

The carbazole alkaloids represent a large and structurally rich family of natural products that are produced by a variety of terrestrial plants.[1] In particular, plants within the Rutaceae family are notable producers of these compounds–with the genus Murraya providing the greatest number of unique structures. Commonly found in the outer Himalayas and on the Indian peninsula, the leaves of M. koenigii (L.) Spreng are, inter alia, extensively utilized as a spice flavoring by the peoples of these areas giving it the name “curry-leaf tree”. Extracts of the plant are also used in local medicine due to their antimicrobial activity.[2]
Within this large family of alkaloids, we were particularly interested in the axially chiral dimeric carbazoles,[3] and most specifically bismurrayaquinone A (1, Figure 1). This unique dimeric carbazolequinone was isolated from the roots of M. koenigii (L.) Spreng by Furukawa and coworkers in 1993[4] and has been the subject of non-stereoselective total syntheses by the groups of both Bringmann and Murphy.[5] Somewhat surprisingly, there has never been an enantioselective synthesis of 1. Bringmann and coworkers did, however, prepare racemic 1 and separated the two enantiomers using preparative HPLC,[5a] and in 2001 reported an approach towards a stereoselective synthesis.[6]
Figure 1.
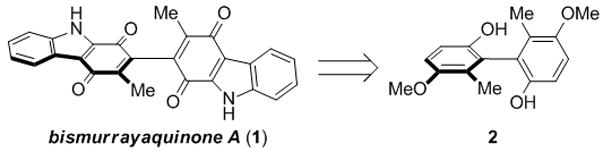
Bismurrayaquinone A (1) and Key Biphenol 2.
Because no enantioselective synthesis of bismurrayaquinone A (1) had been reported, and because there is very little literature data on axially chiral biquinones,[7] we were compelled to explore strategies for the enantioselective synthesis of these molecules. Recently, we reported the development of a new method for synthesizing enantiomerically pure axially chiral biphenols by a method we termed “traceless stereochemical exchange”.[8] This method allows ready access to biphenols of the type represented by structure 2 (Figure 1), and herein we report the use of this method to conduct the first enantioselective synthesis of bismurrayaquinone A (1). These studies have also revealed a fascinating phenomenon related to the configurational stability of chiral biquinones.
Our synthesis of bismurrayaquinone A (1) commenced from commercially available 4-methoxyphenol (3), which was converted into enone 4 following oxidation to the corresponding dimethylketal quinone[9] and subsequent enantioselective conjugate addition of dimethylzinc (Scheme 1).[10]
Scheme 1.
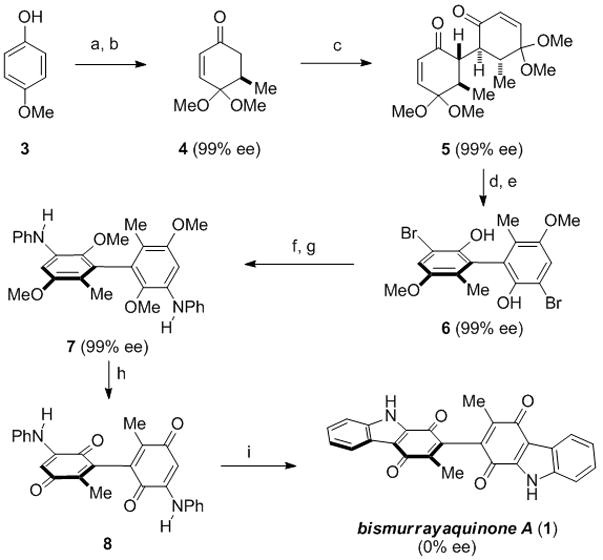
a) PhI(OAc)2, MeOH, 91%; b) Me2Zn, (S,R,R)-phosphoramidite ligand (4.5% mol%), Cu(OTf)2 (2 mol%), 45–52%; c) LDA; CuCl2, 63%; d) BF3•OEt2, toluene, reflux, 87%; e) Br2, 90%; f) MeI, KOH, 84%; g) aniline, Pd(OAc)2 (8 mol%), [HPt-Bu3][BF4] (11 mol%), NaOt-Bu, toluene, reflux, 93%; h) BBr3; SiO2/air, 62%; i) Pd(OAc)2 (0.5 equiv.), Cu(OAc)2, AcOH, reflux, 33%. OAc = acetate, OTf = trifuoromethane sulfonate, LDA = lithium diisopropylamide, (S,R,R)-phosphoramidite ligand = (S,R,R)-(−)-(3,5-Dioxa-4-phosphacyclohepta[2,1-a:3,4-a′]dinaphthalen-4-yl)bis(1-phenylethyl)amine.
