

Abstract

Hip to be square: Styrenes participate in [2+2] cycloadditions upon irradiation with visible light in the presence of an iridium(III) polypyridyl complex. In contrast to previous reports of visible light photoredox catalysis, the mechanism of this process involves photosensitization by energy transfer and not electron transfer.
Keywords: cycloaddition, cyclobutanes, energy transfer, photocatalysis, visible light
The prospect of conducting synthetically useful organic reactions with visible light has attracted significant recent attention from a number of research groups1 including our own.2 These efforts have focused on the utilization of the remarkable photoredox properties of ruthenium3 and iridium4 polypyridyl complexes, and a variety of photocatalytic transformations have been shown to occur upon irradiation with visible light in the presence of these complexes.5 The ability to use visible light rather than the ultraviolet light generally required for traditional organic photochemistry has numerous benefits,6 including: (1) the lower cost and decreased energy demand of visible light sources; (2) the ability to conduct photoreactions without specialized photoreactors or quartz glassware; and (3) the ability to selectively photoexcite the transition metal photocatalyst without inducing undesired radical reactions of photochemically sensitive organic functional groups.
Our research group has been particularly interested in visible light photocatalysis of cycloaddition reactions.2 In our investigations, we have been able to exploit both photoreduction and photooxidation reactions of Ru(bpy)32+ (1a) and related ruthenium(II) chromophores to design [2+2], [3+2], and [4+2] cycloaddition reactions involving radical anion and radical cation intermediates. Collectively, the diversity of products available using this strategy is quite broad; however, the nature of the photoinduced electron-transfer processes that generate the radical ion intermediates necessarily limits the scope of these reactions to either electron-deficient or electron-rich substrates that are amenable to one-electron redox processes. The success of photoredox methods reported from other labs has likewise been dictated by the redox properties of the organic substrates involved.
Recognizing that such electrochemical constraints will be important considerations in the design of any photoredox process, we wondered whether similar transformations could be initiated by energy transfer rather than by an electron-transfer mechanism. Although the quenching of Ru*(bpy)32+ by energy transfer has been documented with a number of organic compounds,7 to the best of our knowledge, there are only two carbon–carbon bond-forming reactions using transition metal photocatalysts that have been demonstrated to proceed via triplet sensitization of an organic substrate. The first is the Ru(bpy)32+-mediated norbornadiene-toquadricyclane valence isomerization studied by Kutal for solar energy storage applications.8 The second is the photocatalytic dimerization of anthracene reported by Castellano to be sensitized by the related ruthenium photocatalyst Ru(dmb)32+ (dmb=4,4'-dimethyl-2,2'-bipyridine).9 Thus, although the utility of UV-absorbing organic chromophores as triplet photosensitizers has been well established for decades, 10 synthetic applications of triplet sensitization with transition metal complexes that absorb in the visible range have not been extensively explored.
 |
(1) |
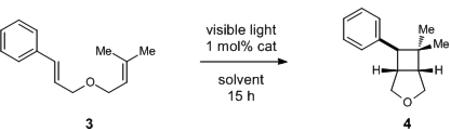
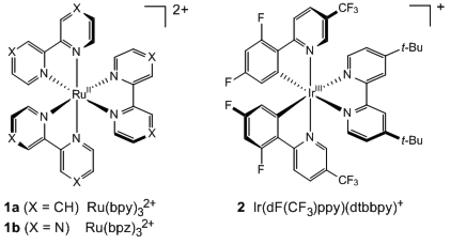
We initiated our investigation by exploring the [2+2] photocycloaddition of styrene 3, a substrate whose oxidation potential (+1.42 V vs SCE)11 has precluded its ability to participate in radical cation cycloadditions previously reported from our lab. Fluorinated iridium complex 2, first reported by Malliaras and Bernhard 12 and subsequently identified by Stephenson 13 as an optimal visible light photocatalyst for Kharasch-type radical additions, does not possess an excited state oxidation potential (+0.89 V)12 sufficient to generate the radical cation of 1. On the other hand, its reported emission maximum at 470 nm corresponds to an ET of 61 kcal/mol. In general, styrenes possess excited state triplet energies (ET) of ~60 kcal/mol. 14 Together, these data suggested that 2 might be capable of sensitizing triplet-state reactions of styrene 3 upon irradiation with visible light.
