

Initial entry of HIV-1 into host cells remains a compelling and yet elusive target for developing agents to prevent infection. This step is mediated by a sequence of interactions of a trimeric gp120/gp41 envelope (Env) protein complex with host cells, including initial gp120 encounter with the cellular receptor CD4 and a chemokine co-receptor usually either CCR5 or CXCR4 [1]. A peptide triazole class of entry inhibitor leads has been shown to bind to gp120 with close to nanomolar affinity, to suppress protein ligand interactions of the Env protein at both its CD4 and co-receptor binding sites and to inhibit cell infection by a broad range of virus subtypes [2]. These inhibitors appear to function mechanistically by conformationally entrapping gp120 in an inactivated state, different from either the flexible ground state of gp120 or the highly structured CD4-activated state. This entrapment effectively halts the entry process at the initial binding stages.
The promising functional activity and unique mode of action of peptide triazoles as HIV-1 entry inhibitors led us to seek potency enhancement by multivalent conjugation. While enhancement of antiviral activity in cell infection assays was indeed achieved, we concomitantly observed the striking ability of the multivalent conjugate as well as the precursor peptide triazole derivative to disrupt the viral particles in the absence of cells. We herein report this unexpected finding, which has significant implications for both prevention and therapeutic applications.
For this study, we synthesized the peptide triazole denoted KR13, composed of the 12-residue amino acid sequence of the previously identified high potency peptide-triazole HNG156 [2d] with a C-terminal extension containing a Cys-SH group (Figure 1A). This cysteine-containing derivative was selected because the introduced SH group facilitates conjugation to the AuNP carriers. In addition, the extension contained β-Ala residues for spacing and a Gln residue for potential side chain modifications. KR13 was prepared by manual solid phase synthesis using Fmoc chemistry on a Rink amide resin at 0.25 mmol scale [2a]. The amino acid sequence of KR13 is RINNIXWSEAMMβAQβAC-NH2, where X is ferrocenyltriazole-Pro. The ferrocenyl group was found in earlier studies [2a, 2c] to lead to optimized peptide triazole potency and was retained here to evaluate the impact of multivalency with this high-efficacy derivative. Direct binding of the peptide triazole to immobilized HIV-1YU2gp120 was measured as previously described [2a] using Surface Plasmon Resonance (SPR) with a Biacore 3000 optical biosensor (GE Healthcare). Steady state analysis was conducted using the method of Morton and coworkers [3] (Figure 1B). KR13 activity was characterized by testing competitive inhibition of soluble CD4 and mAb 17b binding to HIV-1YU2gp120 through Enzyme Linked Immunosorbent Assay (ELISA). The molecular interaction analyses showed that HNG156 analog (KR13) retained high affinity gp120 binding (Figure 1B) and the dual receptor site competition (Figure 1C) functions of HNG156[2d, 2e]. Further, multivalent gold nanoparticle (AuNP) conjugates of KR13 were constructed (Figure 1D) to test the possibility of enhanced antiviral activity by nano-conjugates. The AuNPs were synthesized using a modified citrate reduction method to obtain size-controlled, stable and monodisperse AuNPs [4]. The peptide (KR13) was conjugated to the AuNP using a direct gold-thiol covalent link by incubating the peptide and AuNP at room temperature for 30 minutes. The AuNP-KR13 was purified by filtration and ultracentrifugation and the extent of peptide triazole conjugation on AuNPs was determined using amino acid analysis (Yale University). The size and extent of polydispersity of the AuNP-KR13 conjugates were measured using Transmission Electron Microscopy (TEM) with a JEM 2100 operated at 200kV, and Dynamic Light Scattering (DLS) with a Zetasizer NS90 (Malvern Instruments). The TEM image is shown in Figure 1E. The methodological details are given in the supporting information provided.
Figure 1.