The efficiency of the Feringa conjugate addition in terms of yield dropped from 52% on a 5 gram scale to 45% yield when conducted on 10 grams, but the enantioselectivity was maintained at 99% ee. Gram-scale oxidative dimerization of 4 was conducted under our reported conditions[8] to provide dione 5 in 63% yield (99% ee).[11] Conversion of dione 5 to bromophenol 6 proceeded smoothly after aromatization to 2 (not shown) and regioselective bromination. Our plan at this juncture was to utilize methods for palladium-catalyzed carbazolequinone synthesis to complete the synthesis.[12] Accordingly, we converted bromide 6 to bisaniline 7 through O-methylation followed by an efficient Buchwald–Hartwig amination with aniline (93%) using conditions reported by Bedford and coworkers.[13] Conversion of 7 to the open biquinone 8 was carried out as a prelude to carbazole formation. This transformation was best achieved by global demethylation of 7 with BBr3 to produce the free dihydroquinone, which spontaneously oxidized to biquinone 8 upon stirring the reaction mixture with silica gel in the presence of air (62%). Exposure of biquinone 8 to Knölker’s conditions for Pd(OAc)2-catalyzed carbon–carbon bond formation[12a] generated bismurrayaquinone A (1) in a modest 33% yield. Spectroscopic data [H-NMR, C-NMR] were identical to those reported in the literature,[4,5a] but analysis of the synthetic material revealed it to be racemic.[14]
In order to determine the point at which racemization had occurred we first established that dihydroquinone 7 was optically pure (99% ee). Analysis of quinone 8 shortly after it was isolated indicated that it maintained its optical purity initially, but underwent an interesting N–O tautomerization to form enol 9 after purification (Scheme 2). This tautomerization complicated the analysis of optical purity. We could, however, resolve the enantiomeric tautomers of 9 by HPLC and after 150 hours at room temperature noted the sample was 59% ee. The sample was fully racemic by 500 hours at room temperature. Given the elevated temperatures required for carbazole formation, the 59% ee sample of 9 provided racemic bismurrayaquinone A (1) in 65% yield using Knölker’s conditions for carbazole formation.[12a]
Scheme 2.
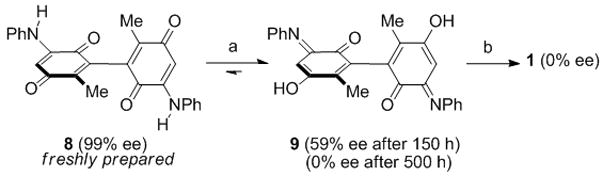
a) room temperature, 150 h, 100% conversion; b) Pd(OAc)2 (0.5 equiv.), Cu(OAc)2, AcOH, reflux, 65%. OAc = acetate.
These findings raised questions regarding the configurational stability of the natural product itself. In their isolation report, Furukawa and coworkers did not record an optical rotation or CD-spectrum of natural bismurrayaquinone A (1),[4] and so it is not known whether 1 exists as a single enantiomer or as a racemate in nature. As mentioned in the introduction, Bringmann and coworkers were able to separate racemic 1 using HPLC. Despite recording CD spectra of their pure enantiomers they did not, however, mention anything regarding the configurational stability of 1. In light of our findings regarding the stability of quinone 8, we anticipated that enantiomerically pure bismurrayaquinone A (1) would most likely undergo rapid racemization, and we next sought to generate optically pure bismurrayaquinone A (1) in order to test this assumption.
