

Mass spectrometry (MS) has revealed the composition, stoichiometry, connectivity, and dynamics of many multiprotein complexes that remain challenging for other structural biology tools.[1] More recently, ion mobility (IM), a gas-phase separation technology that operates to resolve protein ions according to their size and charge,[2] coupled with MS (IM-MS) has been used to generate 3D structure information from such samples.[3] Information from many such gas-phase technologies[4] can be combined to overcome challenging aspects of protein structure characterization. Even though such methods are proving to be useful, their development is not devoid of experimental challenges. Chief among these is establishing a general correlation between gas-phase measurements and protein structures in solution. Several reports have observed significant rearrangements of protein structure upon desolvation and ionization,[5] although recent data suggest that these examples may be in the minority.[6] Despite this, general protocols aimed at protecting protein structure upon the removal of bulk solvent will undoubtedly enable biomolecular structure characterization through gas-phase structural biology approaches, like IM-MS.
Recent efforts to develop such protocols use additives, both in solution prior to ionization[7] and in the gas-phase prior to MS analysis,[8] as a means of stabilizing protein complex ions. We have focused on the former, using Hofmeister-type salt additives, and have recently classified a large number of anions for their ability to stabilize multiprotein structure[9] using measurements of both collision induced unfolding (CIU), where ions are heated with collisions and induced to unfold, and collision induced dissociation (CID), where increased collisional heating leads to the dissociation of assemblies into a highly unfolded monomers and stripped complexes.[10] Our previous data revealed that anions bind to protein complexes during or prior to the nano-electrospray ionization (nESI) process and can stabilize protein ions through dissociation as neutrals, which act to carry away excess energy from the gas-phase protein ions, thus allowing their structures to remain compact – in configurations easily correlated to X-ray and NMR datasets.[9] In this work, we study the influence of cation-based stabilizers, compare these additives to our previous anion dataset, and find dramatic mechanistic differences between the two.
Figs. 1A and 1B show data for tetrameric transthyretin (TTR, 55 kDa). In order to demonstrate the effect of different cations on TTR, a series of tandem mass spectra (showing CID, Fig. 1A) and arrival time distributions (showing CIU, Fig. 1B) of the 14+ charge state of TTR acquired at a trap collision voltage of 60 V and 55 V respectively are shown. For each, all instrument parameters are kept constant and only the composition of the nESI buffer is altered to contain different cationic additives. The peak corresponding to 14+ charge state of TTR isolated for CIU/CID broadens when incubated with added cations, as it contains unresolved peaks corresponding to a range of previously-described adducted forms (Fig S1).[11] In a similar fashion to anions, when cations dissociate from the protein complexes studied here, they do so as neutrals (bound to acetate or hydroxide counter-ions). In Fig. 1A, signals for 14+ TTR and 6+ to 8+ transthyretin monomer are observed at substantially different levels as a function of the cation added, while Fig. 1B reveals strikingly different arrival time distributions for 14+ cation-bound TTR, with different relative abundances for compact (I) and unfolded conformer families (II–IV). These data clearly demonstrate the differential influence of cation additives on protein dissociation and unfolding in the gas-phase.
Figure 1.

(A) MS data for 14+ TTR incubated with 10 acetate-based cations at 60 V trap collision voltage. The data also contain peaks corresponding to 6–8+ charge state monomers, resulting from CID of the complex. (B) IM data for the same ions shown in A, at 55 V trap collision voltage. Four drift time features showing are observed, and labeled from I to IV. Histogram plots charting collision energy (eV*) required to dissociate (C) and unfold (D) 50% of the dimeric protein ion population for BLA and tetrameric protein ion populations for TTR, avidin, ConA and ADH are shown for a range of cation additives. Control data sets are also marked on the plot (NH4+).
