

Abstract
Proteomics analyses were performed on the brains of wild-type (WT) controls and an Alzheimer’s disease (AD) mouse model, APP/PS-1 human double mutant knock in mice. Mice were given either drinking water or water supplemented with N-acetylcysteine (NAC) (2mg/kg body weight) for a period of five months. The time periods of treatment correspond to ages prior to Aβ deposition (i.e., 4–9 months), resembling human mild cognitive impairment (MCI), and after Aβ deposition (i.e., 7–12 months), more closely resembling advancing stages of AD. Substantial differences exist between the proteomes of WT and APP/PS-1 mice at 9 or 12 months, indicating that Aβ deposition and oxidative stress lead to downstream changes in protein expression. Altered proteins are involved in energy-related pathways, excitotoxicity, cell cycle signaling, synaptic abnormalities, and cellular defense and structure. Overall, the proteomic results support the notion that NAC may be beneficial for increasing cellular stress responses in WT mice and for influencing the levels of energy- and mitochondrial related proteins in APP/PS-1 mice.
Keywords: APP/PS1, N-acetylcysteine, protein oxidation, proteomics, oxidative stress, Alzheimer disease, Mild cognitive impairment
Introduction
As the number of persons in the United States directly affected by Alzheimer disease (AD) is expected to increase in the next forty years to an estimated 16–20 million persons [1], the need for preventative or modulatory therapies is pressing. While there are many types of compounds being explored as potential AD treatments, the oxidative stress hypothesis associated with AD has brought to the forefront the possible therapeutic utility of antioxidant compounds. This is based on the observed increase in reactive oxygen species (ROS) in AD brain that leads to downstream increases in protein oxidation [2], DNA and RNA oxidation [3–7], and lipid peroxidation [8–9]. The oxidative stress present in AD brain is also a result of decreased endogenous antioxidant defenses [10–11] and ultimately leads to neuronal death. Because of the profound consequences of oxidative stress in AD brain and reports that high doses of compounds such as alpha-tocopherol (vitamin E) may be effective in delaying disease progression [12], the potential of antioxidant compounds as AD therapeutics seems promising.
Our laboratory has provided substantial evidence that amyloid-beta (Aβ) (1–42), known to be heavily implicated in AD, mediates oxidative stress in AD brain [13]. Aβ(1–42) is derived from the amyloid precursor protein (APP) through cleavage of APP by β- and γ- secretases, in which presenilin 1 (PS-1) is a component of the γ-secretase complex. Genetic mutations in APP, PS-1, and PS-2 are associated with familial AD (FAD) [14]. PS-1 mutations lead to altered processing of APP and thus an increase in Aβ peptides which are present as toxic oligomers or in senile plaques, pathological hallmarks of AD.
A mammalian model of FAD was developed by Borchelt et al. (licensed to Cephalon, Inc.), in which mice were backcrossed to carry the APPNLh/APPNLh × PS-1P264L/PS-1264L mutations in order to humanize the mouse Aβ sequence and to include the PS-1 mutation identified in human AD (APP/PS-1 human double mutant knock-in mice) [15]. APP/PS-1 mice have increased Aβ production and accelerated amyloid deposition [15]. We have shown that neurons from APP/PS-1 mice (generated using the Cre-loc© knock-in technology) compared to wild type (WT) exhibit increased protein oxidation, lipid peroxidation, and susceptibility to oxidation by exogenous oxidants [16]. Furthermore, cerebral amyloid deposition increases in an age-dependent manner in APP/PS-1 mice [17–19]. Oxidative stress levels [as measured by protein carbonyls (PCO), 3-nitrotyrosine (3NT)-modified proteins, markers of protein oxidation, and 4-hydroxynonenal (HNE)-bound proteins, a marker of lipid peroxidation] also increase in brain in an age-dependent manner in APP/PS-1 knock-in mice when compared to WT controls [20].
We recently explored the antioxidant, N-acetylcysteine (NAC), as an AD treatment at different disease stages in an APP/PS-1 human double mutant knock-in mouse model [21]. NAC is currently a FDA-approved drug for acetaminophen-based liver toxicity [22] and heavy metal poisoning. NAC serves as an antioxidant by indirectly increasing intracellular glutathione (GSH) levels and also acts directly as a free radical scavenger. Depletion of GSH is associated with many neurodegenerative disorders, including AD [11, 23–24]; however, dietary or pharmacological agents/mimetics that increase endogenous GSH levels can provide neuroprotection against oxidative stress [7, 13, 25]. That NAC increases endogenous GSH levels and protects brain from protein oxidation against hydroxyl radicals has been previously demonstrated in vivo [26]. NAC has also been shown to be partially protective in brain against peroxynitrite-induced damage [27] and protective against 3-nitropropionic acid [28] and acrolein-induced protein oxidation [29]. Furthermore, NAC restores memory and decreases oxidative stress in aged senescence-prone mice [30].
