz R, Watanabe T, Al-Harrasi A, Fisera L, Hlobilová I, Zahn G, Reiβig HU. Eur J Org Chem. 2005:1003. [Google Scholar]; e) Grover HK, Lebold TP, Kerr MA. Org Lett. 2011;13:220. doi: 10.1021/ol102627e. [DOI] [PubMed] [Google Scholar]; f) Gerasyuto AI, Hsung RP, Sydorenko N, Slafer B. J Org Chem. 2005;70:4248. doi: 10.1021/jo050171s. [DOI] [PubMed] [Google Scholar]
- 6.With the exception of [3 + 3]-cycloaddition reactions, syntheses of 1,2,3,6-tetrahydropyridazine are rare: Xu S, Chen R, Qin Z, Wu G, He Z. Org Lett. 2012;14:996. doi: 10.1021/ol2032569.Na R, Jing C, Xu Q, Jiang H, Wu X, Shi J, Zhong J, Wang M, Benitez D, Tkatchouk E, Goddard WA, Guo H, Kwon O. J Am Chem Soc. 2011;133:13337. doi: 10.1021/ja200231v.Shen L, Sun L, Ye S. J Am Chem Soc. 2011;133:15894. doi: 10.1021/ja206819y.Zhang Q, Yang L, Tong X. J Am Chem Soc. 2010;132:2550. doi: 10.1021/ja100432m.Oelke AJ, France DJ, Hofmann T, Wuitschik G, Ley SV. Angew Chem. 2010;122:6275. doi: 10.1002/anie.201002880.Angew Chem Int Ed. 2010;49:6139.Nair V, Mathew SC, Biju AT, Suresh E. Angew Chem. 2007;119:2116. doi: 10.1002/anie.200604025.Angew Chem Int Ed. 2007;46:2070.Jensen HH, Lyngbye L, Jensen A, Bols M. Chem Eur J. 2002;8:1218. doi: 10.1002/1521-3765(20020301)8:5<1218::aid-chem1218>3.0.co;2-x.
- 7.Enantioselective heteroatom-carbene insertion reactions: Zhu SF, Xu B, Wang GP, Zhou QL. J Am Chem Soc. 2012;134:346. doi: 10.1021/ja2084493.Liu B, Zhu SF, Zhang W, Chen C, Zhou QL. J Am Chem Soc. 2007;129:5834. doi: 10.1021/ja0711765.Moody CJ. Angew Chem. 2007;119:9308.Angew Chem Int Ed. 2007;46:9148.Xu B, Zhu S, Xie X, Shen J, Zhou Q. Angew Chem. 2011;123:11685;. doi: 10.1002/anie.201105485.Angew Chem Int Ed. 2011;50:11483.Lee EC, Fu GC. J Am Chem Soc. 2007;129:12066. doi: 10.1021/ja074483j.
- 8.Non-asymmetric vinylogous O-H insertion: Hansen JH, Davies HML. Chem Sci. 2011;2:457.Davies HML, Yokota Y. Tetrahedron Lett. 2000;41:4851.Sevryugina Y, Weaver B, Hansen J, Thompson J, Davies HML, Petrukhina MA. Organometallics. 2008;27:1750.Asymmetric vinylogous C-H functionalization: Lian Yajing, Davies Huw ML. Org Lett. 2012;14:1934. doi: 10.1021/ol300632p.
- 9.CCDC 881732 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
- 10.See Supporting Information.
- 11.Selected examples of Mannich addition with silicon enolates Akiyama T, Itoh J, Yokota K, Fuchibe K. Angew Chem. 2004;116:1592. doi: 10.1002/anie.200353240.Angew Chem Int Ed. 2004;43:1566.Sugiura M, Kobayashi S. Angew Chem. 2005;117:5306. doi: 10.1002/anie.200500691.Angew Chem Int Ed. 2005;44:5176.Taylor MS, Jacobsen EN. Angew Chem. 2006;118:1550. doi: 10.1002/anie.200503132.Angew Chem Int Ed. 2006;45:1520.Marques MMB. Angew Chem. 2006;118:356.Angew Chem Int Ed. 2006;45:348.
- 12.a) Hashimoto S, Watanabe N, Sato T, Shiro M, Ikegami S. Tetrahedron lett. 1993;34:5109. [Google Scholar]; b) Tsutsui H, Abe T, Nakamura S, Anada M, Hashimoto S. Chem Pharm Bull. 2005;53:1366. doi: 10.1248/cpb.53.1366. [DOI] [PubMed] [Google Scholar]; c) Kitagaki S, Anada M, Kataoka O, Matsuno K, Umeda C, Watanabe N, Hashimoto S. J Am Chem Soc. 1999;121:1417. [Google Scholar]
- 13.a) Wu J, Becerril J, Lian Y, Davies HML, Porco JA, Jr, Panek JS. Angew Chem. 2011;123:6060. doi: 10.1002/anie.201101366. [DOI] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:5938. [Google Scholar]; b) Reddy RP, Lee GH, Davies HML. Org Lett. 2006;8:3437. doi: 10.1021/ol060893l. [DOI] [PubMed] [Google Scholar]; c) Davies HML, Bruzinski PR, Lake DH, Kong N, Fall MJ. J Am Chem Soc. 1996;118:6897. [Google Scholar]; d) Lian Y, Hardcastle KI, Davies HML. Angew Chem. 2011;123:9542. doi: 10.1002/anie.201103568. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2011;50:9370. [Google Scholar]
- 14.Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds. Wiley; New York, NY: 1998. [Google Scholar]
- 15.Use of In(OTf)3 or Cu(OTf)2 gave similar results but required longer reaction times.
- 16.The Z:E ratios of 1b–1e was determined by 1H NMR spectroscopy: 1b = 90:10, 1c = 89:11, 1d = 96:4, 1c = 82:18. See Supporting Information for details. Davies HML, Peng ZQ, Houser JH. Tetrahedron Lett. 1994;35:8939.Ueda Y, Roberge G, Vinet V. Can J Chem. 1984;62:2936.Davies HML, Ahmed G, Churchill MR. J Am Chem Soc. 1996;118:10774.Kundu K, Doyle MP. Tetrahedron: Asymmetry. 2006;17:574.Xu X, Zavalij PY, Hu W, Doyle MP. Angew Chem. 2011;123:11348. doi: 10.1002/anie.201105557.Angew Chem Int Ed. 2011;50:11152.
- 17.CCDC 881733 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif
- 18.1,2,5,6-Tetrahydropyridazines are structural components of potent novel inhibitors of human immunodeficiency virus (HIV) and hepatitis C virus (HCV): Watkins WJ, Ray AS, Chong LS. Curr Opin Drug Discovery Dev. 2010;13:441.Manlove A, Groziak MP. In: Prog Heterocycl Chem. Gordon G, John J, editors. Vol. 21. Elsevier Ltd; Oxford UK: 2009. pp. 375–414.Ruebsam F, et al. Bioorg Med Chem Lett. 2008;18:3616. doi: 10.1016/j.bmcl.2008.04.066.Stefania F, Stefano A, Maria LB, Laura DL, Frauke C, Rosaria G. J Heterocycl Chem. 2009;46:1420.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.