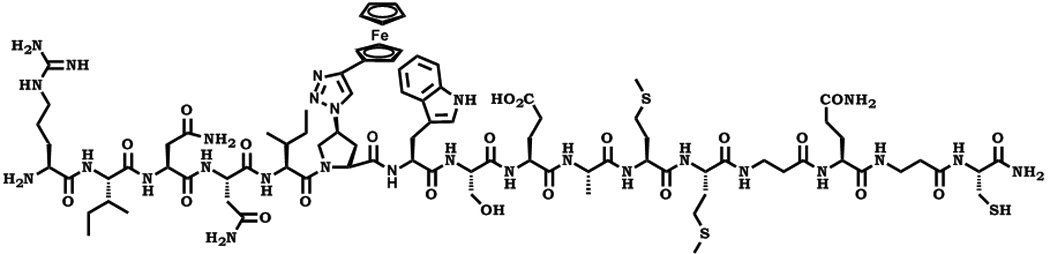
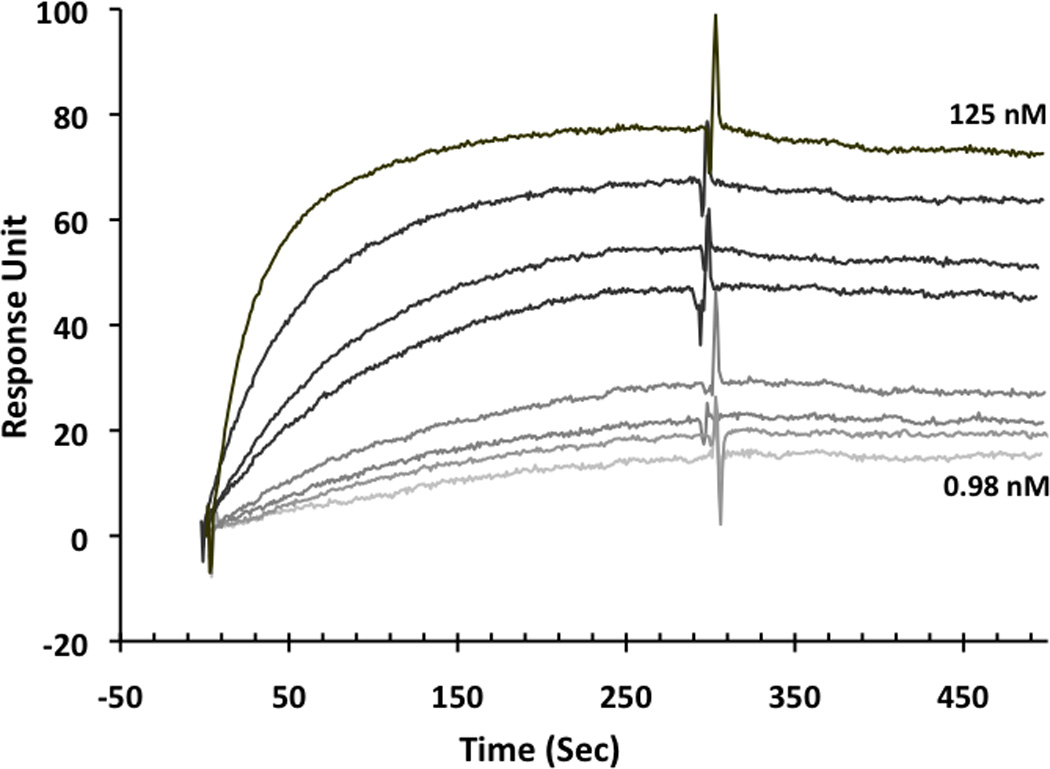
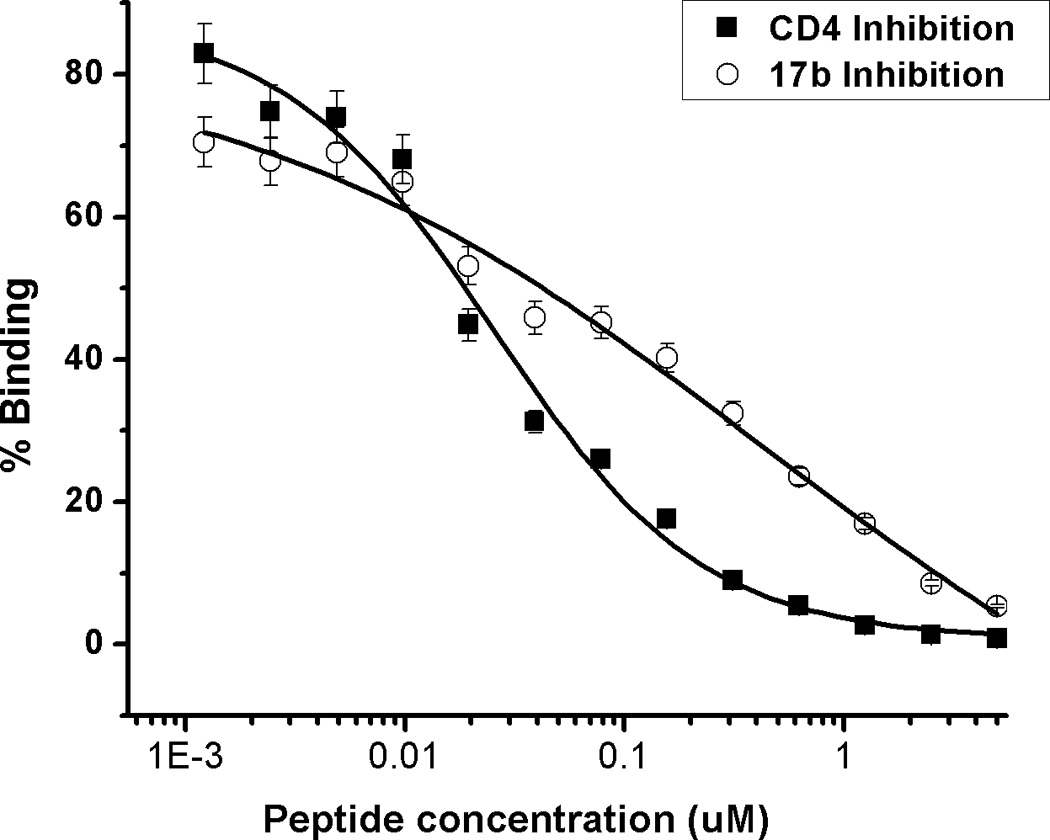
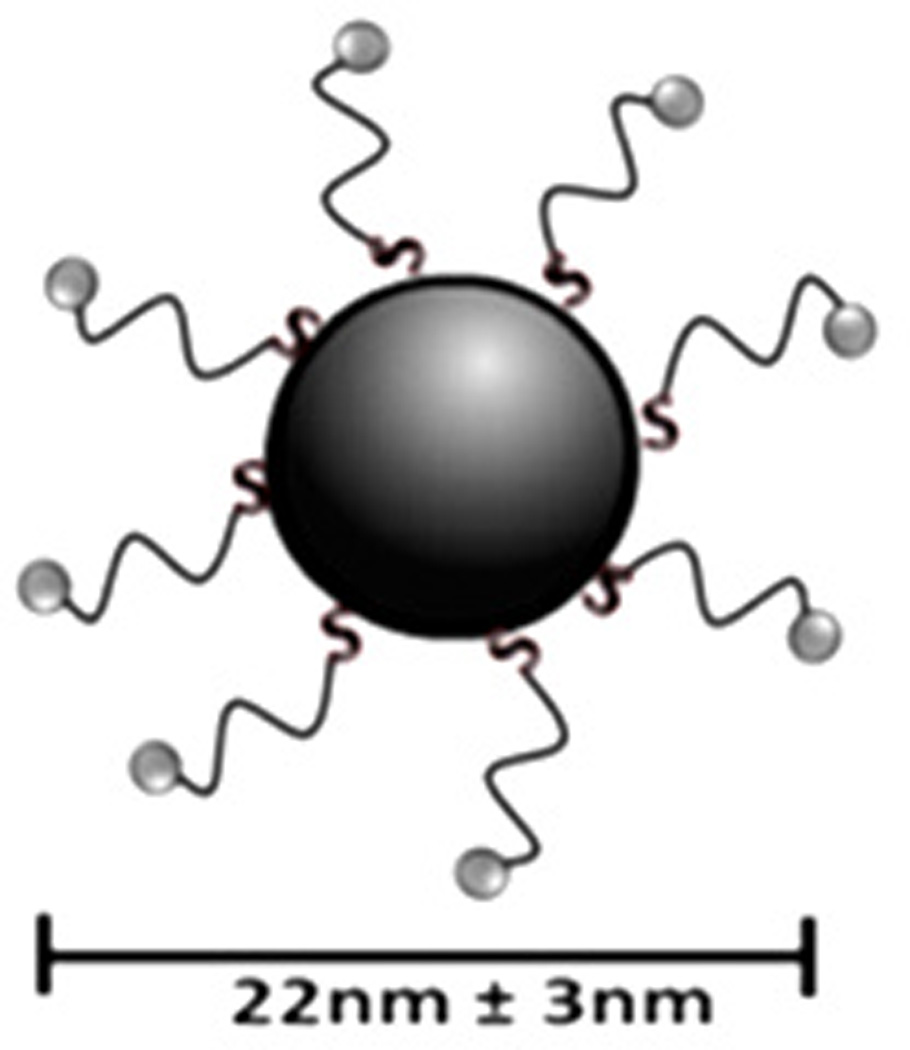
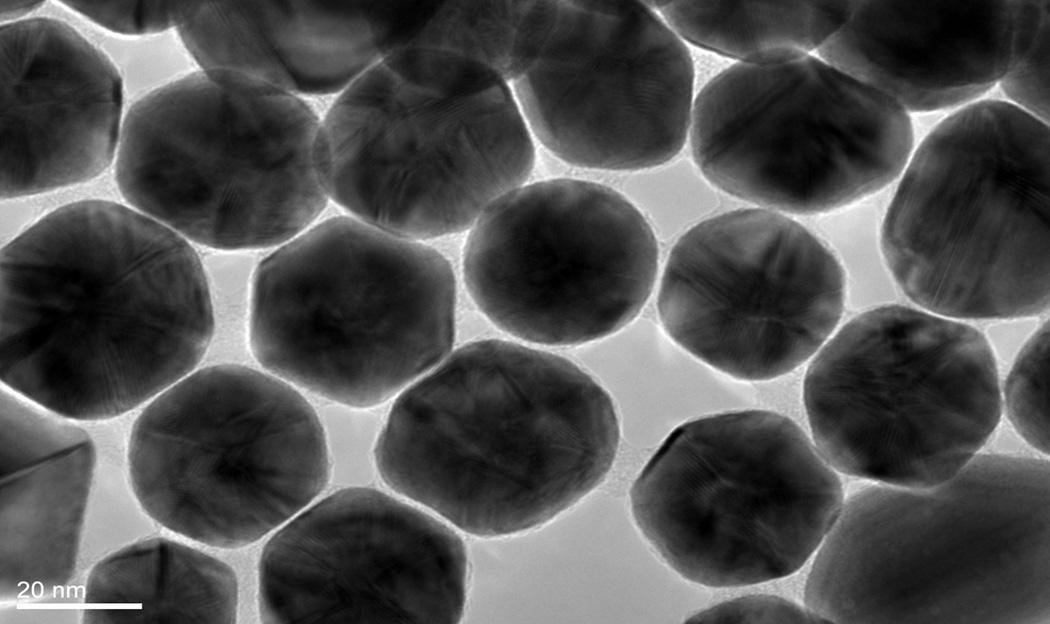
Binding activity of peptide triazole KR13, and the AuNP conjugate derived from this peptide. (A) KR13 primary structure, (B) SPR sensograms of direct binding of KR13 to immobilized gp120. Sensorgram graytones are darker with increasing analyte concentrations; Kd from steady state analysis = 11.3 nM, obtained from the steady state fit [3b] (C) ELISA-derived competition plots for 17b (IC50 = 45.3 ± 1.2 nM) and CD4 (IC50 = 25 ± 