Given that dimethylhydroquinone 7 was configurationally stable, we expected the corresponding carbazole (i.e., 11) to be similarly stable (Scheme 3). Mild oxidative conversion of 7 into 1 would then allow us to follow the racemization of the natural product and provide fundamental insight into these structures. Dihydroquinone 7 proved resistant to palladium-catalyzed carbazole formation under a wide variety of conditions. However, conversion of bromide 6 into chloroaniline 10 through Buchwald–Hartwig amination with 2-chloroaniline allowed the desired carbazole 11 to be formed in good yield (65%, 2 steps from 6).[13,15] Conversion to bismurrayaquinone A (1) was achieved through a rapid and high yielding oxidation with ceric ammonium nitrate (95% yield).
Scheme 3.
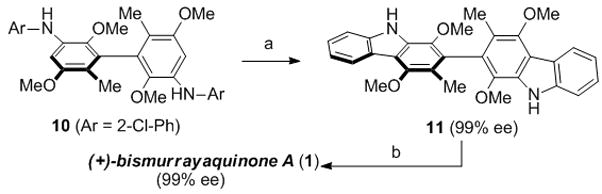
a) Pd(OAc)2 (20 mol%), [HPt-Bu3][BF4] (28 mol%), NaOt-Bu, 160 °C, μwave, 73% b) CAN, MeCN/H2O, 95%. OAc = acetate, CAN = ceric ammonium nitrate.
Much to our surprise, synthetic bismurrayaquinone A (1) was configurationally stable at room temperature and did not tautomerize. We therefore decided to experimentally determine the activation barriers for atropisomerization for a number of biquinones, including the natural product (Figure 2). While bismurrayaquinone A (1) was configurationally stable at room temperature, it racemized within 130 hours when heated to 105 °C and within 51 hours at 110 °C. The activation energy (Ea) for atropisomerization of 1 was determined to be 54 kcal•mol−1 by monitoring the change in enantiomeric excess over time at two different temperatures (see Supporting Information). Unfortunately, open biquinone 8 proved difficult to study due to tautomerization (see Scheme 2), while the diphenyl homolog 12 underwent rapid racemization during its synthesis from the corresponding dihydroquinone. The simplified quinone 13 was more stable than 12, but significantly less so than bismurrayaquinone A (1): racemization was complete after 50 hours at room temperature. The Ea for atropisomerization of 13 was determined to be 13 kcal•mol−1, significantly lower than bismurrayaquinone A (1). The corresponding biphenol 2 required heating to 180 °C in order for racemization to be observed, but competing decomposition afforded data that was unacceptable for calculating the barrier for atropisomerization.
Figure 2.
Racemization studies of biquinones.
A priori rationalization of the relative stability of bismurrayaquinone A (1) compared to the other biquinones studied was challenging. We initially speculated that racemization might be induced photochemically as related phenomena have been reported for several axially chiral biaryls.[16] Racemization of quinone 8 proceeded just as rapidly in the dark as it did in sunlight, however. Lumb and Trauner showed in their elegant biomimetic synthesis of microphyllaquinone that binapthoquinones will undergo a tautomerization/6π-electrocyclization cascade.[17] If such a process was reversible this may promote racemization, but under our neutral conditions we never observed the formation of pyran species that would indicate such a process was operative for 1 or 13.[18] We next turned to molecular modelling of the atropisomerization of 1 and 13, which was carried out by calculating an energy profile with changing dihedral angle about the central C–C bond (Spartan, B3LYP 6–31G* level).[19] Unfortunately, at this level of theory both compounds gave activation barriers of approximately 23–25 kcal•mol−1, values significantly different than the experimentally determined values. We could not make any firm conclusions from these calculations.
Insight came when we succeeded in growing single crystals of bismurrayaquinone A (1) and biquinone 13 that were suitable for X-ray crystallographic analysis.[20] As anticipated each of the biquinones possessed shorter C–O bonds than the corresponding biphenol 2[8] (1.22 Å for 1 and 13 vs 1.37 Å for 2), establishing a rationale for the lower barriers of biquinones compared to biphenols. In general bond lengths for 1 and 13 were similar, but we noted greater out-of-plane distortion for biquinone 13 in comparison to bismurrayaquinone A (1). A view of the X-ray structures of each molecule, with an arrow highlighting the effect of the distortion, clearly shows this interesting phenomenon (Figure 3, atoms from 1 have been removed for clarity).
Figure 3.