Indeed, irradiation of 3 with a 23 W compact fluorescent light bulb in the presence of 1 mol% of iridium complex 2•PF6 resulted in the formation of [2+2] cycloadduct 4 in a wide range of solvents (Table 1, entries 1–7). Consistent with a triplet sensitization mechanism, we observed no dramatic dependence on solvent polarity, whereas we have observed that radical cation processes benefit from the ability of polar solvents to stabilize the charged intermediates. Slightly faster conversion was observed in DMSO (entry 7), and upon optimization we were able to obtain the cycloadduct in good yield at lower reaction concentrations (entry 8). Also consistent with a triplet sensitization mechanism was the observation that metal complexes reported in other visible light photocatalysis applications with triplet state energies lower than that of styrene were ineffective in this transformation (entries 9 and 10).15 Finally, control reactions confirmed that no reaction occurs either in the absence of light or the absence of photocatalyst (entries 11 and 12).16
 |
(2) |
 |
(3) |
Table 1.
Optimization and control studies for photocatalytic [2+2] cycloaddition
| ||||
|---|---|---|---|---|
| entry[a] | catalyst | solvent | conc. (M) | yield[b] |
| 1 | 2•PF6 | CHCl3 | 0.05 M | 22% |
| 2 | 2•PF6 | CH2Cl2 | 0.05 M | 26% |
| 3 | 2•PF6 | THF | 0.05 M | 21% |
| 4 | 2•PF6 | acetone | 0.05 M | 24% |
| 5 | 2•PF6 | MeOH | 0.05 M | 21% |
| 6 | 2•PF6 | MeCN | 0.05 M | 13% |
| 7 | 2•PF6 | DMSO | 0.05 M | 33% |
| 8 | 2•PF6 | DMSO | 0.01 M | 89% (83%)[c] |
| 9 | 1a•(PF6)2 | DMSO | 0.01 M | 0% |
| 10 | 1b•(PF6)2 | DMSO | 0.01 M | 0% |
| 11 | none | DMSO | 0.01 M | 0% |
| 12[d] | 2•PF6 | DMSO | 0.01 M | 0% |
Reactions irradiated using a 23 W compact fluorescent light bulb.
Yields determined by 1H NMR analysis against a calibrated internal standard unless noted.
Isolated yield in parenthesis.
Control reaction conducted in the dark.
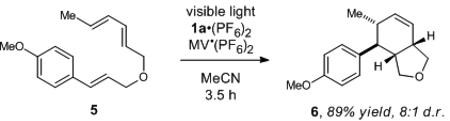
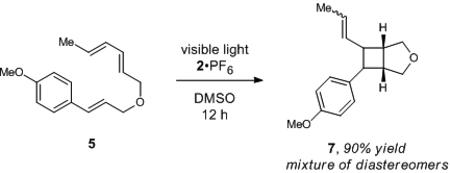
To further differentiate this triplet sensitization mechanism from radical cation cycloadditions, we investigated the reaction of diene substrate 5 under two different sets of conditions. First, Bauld has reported that the radical cation of 5 undergoes efficient intramolecular Diels–Alder cycloaddition, 17 and we recently reported photocatalytic conditions that generate the same radical cation intermediate and produce [4+2] cycloadduct 6 in good yields (eq 2).2h On the other hand, exposure of 5 to iridium catalyst 2 under visible light affords a complex mixture of products that we identified as diastereomeric [2+2] cycloadducts (eq 3),18 with only a trace of the [4+2] cycloadduct. Similar preferences for [2+2] periselectivity in cycloadditions involving dienes with a variety of triplet sensitizers have been previously reported.19
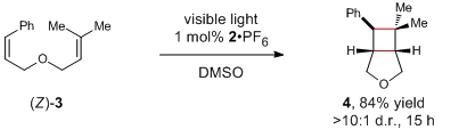
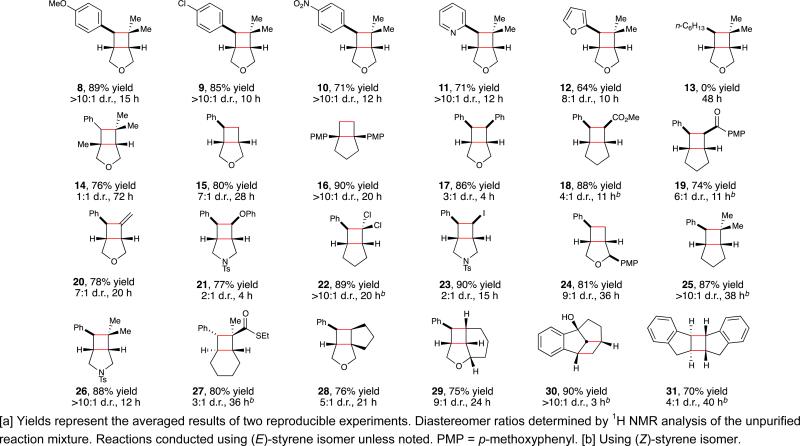
Studies exploring the scope of this process are summarized in Table 2. Several themes emerge from an examination of these data. First, as expected from the propensity of alkene triplets to undergo