4.2 nM) binding to plate-immobilized gp120. (D) Scheme of the AuNP-KR13 conjugate synthesized using thiol linkage; size of AuNP-KR13 measured using DLS (n=3). (E) TEM image of AuNP-KR13.
The HIV-1 viral entry inhibition potencies of KR13 and AuNP-KR13 conjugates were compared using a single-round pseudoviral infection luciferase reporter assay as previously described [2e]. The profiles for inhibition of infection of modified Human Osteosarcoma Cells (HOS.T4.R5) engineered to express CD4 and CCR5 receptor and co-receptor respectively by pseudotyped HIV-1BaL are shown in Figure 2A. Compared to peptide triazole alone, the AuNP-KR13 conjugate exhibited a close to 3-orders of magnitude enhancement of infection inhibition activity, IC50 values were 23 ± 6 nM and 1 ± 0.1 nM, respectively, for KR13 alone and AuNP-KR13 conjugate. This potency enhancement was similar to previous observation of inhibition enhancement with a multivalent AuNP derivative of the CCR5 antagonist TAK-779 AuNPs [5]. The lack of inhibition of cell infection by control VSV-G pseudotype virus (Figure 2A) shows that the viral inhibitions of both KR13 and AuNP-KR13 are specific for HIV-1 envelope. No significant cytotoxicity was observed for either KR13 or AuNP-KR13 (Figure 2B).
Figure 2.