For a more quantitative measurement of stability, we monitored CID and CIU data as a function of collision voltage (Fig. S2). From these data we constructed the histograms shown in Figs. 1C and 1D, which plot the ion energy (eV*) values at which the intensity observed for intact (Itet) and compact (If) tetramer ions decrease by 50% respectively. Data include three tetrameric protein complexes other than TTR, including avidin, concanavalin A (ConA) and alcohol dehydrogenase (ADH), and dimeric β-lactoglobulin A (BLA), screened in the presence of the same 10 acetate-based cations. A number of general trends in protein stability are observed. Firstly, the protein complexes studied here undergo CIU at lower energy relative to CID, as reported previously.[12] We note that following incubation with stabilizing cations, ADH does not appreciably undergo CID even at the highest activation energy attainable (Fig. S3), resulting in the lack of its dissociation data in Fig. 1C. Secondly, cations stabilize gas-phase protein complexes to different degrees. In general, Mg2+ and Ca2+ have a universally stabilizing influence on Itet and If for all protein complexes studied here. Conversely, cations such as K+, Rb+ and TMA+ have a negligible stabilizing effect relative to control (ammonium acetate). Interestingly, TrisH+ exhibits a greater ability to stabilize gas-phase protein complexes than other singly-charged cations studied here.[7a] For example, TrisH+ is the second-most stabilizing cation screened in our BLA dataset (behind Mg2+). In addition, the relative stability of the five complexes studied here are not influenced by cation additives, with BLA requiring the most energy to dissociate and TTR requiring the most energy to unfold under equivalent conditions.[9]
Despite these similarities, we find several significant differences in the stabilization provided by cation additives when compared with our previous anion data. Firstly, cation adducts seem to stabilize protein complexes against CID to a greater extent, on average, than equivalent anions. The ion energy at which CID occurs is raised by 31% by the average cation, while this threshold is increased by only 19% by the average anion. This observation can be extended to include the general stability afforded to complexes bound by the most-stabilizing cations, the stabilities for which are in general much greater than any anion-bound complexes studied to date (Fig. S5). Conversely, anionic adducts are, in general, better stabilizers of gas-phase protein unfolding than cations. Data recorded for cation-adducted protein complexes indicate an average CIU threshold increase of only 26% where anions achieved a 36% increase in stability under similar conditions. It is therefore anticipated that the mechanism of protein structure stabilization for cation-adducted protein ions is dramatically different from their anion counterparts. Fig. 2A shows plots of ion mass as a function of activation voltage for TTR. Previous data for anions showed a preference for complete dissociation of protein-anion adducts at relatively low activation voltages in order stabilize through 'dissociative cooling'.[9] The cation adducts studied here that impart the most protein stabilization, however, tend to remain bound to the protein complex even at large activation voltage values. In further contrast to our studies of protein-anion adducts, CIU and CID stabilities are highly correlated for cation-adducted complexes. The linear relationship between CID and CIU stability thresholds exhibits a R2 of 0.94 (Fig. 2B) compared to anion-based data reported previously (R2=0.55). This further indicates a disparity between the stabilization mechanisms operative for anionic and cationic additives.[9]
Figure 2.

(A) Plot of the measured average mass increase relative to the sequence mass of TTR as a function of trap collision voltage for a range of cation additives. The approximate numbers of cations that stay strongly bound to the protein assembly even at large trap collision voltage are shown on the right. (B) A plot of the average CID versus CIU collision energies (eV*) for the 5 protein complexes studied for each cation additive. The protein-cation complexes have highly-correlated unfolding and dissociation energies (dashed line). (C) Data from Fig. 2B plotted against the charge-per-unit-area of the cations added (vertical axis) illustrate a well-correlated relationship between protein-cation complex stability and the charged area of added cations.
The above differences between anionic and cationic stabilizers inform our mechanistic description of their action, which has been normalized for the relative binding affinities of the cations studied here. Whereas anions perform optimally as stabilizers when they bind to the protein and then dissociate from the complex after relatively minimal activation, the best cationic stabilizers are those that remain bound to the protein assembly in large numbers, even following extensive activation in the gas phase. These highly-stabilizing cations strongly correlate with those that have larger charge-per-unit-area values (Fig. 2C and Fig. S5A). The larger charge-per-unit-area of these cations, much in excess of any anions that we have tested to date (Fig. S5B and Table S1), presumably gives these adducts access to modes of stabilization that rely either upon multidentate interactions within proteins, enabling them to more effectively tether regions of its structure, or by replacing highly-mobile proton charge carriers with less mobile cationic charge carriers that restrict charge mobility and frustrate the Coulombic unfolding of subunits within the complex, which is a critical step in the asymmetric dissociation of non-covalent protein complexes.[13] Although those cations that strongly-stabilize gas-phase protein structure conform to the mechanistic discussion above, evidence of the dissociative-cooling of protein structure is not absent from our cation dataset (Fig. 2A and Fig. 3).