As noted above, NAC provides protection against oxidative stress in the brains of APP/PS-1 human double mutant knock-in mice [21]. In those studies, drinking water was supplemented with NAC and administered daily to WT and APP/PS-1 mice for a period of five months beginning at ages of 4- and 7-months old. These ages correlate with the progression of amyloid deposition which begins at the age of 9 months, with elevated Aβ(1–42) deposition in frank deposits occurring at 12 months [17–19]. By 9 months of age, Aβ(1–42) deposition begins in APP/PS-1 mouse brain [19] and thus mimics pathological conditions of amnestic mild cognitive impairment (MCI), a transitional stage between normal aging, early dementia, and AD [31–32] or early AD. Overall, in this previous study NAC provided neuroprotection against protein oxidation and nitration both prior to amyloid deposition (i.e., 9 month-old animals) and after amyloid deposition (i.e., 12 month-old animals) in APP/PS-1 human double mutant knock-in mice relative to WT [21]. In addition NAC treatment protected against lipid peroxidation when administered in the earlier age group [21]. Thus, NAC may potentially prevent some of the oxidative stress accompanying MCI and early stages of AD as well as reverse oxidative damage found during later stages of AD.
Herein, we seek to gain insights into the mechanisms reflective of Aβ deposition and mechanisms governing the neuroprotection NAC provides against oxidative stress in the brains of APP/PS-1 human mutant double knock-in mice (hereafter referred to as APP/PS-1). To this end, proteomics analyses of the brains from 9- and 12-month old WT & APP/PS-1 mice treated five months in vivo with or without NAC in the drinking water were performed. We hypothesized that (a) the brain proteome of APP/PS-1 mice at 9 months of age (beginning of Aβ deposition) and 12 months of age (frank amyloid deposits) are different from WT mice, and (b) in vivo NAC treatment causes changes in brain protein levels that are consistent with subsequent decreases in oxidative stress. Two-dimensional polyacrylamide gel electrophoresis (2D PAGE) coupled with mass spectrometry (MS) analysis and database searching was used to detect differences in protein expression. The results are discussed with relevance to the effect Aβ deposition has on the brain proteome and with relevance to potential in vivo NAC treatment in MCI and AD.
Experimental Section
Chemicals
Unless otherwise indicated, chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
Animals and NAC Administration
WT and APP/PS-1 human double mutant male mice were housed in the University of Kentucky Central Animal Facility. The APP/PS-1 mice were generated using the Cre-loc© knock-in technology by Cephalon, Inc., (Frazer, PA) and backcrossed to carry the APPNLh/APPNLh × PS-1P264L/PS-1264L mutations in order to humanize the mouse Aβ sequence and to include the PS-1 mutation identified in human AD [33–34]. All animals were ~30 g in size at the start of the experiments and were fed standard Purina rodent laboratory chow ad libitum on a 12 h light/dark cycle. Animals, both WT and APP/PS-1 mice, were provided with either drinking water or a 0.001% solution of NAC in drinking water (pH 7.2) for a period of 5 months. Treatments began with one group of animals at 4 months and a second group of animals at 7 months. These times were chosen based on previous studies [35–36], which correlate with Aβ deposition. As noted, the mice employed begin to deposit Aβ at 9 months of age. Thus, the 4–9 month period investigated brain prior to Aβ deposition. These mice have frank amyloid deposition at 12 months of age. Thus, the 7–12 month period investigated NAC treatment during and after Aβ deposition had begun and plaques had formed. New NAC solutions were provided every other day. Mice consumed ~4 to 5 ml of water per day which amounts to a daily cumulative NAC dose of 2 mg/kg (body weight). Animals from both age groups were sacrificed after 5 months of treatment and brains were flash frozen with liquid nitrogen and stored at −80 °C until further use.