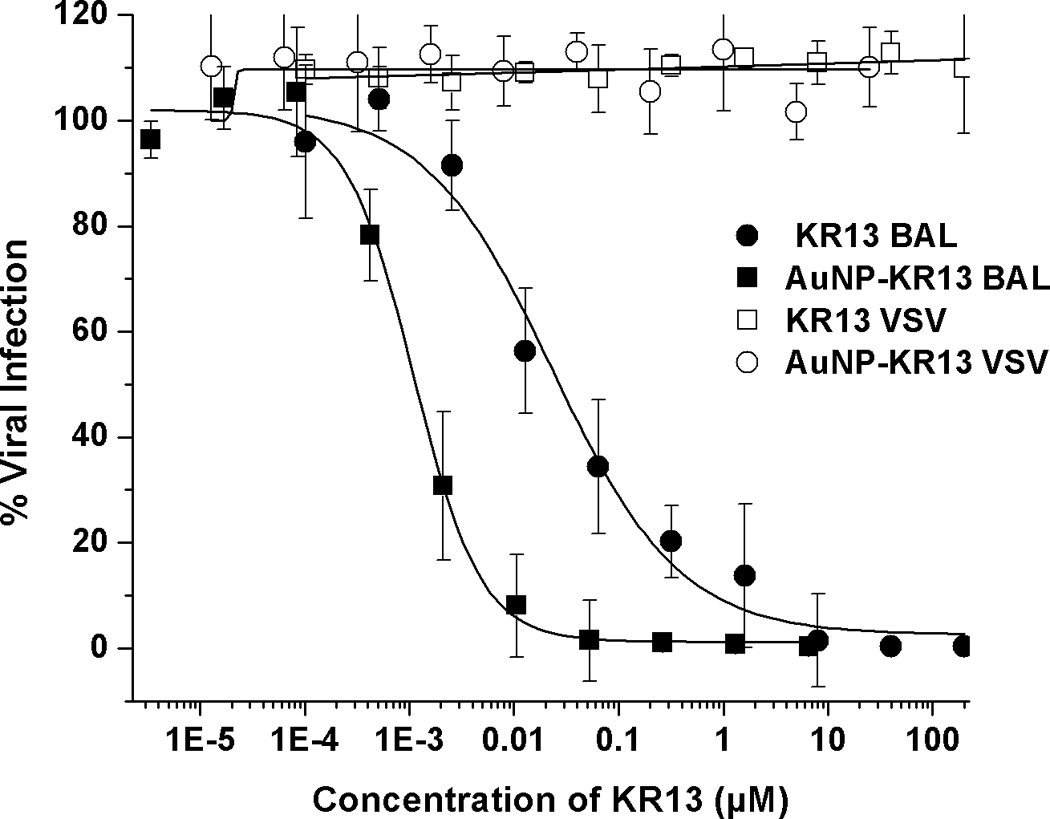

Inhibition of single round cell infection by KR13 (circles) and AuNP-KR13 (squares). (A) Inhibition of infection of HOS.T4.R5 cells by recombinant viruses pseudotyped with the envelope for HIV-1BaL (solid) or with the envelope for VSV-G (open). The data were normalized to 100% infection activity at 0 µM concentration of KR13. The calculated IC50 of KR13 alone was 23 ± 6 nM, n=4; that of the AuNP-KR13 conjugate was 1 ± 0.1 nM n=3. Efficacy parameters were calculated using sigmoidal logistic fit in Origin Pro 8 software. B) Cell toxicity assay using WST-1 reagent; no statistically significant differences measured, P<<<0.05, n =3 using a t test.
We subsequently tested the effects of KR13 and AuNP-KR13 on the virus particle itself by measuring release of the nucleocapsid protein p24. This was done with the initial intent to broaden our understanding of the effects of the inhibitors on the virus in the earliest stages of HIV-1 infection. Capsid protein release from virus pseudoparticles was detected by incubating the inhibitor with purified pseudovirus for 30 minutes at 37 °C followed by detection of release of p24 in the supernatant using western blot analysis with an Odyssey Infrared Imaging System (Li-Cor). For this p24 release analysis, the positive control was 1% Triton X-100 detergent-lysed pseudovirus, and the negative control was mock treated intact virus. Western blot analyses (Figure 3A) revealed concentration dependent p24 release by both KR13 and AuNP-KR13. Quantitation of p24 band intensities, obtained with Image J software, enabled assessment of IC50 values (Figure 3B) and showed that the hierarchy of disruption potencies was similar to that found (Figure 2) for inhibition of cell infection. The absolute IC50 values, obtained for p24 release dose response data fitted using Origin Pro 8, were 866 ± 55 nM and 15.6 ± 2 nM, respectively, for KR13 and the AuNP-KR13.
Figure 3.
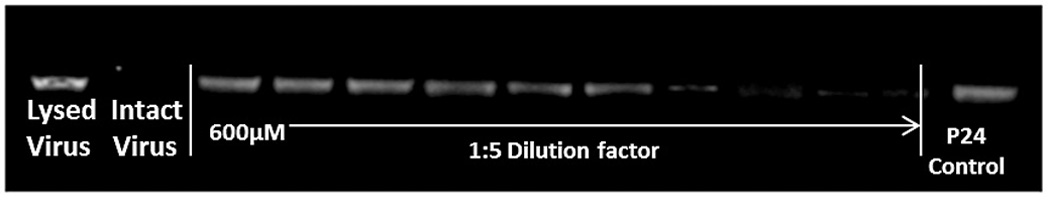


Gag p24 release from HIV1BaL pseudotype virus caused by KR13 and AuNP-KR13. (A) and (B): Western blot gel images showing p24 release as a function of dose of KR13 alone (A) and AuNP-KR13 (B). Controls shown are lysed virus (treated with 1% Triton X-100) intact virus (no treatments) and p24 control (5 µL of 20 µg/ml). The highest concentration of KR13 in both cases is indicated, with subsequent lanes corresponding to sequential 1:5 fold dilutions indicated by arrows. C) Dose response plots of band intensities of the relative release percentage of p24 compared to the lysed virus control for both KR13 alone (circles) and AuNP-KR13 (squares).