 |
(4) |
geometric isomerization, the photocycloaddition is stereoconvergent; the reaction of the (Z) isomer of 3 gave results identical to the reaction of (E)-3 (eq 4). A time course of the reaction confirms that (E)/(Z) photoisomerization of 3 occurs faster than productive cycloaddition to 4 (see Supporting Information). Conversely, cyclic styrenes that are incapable of undergoing this energy-wasting isomerization react much more rapidly than acyclic styrenes (30). The ability of the cyclic constraint to extend the lifetime of the triplet is also presumably responsible for the ability of indene to undergo intermolecular cycloaddition (e.g., 31); acyclic styrenes simply undergo rapid (E) to (Z) isomerization without productive intermolecular cycloaddition. We also observe that the reaction can be successfully initiated with a variety of substituted styrenes regardless of their electronic nature (8–12),20 including both electron-rich and electron-deficient heterostyrene compounds. On the other hand, substrates containing only aliphatic, nonconjugated alkenes are recovered unchanged from these reaction conditions (13), consistent with their significantly higher triplet energies compared to styrenes.21 The diversity of coupling partners suitable for this cycloaddition is extensive and includes aliphatic alkenes, styrenes, enones, enoates, enol ethers, haloalkenes, and allenes (17–23). The functional group compatibility of this process is also good; sulfonamides, unprotected alcohols, carbonyl compounds, and halogens are easily tolerated. Collectively, these data indicate that the range of cyclobutane compounds accessible using this methodology is quite broad and unconstrained by the redox properties of the substrates.
Table 2.
Scope studies for photosensitized [2+2] cycloadditions undervisible light.a
|
The ability to use low-energy, operationally convenient, and readily available visible light in the photosensitization reactions makes these processes more attractive for synthetic applications than UV irradiation. In order to demonstrate this point, we compared the reaction of vinyl iodide 32 under our optimized conditions to photoexcitation by direct irradiation with UV (Scheme 1). We find that 32 is rapidly consumed when irradiated in a Rayonet reactor outfitted with 254 nm lamps; however, the mass recovery of the reaction is poor, consistent with the propensity of high-energy UV radiation to promote uncontrolled radical decomposition processes. Indeed, resubjecting cyclobutane 23 to direct irradiation with 254 nm UV light resulted in its complete decomposition within 1 h. Thus, in addition to showing greater operational convenience than traditional UV photolyses, these conditions also increase the tolerance of photochemical reactions towards photosensitive functional groups such as alkyl iodides.
Scheme 1.
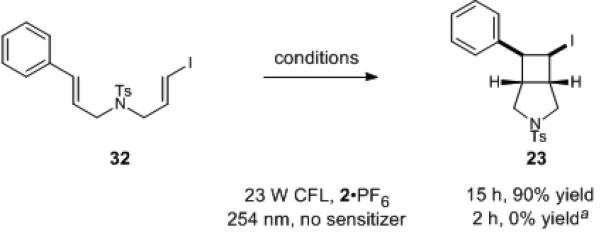
Comparison of visible to UV conditions for cycloaddition of 32. [a] 1H NMR analysis against an internal standard revealed 100% consumption of 32.