Preparation of Brain Homogenate
Brains were homogenized using 20 passes of a Wheaton tissue homogenizer into an ice cold lysing buffer containing 4 µg/ml leupeptin, 4 µg/ml pepstatin, 5 µg/ml aprotinin, 2 mM ethylenediaminetetraacetic acid, 2 mM ethylene glycol-bistetraacetic acid, and 10 mM 4-(2-hydroxyethyl)-1-piperazine-ethanesulfonic acid (pH 7.4). Homogenates were centrifuged at 20000g for 10 min and the pellet was suspended in phosphate-buffered saline. Pellet suspensions were washed twice with PBS by centrifugation at 32000g for 10 min. The supernatants were used in other studies of enzymatic activity [21]. The BCA method was used for protein concentration (Pierce, Rockford, IL).
Brain homogenates (150 µg) were treated with 4× volume of 2N HCl at room temperature for 30 min. For protein precipitation, ice-cold 100% tricholoroacetic acid (TCA) was added to a final concentration of 15% TCA and allowed to react on ice for 10 min. Solutions were centrifuged at 14000g for 5 min at 4 °C, and precipitates were washed with 0.5 mL of 1:1 ethanol:ethyl acetate (v/v) 4× by centrifugation at 14000g for 3 min at 4 °C. The protein pellets were then reconstituted in 200 µL of a rehydration buffer containing 8 M urea, 2 M thiourea, 50 mM dithiothreitol, 2% (w/v) CHAPS, 0.2% (v/v) Biolytes, and bromophenol blue. After one to two hours of mild vortexing, protein samples were ready for the first dimension of isoelectric focusing (IEF).
Two-dimensional Gel Electrophoresis
Protein samples were loaded onto 11 cm, pH 3–10 ReadyIPG Strips (Bio-Rad, Hercules, CA). After one hour, 2 mL of mineral oil was added to protein wells to reduce solvent evaporation. Proteins were taken up into strips by active rehydration at 50 V for 16–18 hours. The conditions for IEF were as follows: 300 V for 1 h, a linear gradient to 8000 V for 5 h and finally, 20,000 V for 1 h. Focused strips were stored at −80 °C until the second dimension of gel electrophoresis was performed. For sodium dodecyl sulfate (SDS) PAGE, thawed strips were equilibrated for 15 min in a buffer containing 50 mM Tris-HCl (pH 6.8), 6 M urea, 1% (w/v) SDS, 30% (v/v) glycerol, and 0.5% dithiothreitol (DTT). Next, strips were reequilibrated in the same buffer containing 4.5% iodoacetamide rather than DTT. Strips were placed into Linear Gradient (8–16%) Precast Criterion Tris-HCl gels (Bio-Rad, Hercules, CA) and run at 200 V for ~65 min.
SYPRO Ruby Staining
Following electrophoresis, gels were removed from the gel cassettes and placed in fixing solution [7% acetic acid (v/v), 10% methanol (v/v), in water] for 45 min on a rocker. Gels were submerged in SYPRO Ruby fluorescent gel stain (Bio-Rad, Hercules, CA) and allowed to shake overnight on a rocker. SYPRO Ruby was then removed and gels were stored in deionized water.
Image and Statistical Analysis
SYPRO Ruby-stained gels were scanned using a STORM phosphoimager (Molecular Dynamics, Sunnyvale, CA) at excitation and emission wavelengths of 470 nm and 618 nm, respectively, and also saved in a TIFF format. PD-Quest (Bio-Rad, Hercules, CA) imaging software was then used to match and align protein spots across the gels from the different treatment groups. In these experiments, a single 2D gel was generated and analyzed for each biological replicate. Protein spots were normalized to the total density detected in each individual gel image. Proteins were considered to be statistically different between treatment groups based on a ≥1.5 fold-change and a p-value <0.05 (using a Student’s t-test). It should be noted that herein we only assessed changes that occur between treatment groups at each timepoint independently (i.e., only 4–9 months or only 7–12 months). For example, in the 4–9 month aged group, we made two way comparisons as follows: a) WT given drinking water (WT H2O) vs. WT given NAC supplemented water (WT NAC), b) WT H2O vs. APP/PS-1 H2O, and c) APP/PS-1 H2O vs. APP/PS-1 NAC. A Student’s t-test was used to determine statistical differences between treatment groups.