A similar relationship of potency enhancement was observed in the pseudoviral infectivity assay (Figure 2). We verified that p24 release induced by KR13 and AuNP-KR13 was specific to the interaction of the peptide with HIV-1 gp120 by showing no effect with VSV-G pseudotyped virus.
The current study has established that modified peptide triazoles that inhibit cell infection by HIV-1 pseudoviruses are also capable of disrupting virus particles in the absence of cells. Results from p24 analysis of treated pseudotyped viruses demonstrated that the intra-virion capsid protein was indeed released in a dose dependent manner by the peptide triazole KR13 (Figure 3A). This release was enhanced by multivalent display of KR13 on gold nanoparticles (Figure 3B). Furthermore, the magnitudes of dose responses of p24 release by the free peptide and AuNP-KR13 had a similar trend (AuNP-KR13 >> KR13) to that observed for inhibition of cell infection by these compounds (Figure 3C). Hence, it appears likely that cell-independent virus particle disruption is at least part of the mechanism of inhibition of cell infection exhibited by the modified peptide triazoles tested. However, for inhibitors that cause rupture, the relative importance of virus rupture and direct receptor binding inhibition in the overall antiviral effect is yet to be determined. Furthermore, at this stage, we cannot define the physical mechanism by which the virus particle is disrupted to release p24. In preliminary dynamic light scattering analysis (Bastian et al., unpublished), peptide-treated pseudovirus preparations appear to have reduced diameter, which could indicate either virion collapse or fragmentation.
Cell-free virucidal action by peptide triazole variants has significant implications for the potential uses of this type of inhibitor in AIDS prevention and treatment. One possible role is as an early-acting microbicide, by taking advantage of the ability of the peptides to destroy the functional virus particle before encounter and consequent infection of host cells. Therapeutically, the peptides could help clear viruses in circulation. Suppressing the amount of circulating virus could decrease virus spread from infected to uninfected individuals. Therapeutic intervention of this type will ultimately benefit from optimized formulations that will enable adequate delivery and metabolic stability.
Observations made in this work suggest that multivalent display of peptide triazoles is likely to be important for the virucidal effect against HIV-1 virus. An impressive enhancement of p24 release was shown with AuNP-KR13 conjugate, which contains an average of 72 moles of peptides attached per mole of nanoparticle. Since the Cys-SH group of KR13 in the AuNP-KR13 conjugate is coordinated to gold, the virucidal functions of peptide and conjugate are unlikely to be due to the free sulfhydryl component per se. On the other hand, KR13 alone has the potential to dimerize to a disulfide crosslinked dimer, and this latter could be the agent causing p24 release in unconjugated KR13. Supporting this idea, we have found that the parent peptide triazole HNG156, while being a strong inhibitor of cell infection by virus, does not appear to elicit significant, dose-dependent p24 release (Supporting Information). We also have found that neither AuNP alone nor AuNP attached to the irrelevant agent thiol-polyethylene glycol can elicit antiviral or virucidal actions against the virus particle (data not shown). The current view is that formation of stable dimers and other well-defined multivalent peptide triazoles will be important objectives to pursue going forward for developing peptide triazoles as virucidal agents.
The ability to exert ligand-specific, cell-free virucidal effects on HIV-1 particles by the gp120 targeting peptide triazoles may be related to the ability to induce Env conformational changes in an intrinsically metastable virus. The HIV-1 infection process is driven by a cell entry machine in which virus and host cell proteins engage in a sequence of interactions and conformational changes leading to virus-host cell fusion. A major, and it would appear, obligatory phase of this process is the large-scale, receptor-induced conformational rearrangement of Env protein that enables exposure of fusogenic components on the virus surface [6]. Prior data have shown that soluble CD4 and CD4-mimicking small molecules, both of which can exert large conformational changes in Env gp120 similar to those induced by cellular CD4, are able to inactivate the virus in a time-dependent manner in the absence of cells [7]. Further, polyvalent CD4 has been observed by cryoelectron tomography to crosslink simian immunodeficiency virus particles with associated virion rupture [8]. The current work builds on these prior observations by showing that modified peptide triazole allosteric inhibitors, an emerging class of gp120 antagonists that exert substantial conformational effects on the Env gp120 protein, can cause cell-free disruption of HIV-1 virus particles leading to their inactivation. In the peptide triazole case, this inactivation is specific for an allosteric site unique to HIV-1 virions and leads to ligand-specific virus particle rupture. Such cell-free virucidal action through specific inhibitor attack on the conformationally metastable viral envelope machine opens up new directions for HIV-1 treatment and prevention.
