

The stereoselective formation of quaternary carbons is one of the most demanding challenges in organic synthesis.1 An especially direct way to construct such stereocenters would be to combine a prochiral tertiary organometallic and a carbon-centered electrophile (Figure 1A). However, this strategy is not mentioned in the numerous reviews of stereoselective synthesis of chiral quaternary carbons,1 and to our knowledge has never been employed in target-directed organic synthesis. This omission undoubtedly derives from the challenge in generating tertiary organometallic intermediates, particularly those containing three alkyl substituents.[2–4] We were recently drawn to explore this undeveloped approach for forming quaternary carbon stereocenters in the context of fashioning the demanding C8–C14 bond and the C8 quaternary stereocenter of rearranged spongian diterpenes such as aplyviolene (4) and dendrillolide A (5) by the reaction of tertiary organocuprate 1 and cyclopentenone 2. This coupling was anticipated to take place from the convex face of nucleophile 1 and from the face of 2 opposite the branched side chain (Figure 1B).[5]
Figure 1.
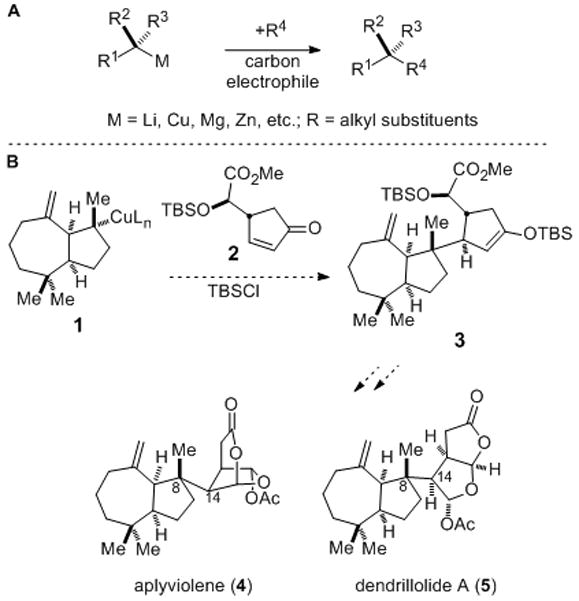
Forming quaternary carbon stereocenters by the reaction of prochiral tertiary organometallics and a carbon-centered electrophile (A) and the potential use this strategy to synthesize rearranged spongian diterpenes such as aplyviolene and dendrillolide A (B).
We report herein that a) unstabilized tertiary organolithium intermediates can be conveniently generated by reductive decyanation of nitrile precursors and that these reagents and their derived cuprates couple in useful yields with carbon-centered electrophiles, b) chiral tertiary organolithium and organocuprate intermediates in the cis-perhydroazulene and cis-perhydropentalene series react with electrophiles with high diastereoselectivity from the ostensibly more-hindered concave face, and c) computational studies that suggest the origin of this unexpected diasteroselectivity.
Reductive lithiation of C–X bonds is widely practiced to generate organolithium intermediates,[6] with reductive lithiation using lithium 4,4′-di-tert-butylbiphenylide (LiDBB) having been employed to produce achiral tertiary organolithium intermediates from chloride, bromide and sulfide precursors.[7–9] We conjectured that chiral tertiary nitriles, which are more readily synthesized than tertiary halides or sulfides, might constitute useful progenitors of chiral tertiary organolithium reagents. Many α-heterosubstituted lithium reagents—including fully substituted ones—have been formed from nitrile precursors and trapped with carbon-centered electrophiles to yield valuable products;[10] moreover, the generation of an assortment of tertiary-benzylic lithium intermediates from benzylic nitrile precursors and their bimolecular trapping has also been reported.[11] Nonetheless, whether less-stable trialkyl tertiary organolithium intermediates could be generated from nitrile precursors and subsequently trapped was unknown, and had been suggested might not be feasible.[11] Encouraged by Rychnovsky’s reductive formation of one such intermediate and its intramolecular trapping,[12] we chose to examine whether unstabilized tertiary lithium reagents and their derived cuprates[13] could be generated from nitrile precursors and trapped in bimolecular reactions with carbon-centered electrophiles.