In-gel Trypsin Digestion
Protein spots of interest were excised from SYPRO Ruby-stained gels with a clean blade and transferred into a 0.5 mL Eppendorf microcentrifuge tube. Excised gel pieces were washed with 0.1 M NH4HCO3 buffer at room temperature for 15 min. Acetonitrile (ACN) was added and allowed to incubate for 15 min. The mixed solution was removed and the gel pieces were allowed to dry in the tubes under a flow hood for 30 min. Next, gel pieces were incubated with 20 mM DTT in 0.1 M NH4HCO3 at 56 °C for 45 min. The DTT solution was removed and gel pieces were incubated with 55 mM iodoacetamide in 0.1 M NH4HCO3 for 15 min. The solution was removed and gel pieces were incubated with 50 mM NH4HCO3 for 15 min, followed by the addition of ACN for 15 min. This mixed solution was removed and gel pieces were allowed to dry for 30 min in a flow hood. A solution of 20 ng/µL modified trypsin (Promega, Madison, WI) in 50 mM NH4HCO3 was added to gel pieces in order to submerge them and were allowed to shake overnight at 37 °C.
Mass Spectrometry and Database Searching
Tryptic solutions were removed from gel pieces and transferred to a new microcentrifuge tube. Additional tryptic peptides were extracted from gel pieces by addition of 5 mM NH4HCO3 for 10 min with sonication. Next, 95% acetonitrile in 1 mM NH4HCO3 was added for another 10 min with sonication. This supernatant was combined with the previous tryptic solution and concentrated to a small volume (~10 µL). C18 ZipTips (Millipore, Billerica, MA) were used to remove salts from samples prior to MS analysis. Samples were loaded into a 96-well plate rack for nanoelectrospray infusion using an Advion Tri-Versa Nanomate (Ithaca, NY). Electrosprayed peptides were analyzed with an LTQ-Orbitrap XL (ThermoScientific, Waltham, MA) mass spectrometer. The Orbitrap was set to acquire a full MS scan at 60,000 resolution and in Data Dependent mode the eight most intense ions were selected for fragmentation and mass analyzed in the Orbitrap at 30,000 resolution. Conditions for fragmentation in the ion trap include a normalized collision energy of 35%, activation time of 30 ms, and selection of only +2 charge states or higher. Total acquisition time was 5 minutes per sample. SEQUEST was used for database searching against the Uniprot SwissProt Database and the International Protein Index (IPI) Mouse Database and included 2 trypsin miscleavages and a fixed carbamidomethyl modification. Filter criteria of returned protein lists included protein probabilities <0.01, peptide XCorr values >1.5 (for +1 charge state), 2.0 (+2 charge state), 2.5 (+3 charge state), and 3.0 (+4 charge state), peptide ΔCN values >0.1, and at least 2 peptides identified for each protein. Protein MW and pI information was also used to assess individual protein identifications based on the location of the excised protein spot from the 2D gel. Only protein spots assigned to a single protein were further considered.
Western Blotting
Samples (100 µg) from the 7–12 month treatment group were incubated with sample loading buffer for protein denaturation and subject to electrophoresis on a 12.5% SDS-polyacrylamide gel. Proteins were transferred to a nitrocellulose membrane at 90 mA/gel over a 2 h period. Nitrocellulose membranes containing the transferred proteins were blocked for 1h in fresh wash buffer [PBS, 150 mM NaCl, 0.05% Tween 20, pH 7.4] containing 3% bovine serum albumin, and incubated with a 1:2000 dilution of anti-mouse enolase monoclonal antibody (Santa Cruz Biotechnologies; Santa Cruz, CA) and anti-mouse actin antibody (Sigma-Aldrich) in wash buffer for 2 h. The membrane was washed 3× in wash buffer and incubated for 1 h with a 1:8000 dilution of anti-mouse IgG horseradish peroxidase secondary antibody diluted in wash buffer. The membrane was washed 3× times in wash buffer and developed using chemiluminescence reageants from an ECL kit (Pierce). Blots were scanned on a phosphorimager and analyzed using ImageQuant 1D software. Actin was probed as a loading control.
Results
Proteomics analyses using 2D PAGE, in-gel trypsin digestion, and MS were performed on entire brain homogenate obtained from WT and APP/PS-1 mice given drinking water or water supplemented with NAC for a period of five months. NAC treatment began at either 4 months or 7 months in order to better understand the mechanisms by which NAC provides neuroprotection against Aβ(1–42)-mediated protein oxidation and nitration and lipid peroxidation prior to and after amyloid deposition. Thus, for the two age groups of mice, (i.e., 9 months and 12 months) differences in global protein expression were assessed. Comparisons were as follows: a) WT H2O vs. WT NAC, b) WT H2O vs. APP/PS-1 H2O, and c) APP/PS-1 H2O vs. APP/PS-1 NAC.