To demonstrate that these conditions are applicable to the preparation of complex natural products, we undertook a concise synthesis of cannabiorcicyclolic acid (37), one of several known cyclobutane-containing cannabinoids (Scheme 2).22 The chromene precursor 35 is easily obtainable by base-catalyzed condensation between phenol 33 and citral (34) in 54% yield. Photocycloaddition sensitized by 2•PF6 affords 86% yield of cyclobutane 36 after 8 h of irradiation with a 23 W compact fluorescent light bulb. In contrast, the direct irradiation of 35 with 254 nm UV light for 5 h gave only 19% yield of the desired cycloadduct with only 9% of the starting chromene remaining. Finally, hydrolysis of the ethyl ester under typical ester hydrolysis conditions affords (±)-cannabiorcicycloic acid in 97% yield.
Scheme 2.
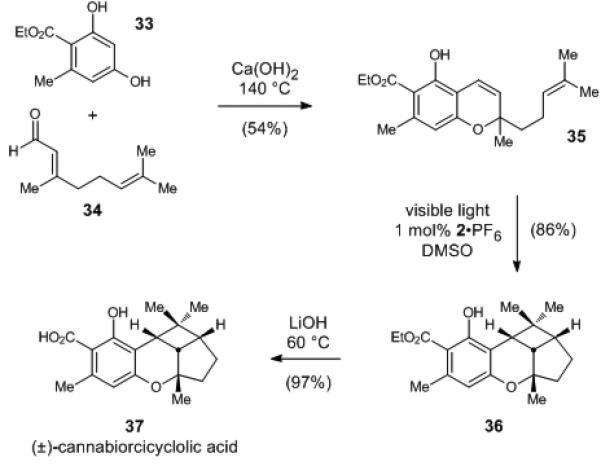
Synthesis of cannabiorcicycloic acid.
The results of this study are significant for a variety of reasons. First, they demonstrate that the class of transition metal photocatalysts that have earned significant recent interest for their ability to promote organic transformations by photoinduced electron transfer can also catalyze reactions by energy transfer processes upon irradiation with household visible light sources. While this energy transfer cycloaddition maintains the same operational facility as previously reported photoredox transformations, the scope of the reaction is not constrained by the electrochemical properties of the substrates, and is instead governed by the relative excited state energies of the catalyst and styrenes. More broadly, these results suggest that a great variety of organic photochemical reactions known to be promoted by direct UV photoexcitation might also be accessible by visible light photocatalysis. Studies to investigate this proposition are currently underway in our laboratory.
Supplementary Material
Acknowledgments
This research was conducted using funds from the NIH (GM095666) and the Sloan Foundation. The NMR facilities at UW-Madison are funded by the NSF (CHE-9208463, CHE-9629688) and NIH (RR08389-01, RR13866-01).
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For recent examples, see: Nicewicz DA, MacMillan DWC. Science. 2008;322:77–80. doi: 10.1126/science.1161976.Narayanam JMR, Tucker JW, Stephenson CRJ. J. Am. Chem. Soc. 2009;131:8756–8757. doi: 10.1021/ja9033582.Zou Y-Q, Lu L-Q, Fu L, Chang N-J, Rong J, Chen J-R, Xiao W-J. Angew. Chem. 2011;123:7309–7313. doi: 10.1002/anie.201102306.Angew. Chem. Int. Ed. 2011;50:7171–7175. doi: 10.1002/anie.201102306.McNally A, Prier CK, MacMillan DWC. Science. 2011;334:1114–1117. doi: 10.1126/science.1213920.Nagib DA, MacMillan DWC. Nature. 2011;480:224–228. doi: 10.1038/nature10647.Kalyani D, McMurtrey KB, Neufeldt SR, Sanford MS. J. Am. Chem. Soc. 2011;133:18566–18569. doi: 10.1021/ja208068w.Freeman DB, Furst L, Condie AG, Stephenson CRJ. Org. Lett. 2012;14:94–97. doi: 10.1021/ol202883v.Tucker JW, Zhang Y, Jamison TF, Stephenson CRJ. Angew. Chem. 2012;124:4220–4223. doi: 10.1002/anie.201200961.Angew. Chem. Int. Ed. 2012;51:4144–4147. doi: 10.1002/anie.201200961.Cherevatskaya M, Neumann M, Füldner S, Harlander C, Kümmel S, Dankesreiter S, Pfitzner A, Zeitler K, König B. Angew. Chem. 2012;124:4138–4142. doi: 10.1002/anie.201108721.Angew. Chem. Int. Ed. 2012;51:4062–4066. doi: 10.1002/anie.201108721.Hari DP, Schroll P, König B. J. Am. Chem. Soc. 2012;134:2958–2961. doi: 10.1021/ja212099r.DiRocco DA, Rovis T. J. Am. Chem. Soc. 2012;134:8094–8097. doi: 10.1021/ja3030164.Maity S, Zhu M, Shinabery RS, Zheng N. Angew. Chem. 2012;124:226–230. doi: 10.1002/anie.201106162.Angew. Chem. Int. Ed. 2012;51:222–226.