In summary, we report the ability of modified peptide triazole inhibitors that target HIV-1 gp120 to physically disrupt virus particles in the absence of host cells. At conditions similar to those at which the peptide triazole “KR13” inhibited HIV-1 BaL pseudovirus infection of HOS.T4.R5 cells, it also caused release of HIV-1 gag p24 when incubated with virus alone. Both inhibition of cell infection and p24 release were enhanced substantially by multivalent display of KR13 on gold nanoparticles. Virucidal function of the modified peptide triazoles argues for their potential use as microbicidal and therapeutic agents to suppress the progression and spread of AIDS. The results also suggest that ligand-specific pathogen rupture may be possible for other viruses, such as influenza, ebola and dengue, that contain metastable prefusion surface protein complexes[9].
Experimental Section
Peptide Triazole and Gold Nanoparticle-Peptide Conjugate (AuNP-KR13) Synthesis
The peptide triazole, KR13 was synthesized by manual solid phase synthesis using Fmoc chemistry on a Rink amide resin at a 0.25mmol scale as shown in Umashankara and coworkers [2e]. For the gold nanoparticles, the citrate reduction method developed by Frens [4] was modified in order to synthesize size-controlled, stable and monodisperse AuNPs (Supporting Information). For 20nm AuNPs, briefly, 1% HAuCl4 (300 µl) was added to 18 µm filtered water (total volume, 30 ml) and heated to 100 °C in an Erlenmeyer flask for 1 hour followed by addition of 1% citric acid (600 µl). The synthesized particles were stabilized using Bis (P-sulfonatophenyl) Phenylphosphine dehydrate dipotassium Salt (BSPP) followed by purification and transfer into phosphate buffer 7.2 using a Millipore 10,000 kDa filter (Supporting Information).
The peptide-nanoparticle conjugation was conducted by adding a predetermined stabilizing concentration of KR13 to the synthesized AuNP and incubating under vigorous stirring at room temperature for 30 minutes. The conjugate was purified by several washes in phosphate buffer (pH 7.2) using ultracentrifugation and further filtration on a 0.2 µm filter (Supporting Information). The conjugation efficiency was calculated using amino acid analysis of the conjugate (Yale University – College of Medicine).
Viral Inhibition Detected Using Luciferase Reporter Assay System
Modified Human Osteosarcoma Cells engineered to express CD4 and CCR5 (HOS.T4.R5), as well as the vector for pNL4-3.Luc R-E, were obtained from Dr. Nathaniel Landau [10]. The recombinant virus consisted of the pro-viral envelope plasmid sequence corresponding to the CCR5 targeting HIV-1BaL strain and the backbone sequence corresponding to an envelope-deficient pNL4-3-Fluc+env− provirus. The single-round pseudoviral infection luciferase reporter assay was conducted as previously described [2e]. Non-linear regression analysis with Origin V.8.1 (Origin Lab) was used to obtain the IC50 values. All experiments were performed at least in triplicate and results were expressed as relative infection with respect to cells infected with virus in the absence of inhibitor (100% infected).
p24 Release Assay
The stability of the VLP during the viral inhibition by KR13 and AuNP-KR13 was compared using a p24 release assay. This assay was conducted under conditions to best mimic the viral assay conditions. An equal volume of intact VLP (HIV-1BaL), purified through the sucrose cushion, was added to a series of samples that contained a 1:3 serial dilution of both AuNP-KR13 conjugates and KR13 alone at working concentrations determined from the viral assay above. Lysed virus (using 1% Triton X-100 followed by heating at 95 °C), intact virus as well as anti-p24 were used as controls. All prepared samples were incubated 30 minutes prior to a clarifying spin. A western blot analysis was conducted on the supernatant for p24 release using rabbit anti-p24 (abcam) and Goat Anti-Rabbit IgG conjugated with IRDye® 800CW (LI-COR Biosciences). The LICOR IR imaging system was used in order to detect the blot, and Image J software tool was used in order to quantify the band intensities.