Figures 1 and 2 a)–c) show examples of typical 2D gel images of proteins isolated from the brains of WT and APP/PS-1 mice given drinking water or NAC-supplemented drinking water from 4–9 or 7–12 months of age. Protein spots that were differentially expressed between the various treatment groups (as described above) and identified with MS are labeled in the figure and listed in Tables 1 and 3. As shown in Tables 1 and 3, the probability for false protein identifications with SEQUEST was <1e-03 for all proteins indicating high confidence in spot assignments. Proteins were identified with at least two different peptide sequences and multiple peptide hits corresponding to every MS/MS event resulting in significant peptide identification. For example, aconitate hydratase was identified with six peptide sequences from 23 multiple MS/MS spectra (Table 1).
Figure 1.
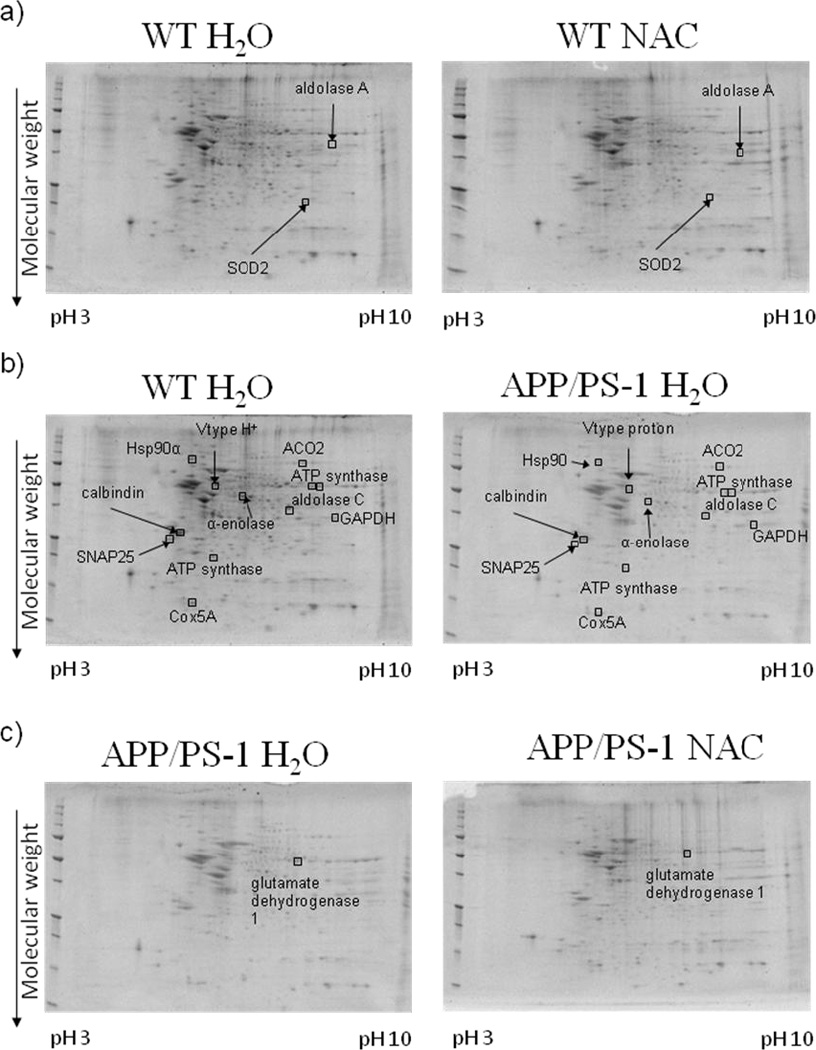
Representative 2D gel images of differentially expressed brain proteins from the 4–9 month treatment groups in comparisons of a) WT mice given normal drinking water or NAC supplemented water, b) WT mice and APP/PS-1 mice given normal drinking water, and c) APP/PS-1 mice given normal drinking water or NAC supplemented water. Differentially expressed proteins (i.e., ratio values of 0.67≤x≤1.5, p-value <0.05 for N = 8 or 9) are labeled in the images.
Figure 2.