- 2.a Ischay MA, Anzovino ME, Du J, Yoon TP. J. Am. Chem. Soc. 2008;130:12886–12887. doi: 10.1021/ja805387f. [DOI] [PubMed] [Google Scholar]; b Ischay MA, Lu Z, Yoon TP. J. Am. Chem. Soc. 2010;132:8572–8574. doi: 10.1021/ja103934y. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lu Z, Shen M, Yoon TP. J. Am. Chem. Soc. 2011;133:1162–1164. doi: 10.1021/ja107849y. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hurtley AE, Cismesia MA, Ischay MA, Yoon TP. Tetrahedron. 2011;67:4442–4448. doi: 10.1016/j.tet.2011.02.066. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lin S, Ischay MA, Fry CG, Yoon TP. J. Am. Chem. Soc. 2011;133:19350–19353. doi: 10.1021/ja2093579. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Tyson EL, Farney EP, Yoon TP. Org. Lett. 2012;14:1110–1113. doi: 10.1021/ol3000298. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Parrish JD, Ischay MA, Lu Z, Guo S, Peters NR, Yoon TP. Org. Lett. 2012;14:1640–1643. doi: 10.1021/ol300428q. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Lin S, Padilla CE, Ischay MA, Yoon TP. Tetrahedron Lett. 2012;53:3073–3076. doi: 10.1016/j.tetlet.2012.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For reviews of the photochemical properties of Ru(bpy)32+ and its derivatives, see: Kalyanasundaram K. Coord. Chem. Rev. 1982;46:159–244.Juris A, Balzani V, Barigelletti F, Campagna S, Belser P, von Zelewsky A. Coord. Chem. Rev. 1988;84:85–277.
- 4.For a review of the photochemical properties of iridium polypyridyl complexes, see: Flamigni L, Barbieri A, Sabatini C, Ventura B, Barigelletti F. Top. Curr. Chem. 2007;281:143–203.
- 5.For recent reviews, see: Zeitler K. Angew. Chem. 2008;121:9969–9974.Angew. Chem. Int. Ed. 2009;48:9785–9789. doi: 10.1002/anie.200904056.Yoon TP, Ischay MA, Du J. Nat. Chem. 2010;2:527–532. doi: 10.1038/nchem.687.Narayanam JMR, Stephenson CRJ. Chem. Soc. Rev. 2011;40:102–113. doi: 10.1039/b913880n.Teply F. Collect. Czech. Chem. Commun. 2011;76:859–917.Tucker JW, Stephenson CRJ. J. Org. Chem. 2012;77:1617–1622. doi: 10.1021/jo202538x.