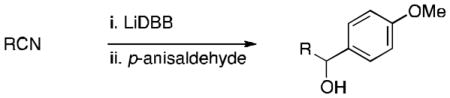
Initially we investigated the formation of simple tertiary organolithium intermediates from six tertiary nitrile precursors. After considerable optimization, the following procedure was found to be effective: the tertiary nitrile (1.5 equiv) was added rapidly to 3 equiv of freshly prepared LiDBB[14] in THF at −78 °C, followed 1 min later by the addition of 1.0 equiv of an aldehyde. A variety of neopentylic alcohols could be prepared in this way in useful yields, with the presence of alkene, alkyl ether, triisopropylsilyl ether (but not tert-butyldimethylsilyl ether), and electron-rich aromatic rings being tolerated (Table 1).
Table 1.
Trapping of tertiary organolithium intermediates derived from nitriles with p-anisaldehyde.a
| ||
|---|---|---|
| Entry | R | Yield [%] |
| 1 | tBu | 71 |
| 2 |
|
73 |
| 3 |

|
66 |
| 4 |
|
71 |
| 5 |
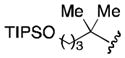
|
61 |
| 6 |
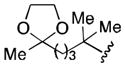
|
59 |
| 7 |
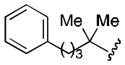
|
70 |
Conditions: 1.5 equiv nitrile, 3.0 equiv LiDBB, −78 °C, 1 min; 1 equiv aldehyde. LiDBB = lithium di-tert-butylbiphenylide. TIPS = triisopropylsilyl.
The conversion of reductively generated tertiary organolithium intermediates to tertiary organocuprates, and the use of the latter in conjugate reactions, was investigated next. After exploring the formation and reactivity of various lithium and dilithium organocuprates, we settled on dilithium cyanocuprates containing a “non-transferable” trimethylsilylmethyl substituent.[15] In the optimized procedure, a THF solution of Me3SiCH2CuCNLi (generated from trimethylsilylmethyllithium and copper cyanide) was added rapidly to the freshly prepared tertiary organolithium intermediate at −78 °C, and, after 5 min, a THF solution of an enone and a trialkylsilyl chloride was added.[16] In this way, eight diverse 3-substituted cyclohexanones 6 were formed in yields of 56–75% (Table 2). In addition, three trans-cyclopentenoxysilane adducts 7 were prepared in good yields (51–81%) and >20:1 diastereoselectivities by the reaction of tertiary organocuprate intermediates with cyclopentenone 2.
Table 2.
1,4-Addition of tertiary organocuprates derived from tertiary nitriles to cyclohex-2-en-1-one or enone 2.
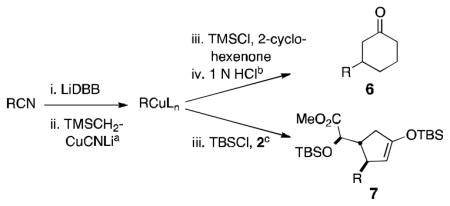
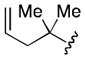
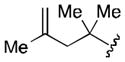
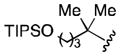
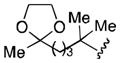
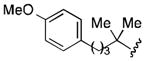
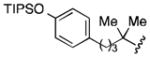
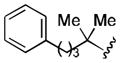
Conditions: 2.0 equiv nitrile, 4.0 equiv LiDBB, −78 °C, 30 sec; 3.1 equiv TMSCH2CuCNLi, 5 min.
Conditions: 1.0 equiv 2-cyclohexenone, 5.0 equiv TMSCl, −78 °C, 1 h; 1 N HCl, RT.
Conditions: 1.0 equiv 2, 2.0 equiv TBSCl, −78 °C, 1 h.
Isolated as the silyl enol ether.
2.5 equiv of TMSCH2CuCNLi used in a.
2.0 equiv of TMSCH2CuCNLi used in a and 5.0 equiv of TBSCl in c. TMS = Trimethylsilyl. TBS = tert-butyldimethylsilyl.