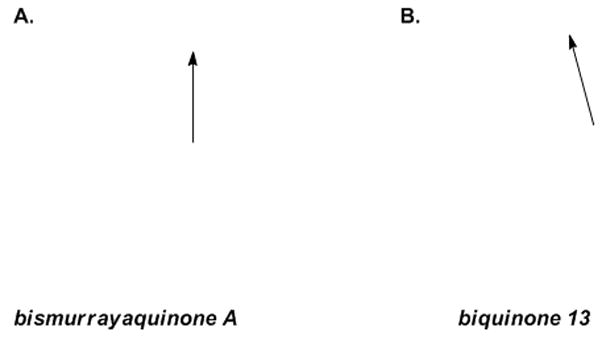

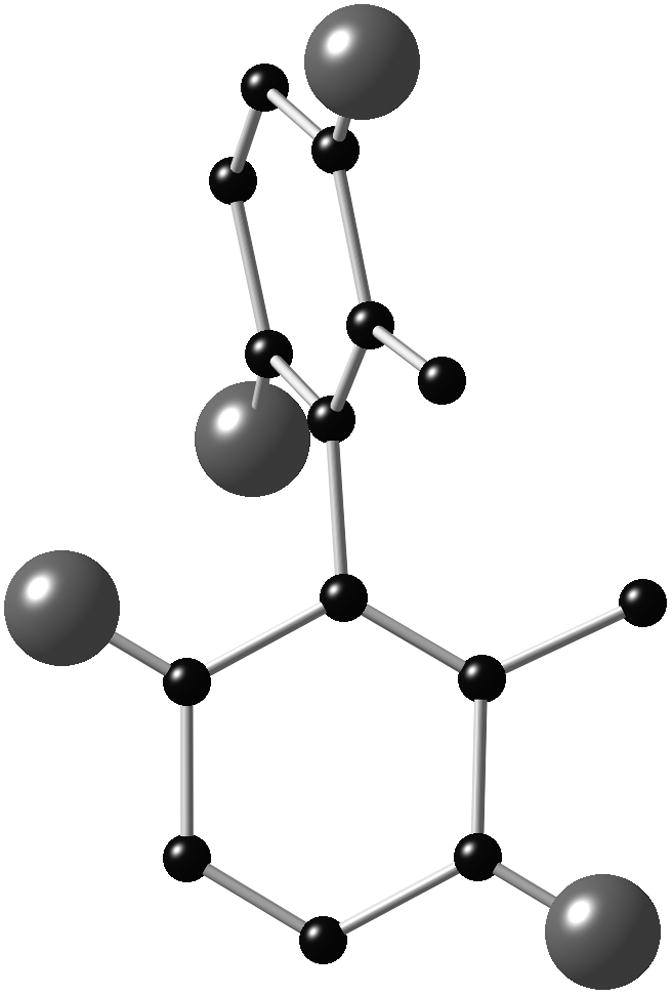
X-ray structures of bismurrayaquinone A (1, A) and biquinone 13 (B), with some atoms removed for clarity.
Out-of-plane distortion of the type exhibited by biquinone 13 has been proposed by Ling and Harris to explain observed trends in the activation parameters for the racemization of 2,2′-di-iodobiphenyls.[21] These deformations lead to significant ground-state destabilization and allow the substituents on either side of the central carbon–carbon bond to pass one another sequentially during atropisomerization. Consequently, more easily accommodated out-of-plane distortion should lead to lower barriers to atropisomerization; an outcome consistent with our experimental observations in the current study. Related distortions are seen in the X-ray structure of gossypolone,[22] a 2,2′-binaphthoquinone that racemizes at room temperature.[7a,b]
Lastly, our X-ray structure of bismurrayaquinone A (1) enabled us to confirm its absolute configuration though Bijvoet Pair analysis.[23] Thus, the sequences outlined herein provided (R)- or M-(+)-bismurrayaquinone A. We also prepared (S)- or P(−)-bismurrayaquinone A through an analogous sequence, employing ent-4. Curiously, however, the CD spectrum we recorded for M-1 and P-1 displayed the opposite shape to that predicted by the calculations reported by Bringmann and coworkers in 1995.[5a] In their manuscript, Bringmann and coworkers note that application of the standard exciton chirality method[24] was not possible, and so made recourse to an alternative method involving semi-empirical AM1 and CNDO calculations to predict the CD spectrum of each enantiomer. Advances in computational techniques, however, have now allowed a recalculation of the CD spectrum using the TDCAM-B3LYP/TZVP method.[25] These new results support our X-ray analysis that (+)-bismurrayaquinone A possesses the (R)- or M-configuration. Despite the fact that any definitive statement as to the naturally occurring configuration of bismurrayaquinone A (1) will require re-isolation from the curry-leaf tree, our work and that of Bringmann serves to highlight the impact synthetic chemistry can have on fundamental questions of structure and stereochemistry.