Figure 3.
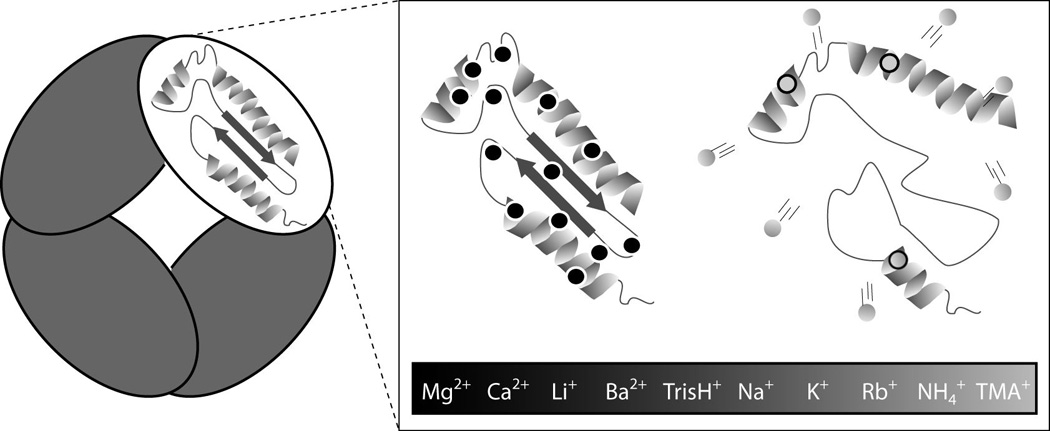
A mechanistic diagram of protein structure stabilization through bound cations, summarizing our current data set. Two models are shown: The cations of high charge density (black) that bind in large numbers to protein complexes will retain their binding position within the protein sequence and become less mobile as charge carriers. Conversely, cations of low charge density (grey) dissociate readily and bind in smaller numbers to proteins, weakening their ability to enhance stability.
In summary, these data present the first mechanistic description of additive cation stabilizers that cover a broad range of both cationic additives and multiprotein complexes. We observe that, in general, cations of high charge-per-unit-area stabilize proteins in a complimentary and significantly different way relative to most anions,[9] and we plan to exploit this in the future by using salt additives that are tailored to take advantage of both cationic and anionic protein adducts to improve protein structural stability. We believe that such additives are critical for IM-MS to fully-realize its potential as a high-throughput method for discovering multiprotein topology and structure, and as a means of elucidating the critical role of surfactant molecules in stabilizing gas-phase membrane protein complexes.[14]
EXPERIMENTAL METHODS
Materials
Proteins Avidin (egg white), TTR (human), Con A (jack bean), ADH (yeast), BLA, (bovine), and salts (acetate anion with ammonium, tetramethylammonium (TMA), sodium, potassium, rubidium, lithium, Tris (2-Amino-2-hydroxymethyl-propane-1,3-diol), calcium, barium, and magnesium counterions) were purchased from Sigma (St. Louis, MO, USA). All protein samples were buffer exchanged into 100 mM ammonium acetate at pH 7 using Micro Bio-Spin 6 columns (Bio-Rad, Hercules, CA) and prepared to a final concentration of 5 µM (avidin, TTR, ConA, ADH) or 10 µM (BLA). In order to study the influence of different salts on protein stability without significantly altering buffer capacity or solution pH, the salts were prepared as stock solutions in 100 mM ammonium acetate at a concentration of 20 mM, each of which was then added to the protein solution. Final solutions contained added salt concentrations of 2 mM for avidin, TTR, ConA, ADH and 0.5 mM for BLA samples. The total salt and protein concentrations listed above were chosen primarily to avoid ion suppression effects.