To explore whether this chemistry could be used to form quaternary stereocenters, we examined its utility in the pivotal fragment coupling step of the projected total syntheses of aplyviolene (4) and dendrillolide A (5) depicted in Figure 1B. Salient results of our investigation of the reactivity of tertiary organolithium and organocuprate reagents generated from cis-perhydroazulene nitriles 8 and 9 are summarized in Scheme 1.[17] We were delighted to find that the organocuprate intermediate derived from nitrile 8 reacted with cyclopentenone 2 to give a single coupled product 10 in 70% yield. To our surprise, the relative configuration of this product showed that electrophilic addition had taken place with high stereoselectivity from the concave face of the cis-perhydroazulene nucleophile.[18] To gain insight into the possible origin of this unexpected diastereoselection, the lithium reagent generated from nitrile 8 was quenched with methanol to give largely one hydrocarbon product; 1H NMR nOe analysis of the derived ketone 11 established that protonation of the tertiary organolithium intermediate also took place with high stereoselectivity from the concave face.[19] In a similar fashion, carboxylation of the lithium reagent generated from nitrile 8, or from epimeric nitrile 9, took place from the concave face to give carboxylic acid 12 in >20:1 diastereoselectivity. The selective formation of carboxylic acid 14 from saturated nitrile precursor 13 shows that the exomethylene group plays at most a minor role in the facial selectivity of the reaction of chiral tertiary lithium reagents in this series.[20]
Scheme 1.
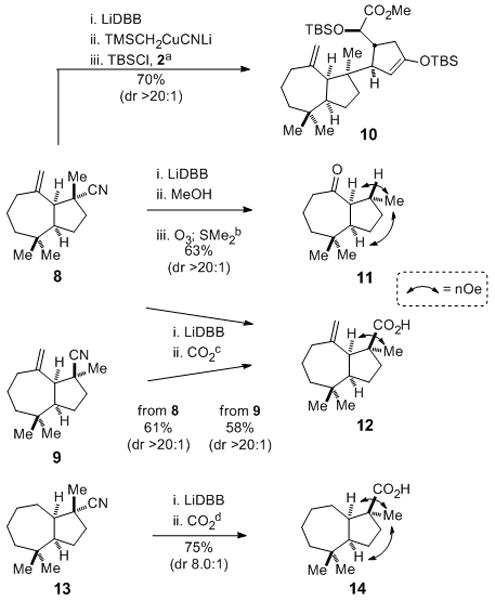
Reductive lithiation/electrophilic trapping of chiral cis-perhydroazulene nitriles 8, 9, and 13: [a] 8 (2.0 equiv), THF, −78 °C; LiDBB (4.0 equiv), 30 sec; TMSCH2CuCNLi (2.0 equiv), 5 min; 1.0 equiv 2, TBSCl (5.0 equiv), in THF, 1 h. [b] 8, THF, −78 °C; LiDBB (2.2 equiv), 30 sec; MeOH; O3, CH2Cl2: MeOH, 78 °C; dimethyl sulfide. [c] 8 or 9, THF, −78 °C; LiDBB (2.2 equiv), 30 sec; CO2; 1 N HCl. [d] as c but with 13.

Reductive decyanation was also used to generate analogous unstabilized tertiary organolithium and organocuprate intermediates in the cis-perhydropentalene series (Scheme 2). Reductive lithiation of cis-perhydropentalene nitrile 15 and quenching with methanol or carbon dioxide gave hydrocarbon 16 (dr = 2.6:1) or carboxylic acid 17 (dr = 4.1:1), with the major isomer in each case arising from preferential reaction from the concave face of the cis-perhydropentalene nucleophile.[21 , 22] In addition, nitrile 15 was converted to an organocuprate and coupled with methyl vinyl ketone to give 18, again with reaction occurring preferentially from the concave face.[23,24]
Scheme 2.
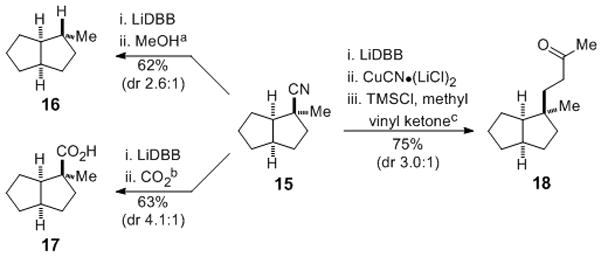
Reductive lithiation and reactivity of cis-perhydropentalene nitrile 15: [a] 15, THF, −78 °C; LiDBB (2.2 equiv), 30 sec; MeOH. [b] 15, THF, −78 °C; LiDBB (2.2 equiv), 30 sec; CO2; 1 N HCl. [c] 15 (4.0 equiv), THF, −78°C; LiDBB (8.0 equiv), 30 sec; CuCN-(LiCl)2 (2.0 equiv), 5 min; TMSCl (5.0 equiv); methyl vinyl ketone in THF, 1 h; 1 N HCl.