Supplementary Material
Acknowledgements
Special Thanks to Kevin Freedman for TEM image processing.
This work is supported by:
NSF CBET – 0853680
NIH – 5 POI GM 56550-13
IPM/USAID GPO-A-00-05-00041-00
References
- 1.a Dalgleish AG, Beverley PCL, Clapham PR, Crawford DH, Greaves MF, Weiss RA. Nature. 1984;312:763–767. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]; b Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. Nature. 1984;312:767–768. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]; c Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. Nature. 1996;384:179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]; d Furuta RA, Wild CT, Weng Y, Weiss CD. Nat Struct Biol. 1998;5:276–279. doi: 10.1038/nsb0498-276. [DOI] [PubMed] [Google Scholar]
- 2.a Gopi HN, Tirupula KC, Baxter S, Ajith S, Chaiken IM. ChemMedChem. 2006;1:54–57. doi: 10.1002/cmdc.200500037. [DOI] [PubMed] [Google Scholar]; b Cocklin S, Gopi H, Querido B, Nimmagadda M, Kuriakose S, Cicala C, Ajith S, Baxter S, Arthos J, Martin-Garcia J, Chaiken IM. J Virol. 2007;81:3645–3648. doi: 10.1128/JVI.01778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Gopi H, Umashankara M, Pirrone V, LaLonde J, Madani N, Tuzer F, Baxter S, Zentner I, Cocklin S, Jawanda N, Miller SR, Schon A, Klein JC, Freire E, Krebs FC, Smith AB, Sodroski J, Chaiken I. J Med Chem. 2008;51:2638–2647. doi: 10.1021/jm070814r. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gopi H, Cocklin S, Pirrone V, McFadden K, Tuzer F, Zentner I, Ajith S, Baxter S, Jawanda N, Krebs FC, Chaiken IM. J Mol Recognit. 2009;22:169–174. doi: 10.1002/jmr.892. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Umashankara M, McFadden K, Zentner I, Schon A, Rajagopal S, Tuzer F, Kuriakose SA, Contarino M, Lalonde J, Freire E, Chaiken I. ChemMedChem. 2010;5:1871–1879. doi: 10.1002/cmdc.201000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a Myszka DG, He X, Dembo M, Morton TA, Goldstein B. Biophys J. 1998;75:583–594. doi: 10.1016/S0006-3495(98)77549-6. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Morton TA, Bennett DB, Appelbaum ER, Cusimano DM, Johanson KO, Matico RE, Young PR, Doyle M, Chaiken IM. J Mol Recognit. 1994;7:47–55. doi: 10.1002/jmr.300070107. [DOI] [PubMed] [Google Scholar]
- 4.Frens G. Nature (London), Phys. Sci. 1973;241(2):20–22. [Google Scholar]
- 5.Bowman MC, Ballard TE, Ackerson CJ, Feldheim DL, Margolis DM, Melander C. J Am Chem Soc. 2008;130:6896–6897. doi: 10.1021/ja710321g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a Sattentau QJ, Moore JP. Philos Trans Roy Soc B. 1993;342:59–66. doi: 10.1098/rstb.1993.0136. [DOI] [PubMed] [Google Scholar]; b Fass D. Adv Protein Chem. 2003;64:325–362. doi: 10.1016/s0065-3233(03)01009-x. [DOI] [PubMed] [Google Scholar]; c Liu S, Zhao Q, Jiang S. Peptides. 2003;24:1303–1313. doi: 10.1016/j.peptides.2003.07.013. [DOI] [PubMed] [Google Scholar]; d Cerutti N, Mendelow BV, Napier GB, Papathanasopoulos MA, Killick M, Khati M, Stevens W, Capovilla A. J Biol Chem. 2010;285:25743–25752. doi: 10.1074/jbc.M110.144121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haim H, Si Z, Madani N, Wang L, Courter JR, Princiotto A, Kassa A, DeGrace M, McGee-Estrada K, Mefford M, Gabuzda D, Smith AB, 3rd, Sodroski J. PLoS Pathog. 2009;5:e1000360. doi: 10.1371/journal.ppat.1000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bennett A, Liu J, Van Ryk D, Bliss D, Arthos J, Henderson RM, Subramaniam S. J Biol Chem. 2007;282:27754–27759. doi: 10.1074/jbc.M702025200. [DOI] [PubMed] [Google Scholar]
- 9.Kielian M, Rey FA. Nat Rev Microbiol. 2006;4:67–76. doi: 10.1038/nrmicro1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connor RI, Chen BK, Choe S, Landau NR. Virology. 1995;206:935–944. doi: 10.1006/viro.1995.1016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.