Ion Mobility-Mass Spectrometry and CIU/CID analysis
Approx. ~5 µL of sample was injected into a quadrupole-ion mobility-time-of–flight mass spectrometer (Synapt G2 HDMS, Waters, Milford MA, USA).[15] Protein ions were generated using a nESI source and optimized to allow transmission of non-covalent protein complexes.[16] The traveling-wave IM separator was operated at a pressure of ~ 3.5 mbar, and a 40 V wave height traveling at 800–1000 m/s to generate IM separation. Collisional activation in the ion trap prior to IM was used to perform CIU and CID experiments. Ions were selected in the quadrupole mass filter at a m/z corresponding to 16+ charge state of Avidin, 19+ of ConA, 14+ of TTR, 24+ of ADH tetramers and 11+ BLA dimers. Charge states were chosen based on their intensity across each solution state studied, and control IM data were collected to rule out overlapping oligomers at this m/z. Trap collision voltage was incremented in 5 V steps. Data analysis and normalization were carried out in a manner identical to our previous report.[9] Some figures contain axes labeled in collision energy (units of eV*). The axis is a normalized version of ion kinetic energy appropriate for making stability comparisons across large mass ranges.[9]
ACKNOWLEDGMENTS
This work is supported by the National Institutes of Health (1-R01-GM-095832-01) and by University of Michigan startup funds.
Footnotes
Supporting Information Available. Additional mass spectra (Fig. S1), a workflow diagram (Fig. S2), CID data for ADH (Fig. S3), details on our data normalization procedures (Fig. S4), and mechanistic information (Fig S5 & Table S1) are available.
References
- 1.a) Heck AJR. Nat. Methods. 2008;5:927–933. doi: 10.1038/nmeth.1265. [DOI] [PubMed] [Google Scholar]; b) Sharon M, Robinson CV. Annu. Rev. Biochem. Vol. 76. Palo Alto: Annual Reviews; 2007. pp. 167–193. [DOI] [PubMed] [Google Scholar]
- 2.a) Pukala TL, Ruotolo BT, Zhou M, Politis A, Stefanescu R, Leary JA, Robinson CV. Structure. 2009;17:1235–1243. doi: 10.1016/j.str.2009.07.013. [DOI] [PubMed] [Google Scholar]; b) Politis A, Park AY, Hyung SJ, Barsky D, Ruotolo BT, Robinson CV. PLoS One. 2010;5:e12080. doi: 10.1371/journal.pone.0012080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Ruotolo BT, Robinson CV. Curr Opin Chem Biol. 2006;10:402–408. doi: 10.1016/j.cbpa.2006.08.020. [DOI] [PubMed] [Google Scholar]; b) Wyttenbach T, Bowers MT. Annu. Rev. Phys. Chem. Vol. 58. Palo Alto: Annual Reviews; 2007. pp. 511–533. [DOI] [PubMed] [Google Scholar]; c) Zhong Y, Hyung SJ, Ruotolo BT. Expert Review of Protemics. 2012;9:47–58. doi: 10.1586/epr.11.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Breuker K, McLafferty FW. Proc. Natl. Acad. Sci. U. S. A. 2008;105:18145–18152. doi: 10.1073/pnas.0807005105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Green MK, Lebrilla CB. Mass Spectrom. Rev. 1997;16:53–71. doi: 10.1002/(SICI)1098-2787(1997)16:2<53::AID-MAS1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]; c) Benesch JLP, Ruotolo BT, Simmons DA, Robinson CV. Chem. Rev. 2007;107:3544–3567. doi: 10.1021/cr068289b. [DOI] [PubMed] [Google Scholar]