The results summarized in Schemes 1 and 2 show that the tertiary lithium and cuprate reagents in these cis-bicyclic ring systems react with electrophiles preferentially from the concave face in an apparently electrophile-independent fashion. As organolithium intermediates typically undergo protonation by SE2ret pathways,[3,25] the observation that the lithium reagents formed from nitriles 8, 9, and 15 protonate from the concave face indicates that lithium preferentially resides on the concave face of these cis-bicyclic ring systems. Computational studies were undertaken to gain further insight into the origin of this preference.
To this aim, the thermodynamic stability of different intermediates occurring after the proposed decyanation and reduction of the radical species with LiDBB, were evaluated at the B3LYP/6-31+G(d,p) level.[26] Both naked carbanionic (8-an, 15-an) and organolithium (8-Li, 15-Li) species derived from cis-perhydroazulene and cis-perhydropentalene precursors 8 and 15, respectively, were considered. In accordance with the experimental conditions and related computational[27] and X-ray[28] data, only monomeric species were considered and lithium was solvated with three discrete dimethyl ether molecules. Both epimers at the reacting center (labelled as α,β) and all possible ring conformations were considered, in order to obtain theoretical diastereomeric ratios based on Boltzmann distributions at 298 K estimated from relative enthalpies (ΔH) of these intermediates.[29] Activation enthalpies (ΔH‡) for the pyramidal inversion of naked carbanions (8-inv, 15-inv), were also calculated. For comparison, the relative enthalpies of epimeric carboxylated (12-CO2−, 17-CO2−) and protonated products, expressed as diastereomeric ratios, were also calculated.[30]
In good agreement with experimental observations, these calculations revealed a greater stability of those species bearing either the lone-electron pair or the lithium atom on the concave faces (>1.7 and >0.5 kcal mol−1 for perhydroazulene and perhydropentalene derivatives, respectively, Table S1, supporting information). The neutralization of the negative charge with bulky solvated Li cations provides diastereomeric ratios closer to the experimental data, proving to be a more realistic model. Conversely, values derived from naked anions look slightly overestimated, in particular for perhydropentalene 15-an. In addition, the calculated lowest barriers for the epimerization of the carbanionic species were perfectly feasible under the reaction conditions (4.0 and 9.1 kcal mol−1 for 8-inv, 15-inv, respectively). As the formation of contact ion pairs (i.e. organolithium) are likely nearly barrierless processes, it is conceivable that the rapid equilibration of such metalated species can also occur through the aforementioned inversion transition structures.
These studies support the notion that observed selectivity is independent of the ground state energy of the diastereomeric products. Whereas the epimeric protonated products showed similar calculated energies, the, bulkier CO2− moieties are preferentially positioned on the less-hindered convex face by 2.3 and 0.7 kcal mol−1 for the cis-perhydroazulene and cis-perhydropentalene carboxylate products, respectively (Table S1). Although the [Li(OMe2)3]+ units are indeed bulkier than CO2−, they are placed at a much larger distance (~2.1 Å) than the covalent C–C bond formed in the product (~1.6 Å), minimizing steric interactions.
The observed stereoselectivities can be easily rationalized by torsional strain considerations (Figure 2). As seen in the minimum energy structures of cis-perhydroazulene organolithium epimers 8-Li-α and 8-Li-β depicted in Figure 2, in each epimer the methyl substituent at the reacting center resides in an equatorial position in order to avoid syn-pentane repulsions with the fused ring. In addition, the seven-membered and five-membered rings adopt nearly optimal conformations in both epimeric intermediates. However, there are significant differences in the dihedral angles around the bridgehead carbons in the two epimers. Inspection of Newman projections along the C1–C2 and C3–C4 bonds of the lithiated five-membered ring (labelled with blue and red dots in Figure 2 insets, respectively) indicates that β epimers are more eclipsed then the α ones, reflected by smaller dihedral angles.[31] Such effects have been reported previously to govern the stereoselectivities in a wide variety of situations.[32] Eclipsing and staggering around the bonds attached to the two bridgehead carbons of 8-Li are correlated. Thus, C3–C4 is eclipsed and C1–C2 is staggered in less stable 8-Li-β, while C3–C4 is staggered and C1–C2 is eclipsed in more stable 8-Li-α. As the C2–Li bond is quite long in these intermediates, it is unlikely that the eclipsing involving these atoms contributes significantly to instability. Instead, the conformation around the C3–C4 bond appears to be more important.
Figure 2.