- 6.a Esser P, Pohlmann B, Scharf HD. Angew. Chem. 1994;106:2093–2108. [Google Scholar]; Angew. Chem. Int. Ed. 1994;33:2009–2023. [Google Scholar]; b Ciana C-L, Bochet CG. Chimia. 2007;61:650–654. [Google Scholar]; c Oelgemöller M, Jung C, Mattay J. Pure Appl. Chem. 2007;79:1939–1947. [Google Scholar]; d Haggiage E, Coyle EE, Joyce K, Oelgemöller M. Green Chem. 2009;11:318–321. [Google Scholar]; e Hoffman N. ChemSusChem. 2012;5:352–371. doi: 10.1002/cssc.201100286. [DOI] [PubMed] [Google Scholar]
- 7.Wrighton M, Markham J. J. Phys. Chem. 1973;77:3042–3044. [Google Scholar]
- 8.Ikezawa H, Kutal C, Yasufuku K, Yamazaki H. J. Am. Chem. Soc. 1986;108:1589–1594. [Google Scholar]
- 9.Islangulov RR, Castellano FN. Angew. Chem. 2006;118:6103–6105. doi: 10.1002/anie.200601615. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2006;45:5957–5959. doi: 10.1002/anie.200601615. [DOI] [PubMed] [Google Scholar]
- 10.For reviews of energy transfer photosensitization, see: Turro NJ. J. Chem. Ed. 1966;43:13–16.Dilling WL. Chem. Rev. 1969;69:845–877.Turro NJ. Modern Molecular Photochemistry. Benjamin/Cummings; California: 1978. pp. 296–359.Albini A. Synthesis. 1981:249–264.
- 11.Value refers to the peak oxidation potential measured for 3 at a scan rate of 100 V/s in MeCN.
- 12.Lowry MS, Goldsmith JI, Slinker JD, Rohl R, Pascal RA, Jr., Malliaras GG, Bernhard S. Chem. Mater. 2005;17:5712–5719. [Google Scholar]
- 13.a Nguyen JD, Tucker JW, Konieczynska MD, Stephenson CRJ. J. Am. Chem. Soc. 2011;133:4160–4163. doi: 10.1021/ja108560e. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wallentin C-J, Nguyen JD, Finkbeiner P, Stephenson CRJ. J. Am. Chem. Soc. 2012;134:8875–8884. doi: 10.1021/ja300798k. [DOI] [PubMed] [Google Scholar]
- 14.a Lamola AA, Hammond GS. J. Chem. Phys. 1965;43:2129–2135. [Google Scholar]; b Ni T, Caldwell RA, Melton LA. J. Am. Chem. Soc. 1989;111:457–464. [Google Scholar]
- 15.ET values for 1a (46.8 kcal/mol) and 1b (47.4 kcal/mol) were calculated from their emission maxima as reported in reference 3b.
- 16.The experiments summarized in Table 1 were conducted open to ambient atmosphere. We observe no significant difference in the rate of the reaction under rigorously degassed, anaerobic conditions.
- 17.Kim T, Mirafzal GA, Liu J, Bauld NL. J. Am. Chem. Soc. 1993;115:7653–7664. [Google Scholar]
- 18.The [2+2] cycloadducts were formed as an inseparable mixture of geometrical and stereochemical isomers. The cyclobutane structures were assigned upon hydrogenation of the alkenes to converge the alkene isomers. See Supporting Information for details.
- 19.a Hammond GS, Turro NJ, Fischer A. J. Am. Chem. Soc. 1961;83:4674–4675. [Google Scholar]; b Liu RSH, Turro NJ, Hammond GS. J. Am. Chem. Soc. 1965;87:3406–3412. [Google Scholar]
- 20.This lack of sensitivity to electronic perturbation provides further evidence against an electron-transfer mechanism. The peak potential for oxidation of the p-nitrostyrene precursor to 10 was measured to be +1.68 V vs SCE, while the excited state oxidation potential of 2 is +1.21 V. Formation of the styrene radical cation would be thermodynamically unreasonable.
- 21.ET for simple unstrained aliphatic alkenes ranges from 76–84 kcal/mol. See reference 14b for representative values.
- 22.a Iwata N, Kitanaka S. Chem. Pharm. Bull. 2011;59:1409–1412. doi: 10.1248/cpb.59.1409. [DOI] [PubMed] [Google Scholar]; b Qin C, Mei Y, Zhou X, Huang PA,S. China J. Chin. Mater. Med. 2010;35:2568–2571. [PubMed] [Google Scholar]; c Mechoulam R, McCallum NK, Burstein S. Chem. Rev. 1976;76:75–112. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.