In conclusion, we have completed the first enantioselective synthesis of bismurrayaquinone A (1) through a concise strategy that employs traceless stereochemical transfer. The approach utilized axially chiral bromide 6 as a late-stage precursor that allowed for a bidirectional palladium-catalyzed carbazole synthesis. Future elaboration of this useful biphenyl core will allow for the synthesis and biological evaluation of numerous bismurrayaquinone A analogs. The activation parameter for atropisomerisation of bismurrayaquinone A (1) was experimentally determined and a rationale based upon X-ray data analysis has been proposed to account for the observed differences in barriers between related compounds. Further detail of our studies into the atropisomerism of biquinones will be reported in due course.
Supplementary Material
Acknowledgments
We thank Reed Larson (NU) for assistance in X-ray crystallographic analysis. We gratefully thank Prof. Dr. Gerhard Bringmann and Dr. Torsten Bruhn (University of Würzburg) for CD spectra calculations.
We thank Professor Gerhard Bringmann and Dr. Torsten Bruhn (University of Würzburg) for conducting these calculations. Details are contained within the Supporting Information.
Footnotes
This work was supported by the National Institutes of Health (1R01GM085322), Northwestern University (NU) and Amgen.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.For relevant reviews on the isolation, biological activity and synthesis of carbazole alkaloids, see:. Knölker HJ, Reddy KR. Chem Rev. 2002;102:4303–4427. doi: 10.1021/cr020059j.Knölker HJ. To Curr Chem. 2005;244:115–148.
- 2.Ramsewak RS, Nair MG, Strasburg GM, DeWitt DL, Nitiss JL. J Agric Food Chem. 1999;44:444–447. doi: 10.1021/jf9805808. [DOI] [PubMed] [Google Scholar]
- 3.For a review of biarylic biscarbazole aklaoids, see: Tasler S, Bringmann G. Chem Rec. 2002;2:114–127. doi: 10.1002/tcr.10014.The following reviews on axially chiral biaryl compounds also contains a wealth of interesting information, see; Bringmann G, Mortimer AJP, Keller PA, Gresser MJ, Garner J, Breuning M. Angew Chem. 2005;117:5518–5563. doi: 10.1002/anie.200462661.Angew Chem, Int Ed. 2005;44:5384–5427. doi: 10.1002/anie.200462661.Bringmann G, Gulder T, Gulder TAM, Breuning M. Chem Rev. 2011;111:563–639. doi: 10.1021/cr100155e.
- 4.Ito C, Thoyama Y, Omura M, Kajiura I, Furukawa H. Chem Pharm Bull. 1993;41:296–2100. [Google Scholar]
- 5.Bringmann G, Ledermann A, Stahl M, Gulden KP. Tetrahedron. 1995;51:9353–9360.Murphy WS, Bertrand M. J Chem Soc, Perkin Trans 1. 1998:4115–4119.Bringmann G, Tasler S. Tetrahedron. 2001;57:331–343.Harrity and coworkers reported the synthesis of racemic N-Me-bismurrayaquinone, see: Davies MW, Johnson CN, Harrity JPA. J Org Chem. 2001;66:3525–3532. doi: 10.1021/jo010098f.
- 6.Bringmann G, Tasler S, Endress H, Mühlbacher Jörg. Chem Commun. 2001:761–762. [Google Scholar]
- 7.The structurally related biquinone, gossypolone, has been shown to racemize at room temperature, see: Dao VT, Dowd MK, Martin MT, Gaspard C, Mayer M, Michelot RJ. Eur J Med Chem. 2004;39:619–624. doi: 10.1016/j.ejmech.2004.04.001.Zhang L, Jiang H, Cao X, Zhao H, Wang F, Cui Y, Jiang B. Eur J Med Chem. 2009;44:3961–3972. doi: 10.1016/j.ejmech.2009.04.025.For related phenomena observed for arylquinones, see: Maharoof USM, Sulikowski GA. Tetrahedron Lett. 2003;44:9021–9023.