Optimized structures of solvated organolithium intermediates derived from perhydroazulene nitrile 8 after reductive lithiation. The Newman projections of interest (shown as insets) are viewed from the C1→C2 and C3→C4 directions. Relative enthalpies calculated at the B3LYP/6-31+G(d,p) level are displayed.
In summary, we have developed a general procedure for the synthesis of tertiary trialkyl-substituted organolithiums and organocuprates and employed these intermediates in the stereocontrolled construction of quaternary carbon stereocenters. Tertiary cis-perhydroazulene and cis-perhydropentalene lithium and cuprate intermediates react with carbon electrophiles with high diastereoselectivity from the sterically more-hindered concave face. Theoretical studies suggest that the thermodynamic preference for the residence of both a naked carbanion or lithium species on the concave faces of such systems dictate the observed stereoselectivity. Differential torsional strain occurring at the bridgehead atoms towards the five-membered ring is at the origin of this stability pattern. The application of prochiral tertiary organometallic and related radical intermediates[22] for fragment coupling in the stereocontrolled synthesis of natural products containing quaternary carbon stereocenters is under current investigation.
Supplementary Material
Footnotes
We thank Dr. Joe Ziller, University of California, Irvine, for the single-crystal X-ray analyses and Dr. John Greaves, University of California, Irvine, for mass spectrometric analyses. This research was supported by the NIH Neurological Disorders & Stroke Institute (Grant NS-12389), the NIH National Institutes of General Medical Sciences (Grant GM-098601), NIH and MECD postdoctoral fellowships for M.J.S. (CA-138084) and G.J.O (EX2010-1063). NMR spectra, mass spectra, and the X-ray analyses were obtained at UC Irvine using instrumentation acquired with the assistance of NSF and NIH Shared Instrumentation grants. Computations were performed on the National Science Foundation Terascale Computing System at the National Center for Supercomputing Applications (NCSA), San Diego Supercomputing Center (SDSC), the California NanoSystems Institute clusters, and the UCLA Hofman2 cluster at IDRE. We thank Dr. Nathan E. Genung for experimental assistance. Unrestricted funds from Amgen and Merck are also gratefully acknowledged.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Dr. Martin J. Schnermann, Department of Chemistry, University of California, Irvine, 1102 Natural Sciences II, Irvine, CA 92697-2025
Nicholas L. Untiedt, Department of Chemistry, University of California, Irvine, 1102 Natural Sciences II, Irvine, CA 92697-2025
Dr. Gonzalo Jiménez–Osés, Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, CA 90095-1569
Prof. Kendall N. Houk, Email: houk@chem.ucla.edu, Department of Chemistry and Biochemistry, University of California, Los Angeles, Los Angeles, CA 90095-1569.
Prof. Larry E. Overman, Email: leoverma@uci.edu, Department of Chemistry, University of California, Irvine, 1102 Natural Sciences II, Irvine, CA 92697-2025.
References
- 1.For recent reviews, see: Das JP, Marek I. Chem Commun. 2011;47:4593. doi: 10.1039/c0cc05222a.Hawner C, Alexakis A. Chem Commun. 2010;46:7295. doi: 10.1039/c0cc02309d.Cozzi PG, Hilgraf R, Zimmermann N. Eur J Org Chem. 2007:5969.Christoffers J, Baro A, editors. Quaternary Stereocenters–Challenges and Solutions for Organic Synthesis. Wiley–VCH; Weinheim: 2005. Douglas CJ, Overman LE. Proc Natl Acad Sci USA. 2004;101:5363. doi: 10.1073/pnas.0307113101.Ramon DJ, Yus M. Curr Org Chem. 2004;8:149.
- 2.The generation, configurational stability and steroselectivity of the reaction of chiral lithium reagents that contain at least one stabilizing substituent (heteroatom or aryl) has received considerable study.[3] To our knowledge, there are no such reports for tertiary lithium reagents containing three different alkyl substituents with the exception of cyclopropyl and cyclobutyl examples.[4]
- 3.For reviews and leading references, see: Gawley RE. Top Stereochem. 2010;26:93. doi: 10.1002/9783906390628.ch3.Clayden J. Organolithiums: Selectivity for Synthesis. Pergamon; Oxford: 2002. pp. 169–335.Basu A, Thayumanavan S. Angew Chem Int Ed. 2002;41:716. doi: 10.1002/1521-3773(20020301)41:5<716::aid-anie716>3.0.co;2-z.Angew Chem. 2002;114:740.