- 5.a) Barran PE, Jurneczko E. Analyst. 2011;136:20–28. doi: 10.1039/c0an00373e. [DOI] [PubMed] [Google Scholar]; b) Hogan CJ, Jr, Ruotolo BT, Robinson CV, Fernandez de la Mora J. J Phys Chem B. 2011;115:3614–3621. doi: 10.1021/jp109172k. [DOI] [PubMed] [Google Scholar]
- 6.Benesch JLP, Ruotolo BT. Curr. Opin. Struct. Biol. 2011;21:641–649. doi: 10.1016/j.sbi.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Freeke J, Robinson CV, Ruotolo BT. International Journal of Mass Spectrometry. 2010;298:91–98. [Google Scholar]; b) Flick TG, Merenbloom SI, Williams ER. Analytical Chemistry. 2011;83:2210–2214. doi: 10.1021/ac1031012. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Merenbloom S, Flick T, Daly M, Williams E. J. Am. Soc. Mass Spectrom. 2011;22:1978–1990. doi: 10.1007/s13361-011-0238-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bagal D, Kitova EN, Liu L, El-Hawiet A, Schnier PD, Klassen JS. Analytical Chemistry. 2009;81:7801–7806. doi: 10.1021/ac900611a. [DOI] [PubMed] [Google Scholar]
- 9.Han LJ, Hyung SJ, Mayers JJS, Ruotolo BT. J. Am. Chem. Soc. 2011;133:11358–11367. doi: 10.1021/ja203527a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Benesch JLP. Journal of the American Society for Mass Spectrometry. 2009;20:341–348. doi: 10.1016/j.jasms.2008.11.014. [DOI] [PubMed] [Google Scholar]
- 11.Verkerk UH, Kebarle P. J. Am. Soc. Mass Spectrom. 2005;16:1325–1341. doi: 10.1016/j.jasms.2005.03.018. [DOI] [PubMed] [Google Scholar]
- 12.a) Ruotolo BT, Hyung SJ, Robinson PM, Giles K, Bateman RH, Robinson CV. Angewandte Chemie-International Edition. 2007;46:8001–8004. doi: 10.1002/anie.200702161. [DOI] [PubMed] [Google Scholar]; b) Hyung SJ, Robinson CV, Ruotolo BT. Chem. Biol. 2009;16:382–390. doi: 10.1016/j.chembiol.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 13.a) Jurchen JC, Williams ER. J. Am. Chem. Soc. 2003;125:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Felitsyn N, Kitova EN, Klassen JS. Anal. Chem. 2001;73:4647–4661. doi: 10.1021/ac0103975. [DOI] [PubMed] [Google Scholar]; c) Bornschein RE, Hyung SJ, Ruotolo BT. J. Am. Soc. Mass Spectrom. 2011;22:1690–1698. doi: 10.1007/s13361-011-0204-y. [DOI] [PubMed] [Google Scholar]; d) Erba EB, Ruotolo BT, Barsky D, Robinson CV. Anal. Chem. 2010;82:9702–9710. doi: 10.1021/ac101778e. [DOI] [PubMed] [Google Scholar]; e) Pagel K, Hyung SJ, Ruotolo BT, Robinson CV. Anal. Chem. 2010;82:5363–5372. doi: 10.1021/ac101121r. [DOI] [PubMed] [Google Scholar]
- 14.Zhou M, Morgner N, Barrera NP, Politis A, Isaacson SC, Matak-Vinkovic D, Murata T, Bernal RA, Stock D, Robinson CV. Science. 2011;334:380–385. doi: 10.1126/science.1210148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Zhong Y, Hyung SJ, Ruotolo BT. Analyst. 2011;136:3534–3541. doi: 10.1039/c0an00987c. [DOI] [PubMed] [Google Scholar]; b) Giles K, Williams JP, Campuzano I. Rapid Commun. Mass Spectrom. 2011;25:1559–1566. doi: 10.1002/rcm.5013. [DOI] [PubMed] [Google Scholar]
- 16.a) Ruotolo BT, Benesch JLP, Sandercock AM, Hyung SJ, Robinson CV. Nature Protocols. 2008;3:1139–1152. doi: 10.1038/nprot.2008.78. [DOI] [PubMed] [Google Scholar]; b) Hernandez H, Robinson CV. Nat Protoc. 2007;2:715–726. doi: 10.1038/nprot.2007.73. [DOI] [PubMed] [Google Scholar]