- 8.Guo F, Konkol LC, Thomson RJ. J Am Chem Soc. 2011;133:18–20. doi: 10.1021/ja108717r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prepared according to the method of Pelter and Elgendy, see: Pelter A, Elgendy S. Tetrahedron Lett. 1988;29:677–680.
- 10.Imbos R, Brilman MHG, Pineschi M, Feringa BL. Org Lett. 1999;1:623–625. [Google Scholar]
- 11.The enantiomeric excess for all chiral molecules was determined by HPLC analysis. See Supporting Information for details.
- 12.a) Knölker HJ, Reddy KR, Wagner A. Tetrahedron Lett. 1998;39:8267–8270. [Google Scholar]; b) Liégault B, Lee D, Huestis MP, Stuart DR, Fangou K. J Org Chem. 2008;73:5022–5028. doi: 10.1021/jo800596m. [DOI] [PubMed] [Google Scholar]
- 13.Bedford RB, Betham M. J Org Chem. 2006;71:9403–9410. doi: 10.1021/jo061749g. [DOI] [PubMed] [Google Scholar]
- 14.This was initially concluded based upon a flat CD-spectrum, but later confirmed by HPLC analysis. See Supporting Information for details.
- 15.Bedford RB, Betham M, Charmant JPH, Weeks AL. Tetrahedron. 2008;64:6038–6050.Campeau LC, Parisien M, Jean A, Fagnou K. J Am Chem Soc. 2006;128:581–590. doi: 10.1021/ja055819x.For the successful implementation of a similar carbazole formation in natural product synthesis, see: Bouclé S, Guillard J. Synthesis. 2011:1616–1620.Sánchez JD, Avendaño C, Menéndez JC. SynLett. 2008:1371–1375.
- 16.For some key examples, see: Mislow K, Gordon AJ. J Am Chem Soc. 1963;85:3521–3521.Zimmerman HE, Crumrine DS. J Am Chem Soc. 1972;94:498–506.Zhang M, Schuster GB. J Phys Chem. 1992;96:3063–3067.Hattori T, Shimazumi Y, Goto H, Yamabe O, Morohashi N, Kawai W, Miyano S. J Org Chem. 2003;68:2099–2108. doi: 10.1021/jo026747k.
- 17.Lumb JP, Trauner D. Org Lett. 2005;7:5865–5868. doi: 10.1021/ol052472u. [DOI] [PubMed] [Google Scholar]
- 18.A 3,3′-diphenyl substituted biquinone that cannot tautomerize also racemized at room temperature, providing further evidence against such a mechanism.
- 19.Details are provided in the Supporting Information.
- 20.CCDC 829781–829784 and CCDC 835897 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. Supporting Information for this manuscript contains additional details on the assignment of absolute strereochemistry through X-ray analysis.
- 21.Ling CCK, Harris MM. J Chem Soc. 1964:1825–1835. [Google Scholar]
- 22.X-ray structure: Talipov SA, Ibragimov BT, Beketov KM, Yu Sh, Islambekov, Mardanov RG, Ismailov AI. Khim Prir Soedin. 1995:814–819.Computations: Beisel CL, Dowd MK, Reilly PJ. J Mol Struct: THEOCHEM. 2005;730:51–58.The related structure of 6,6′-dimethoxygossypolone displays similar deformations, see: Zelaya CA, Stevens ED, Dowd MK. Acta Crystallogr, Sect C: Cryst Struc Commun. 2010;66:517–520. doi: 10.1107/S0108270110036541.
- 23.Hooft RWW, Straver LH, Spek AL. J Appl Cryst. 2008;41:96–103. doi: 10.1107/S0021889807059870. This assignment is consistent with the configuration established for biphenol 2 and bromide S2. See the Supporting Information for additional details. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harada N, Nakanishi K. Circular Dichroic Spectroscopy - Exciton Coupling in Organic Stereochemistry. University Science Books; Mill Valley, CA: Oxford University Press; Oxford: 1983. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.