- 4.The reaction of a few tertiary cyclopropyl and cyclobutyl lithium and magnesium intermediates with carbon electrophiles to form new quaternary carbon stereocenters has been reported, see: Gaoni Y. Tetrahedron. 1989;45:2819.Mandelt K, Meyer-Wilmes I, Fitjer L. Tetrahedron. 2004;60:11587.Hodgson DM, Chung YK, Nuzzo I, Freixas G, Kulikiewicz KK, Cleator E, Paris JM. J Am Chem Soc. 2007;129:4456. doi: 10.1021/ja0672932.
- 5.The tetrasubstituted lithium enolate analogue of 1 is known to react with cyclopentenones analogous to 2 exclusively from the convex face, see: Lebsack AD, Overman LE, Valentekovich RJ. J Am Chem Soc. 2001;123:4851. doi: 10.1021/ja015802o.Schnermann MJ, Overman LE. J Am Chem Soc. 2011;133:16425. doi: 10.1021/ja208018s.
- 6.For a general review of reductive lithiation, see: ref 3b, pp. 149–165.
- 7.From tertiary chlorides and bromides: Freeman PK, Hutchinson LL. Tetrahedron Lett. 1976;17:1849.Freeman PK, Hutchinson LL. J Org Chem. 1980;45:1924.Rawson DJ, Meyers AI. Tetrahedron Lett. 1991;32:2095.Hong S, Corey EJ. J Am Chem Soc. 2006;128:1346. doi: 10.1021/ja057483x.aryl sulfides: Cherkauskas JP, Cohen T. J Org Chem. 1992;57:6.Mudryk B, Cohen T. J Am Chem Soc. 1993;115:7932.Ivanov R, Marek I, Cohen T. Tetrahedron Lett. 2010;51:174.aryl selenides: Krief A, Nazih A, Hobe M. Tetrahedron Lett. 1995;36:8111.
- 8.Remarkable progress has been recorded in recent years by Knochel and coworkers in generating and trapping polyfunctional aromatic and primary and secondary organomagnesium and organozinc reagents by exchange reactions.[9] However, such reactions would be problematic with tertiary halide precursors.
- 9.a) Knochel P, Krasovskiy A, Sapountzis I. In: Handbook of Functionalized Organometallics. Knochel P, editor. Vol. 1. Wiley–VCH; Weinheim: 2005. pp. 109–172. [Google Scholar]; b) Knochel P, Leuser H, Gong L-Z, Perrone S, Kneisel FF. In: Handbook of Functionalized Organometallics. Knochel P, editor. Vol. 1. Wiley–VCH; Weinheim: 2005. pp. 251–346. [Google Scholar]
- 10.a) Zeller E, Grierson DS. Heterocycles. 1988;27:1575. [Google Scholar]; b) Guijarro D, Yus M. Tetrahedron. 1994;50:3447. [Google Scholar]; c) Rychnovsky SD, Hata T, Kim AI, Buckmelter AJ. Org Lett. 2001;3:807. doi: 10.1021/ol006866r. [DOI] [PubMed] [Google Scholar]; d) Perry MA, Morrin MD, Slafer BW, Rychnovsky SD. J Org Chem. 2012;77:3390. doi: 10.1021/jo300161x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsao JP, Tsai TY, Chen IC, Liu HJ, Zhu JL, Tsao SW. Synthesis. 2010:4242. [Google Scholar]
- 12.La Cruz TE, Rychnovsky SD. Chem Commun. 2004:168. doi: 10.1039/b314358a. [DOI] [PubMed] [Google Scholar]
- 13.A number of achiral tertiary organocuprates (generally tert-butyl) have been generated from lithium, zinc and magnesium precursors. For several more complex examples, see: Guijarro A, Rosenberg DM, Rieke RR. J Am Chem Soc. 1999;121:4155.Rieke RD, Hanson MV. Tetrahedron. 1997;53:1925.Lipshutz BH, Sengupta S. Org React. 1992;41:135.d) Reference 7d.
- 14.Mudryk B, Cohen T. Org Synth. 1995;72:173. [Google Scholar]
- 15.Bertz SH, Eriksson M, Miao GB, Snyder JP. J Am Chem Soc. 1996;118:10906. [Google Scholar]
- 16.The use of a silyl chloride was required in order to achieve synthetically useful yields of the 1,4 adduct. For a discussion on the current understanding of the role of silyl chlorides in cuprate reactions and leading references, see: Yoshaji N, Nakamura E. Chem Rev. 2012;112:2339. doi: 10.1021/cr200241f.
- 17.For details of the synthesis of these nitriles, see the supporting information.
- 18.The relative configuration of 10 was determined unambiguously by conversion to crystalline 19, the relative configuration of which was secured by crystallographic analysis: CDCC 885481.
- 19.Identical results were realized with epimeric nitrile 9; see supporting information for details.
- 20.See supporting information for the preparation of nitrile 13.
- 21.The synthesis of nitrile 15 and the methyl epimer of product 18 is reported in the accompanying paper.[22]
- 22.Schnermann MJ, Overman LE. following communication in this issue.
- 23.In this instance, the trimethylsilylmethyl ligand underwent competitive addition to methyl vinyl ketone and a homocuprate was employed.
- 24.The impact of additives (TMEDA, HMPA, MgBr2, ZnCl2) and reaction temperature was examined in the protonation of the organolithium intermediates derived from nitriles 9 and 15. Diastereoselection was not significantly modified in any of these efforts, although the scope of these studies was limited by the highly reactive nature of the tertiary organolithium intermediates and the requirement that THF be used as the solvent for the arene-radical anion-mediated lithiation.[14]
- 25.a) To our knowledge, no information is currently available on the configurational stability of tertiary organolithiums having only alkyl substituents. The stereochemistry of protonation and methylation of 4-tert-butyl-1-phenylcyclohexyllithium has been investigated, see: Keys BA, Eliel EL, Juristi E. Isr J Chem. 1989;29:171.
- 26.a) Becke AD. J Chem Phys. 1993;98:5648. [Google Scholar]; b) Lee C, Yang W, Parr RG. Phys Rev. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 27.a) Capó M, Saá JM. J Am Chem Soc. 2004;126:16738. doi: 10.1021/ja045203s. [DOI] [PubMed] [Google Scholar]; b) Lange H, Huenerbein R, Fröhlich R, Grimme S, Hoppe D. Chem Asian J. 2008;43:78. doi: 10.1002/asia.200700261. [DOI] [PubMed] [Google Scholar]
- 28.a) Hoppe I, Marsch M, Harms K, Boche G, Hoppe D. Angew Chem Int Ed. 1995;34:2158. [Google Scholar]; Angew Chem. 1995;107:2328. [Google Scholar]; b) Zarges W, Marsch M, Harms K, Koch W, Frenking G, Boche G. Chem Ber. 1991;124:543. [Google Scholar]; c) Ly HV, Konu J, Parvez M, Roesler R. Dalton Trans. 2008:3454. doi: 10.1039/b802930j. [DOI] [PubMed] [Google Scholar]
- 29.The structures of each of the two epimers of the same complex are very similar; thus, it was assumed that temperature and entropy effects are negligibly small and thus ΔΔG ≈ ΔΔH.
- 30.See supporting information for further computational details and full descriptions of the calculated structures.
- 31.Newman projections of perhydropentalene lithiums 8-Li-α and 8-Li-β can be found in supporting information.
- 32.a) Wang H, Kohler P, Overman LE, Houk KN. submitted. [Google Scholar]; b) Caramella P, Rondan NG, Paddon MN, Houk KN. J Am Chem Soc. 1981;103:2438. [Google Scholar]; c) Rondan NG, Paddon MN, Caramella P, Mareda J, Mueller PH, Houk KN. J Am Chem Soc. 1982;104:4974. [Google Scholar]; d) Paddon MN, Rondan NG, Houk KN. J Am Chem Soc. 1982;104:7162. [Google Scholar]; e) Lucero MJ, Houk KN. J Org Chem. 1998;63:6973. doi: 10.1021/jo980760g. [DOI] [PubMed] [Google Scholar]; f) Cheong PH, Yun H, Danishefsky SJ, Houk KN. Org Lett. 2006;8:1513. doi: 10.1021/ol052862g. [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Martinelli MJ, Peterson BC, Khau VV, Hutchinson DR, Leanna MR, Audia JE, Droste JJ, Wu YD, Houk KN. J Org Chem. 1994;59:2204. [Google Scholar]; h) Houk KN, Paddon-Row MN, Rondan NG, Wu YD, Brown FK, Spellmeyer DC, Metz JT, Li Y, Loncharich RJ. Science. 1986;231:1108. doi: 10.1126/science.3945819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.