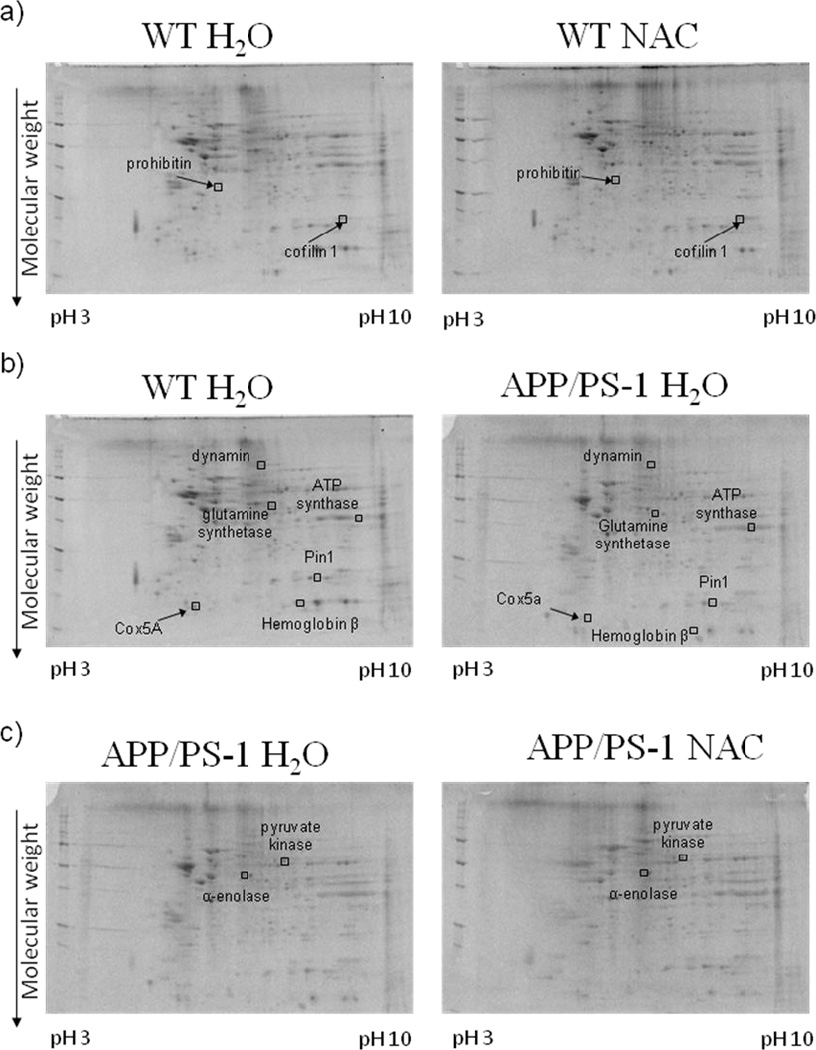
Representative 2D gel images of differentially expressed brain proteins from the 7–12 month treatment groups in comparisons of a) WT mice given normal drinking water or NAC supplemented water, b) WT mice and APP/PS-1 mice given normal drinking water, and c) APP/PS-1 mice given normal drinking water or NAC supplemented water. Differentially expressed proteins (i.e., ratio values of 0.67≤x≤1.5, p-value <0.05 for N = 6) are labeled in the images.
Table 1.
Proteomic Identification of Differentially Expressed Brain Proteins in 4–9 mos. Treatment groups.
| Protein Identified | Accession Numbera |
MW (kDa) |
pI | peptides (hits)b |
Probabilityc |
|---|---|---|---|---|---|
| Fructose-bisphosphate aldolase A | P05064 | 39332 | 8.0 4 |
3 (15) | 2.00E-08 |
| Superoxide dismutase 2 | P09671 | 24589 | 8.8 0 |
2 (7) | 1.00E-06 |
| Aconitate hydratase, mitochondrial | Q99KI0 | 85411 | 7.8 0 |
6 (23) | 8.00E-07 |
| Alpha-enolase | P17182 | 47141 | 6.3 7 |
2 (6) | 1.00E-05 |
| ATP synthase subunit alpha, mitochondrial | Q03265 | 59753 | 9.2 2 |
4 (9) | 1.00E-07 |
| ATP synthase subunit alpha, mitochondrial | Q03265 | 59753 | 9.2 2 |
3 (12) | 2.00E-07 |
| ATP synthase subunit d, mitochondrial | Q9DCX2 | 18739 | 5.4 1 |
3 (9) | 5.00E-07 |
| Calbindin | P12658 | 29976 | 4.5 6 |
2 (5) | 2.00E-04 |
| Cytochrome c oxidase subunit 5A, mitochondrial | P12787 | 16092 | 6.0 9 |
4 (8) | 2.00E-05 |
| Fructose-bisphosphate aldolase C | P05063 | 39371 | 6.7 4 |
4 (14) | 3.00E-08 |
| Glyceraldehyde-3-phosphate dehydrogenase | P16858 | 35788 | 8.3 3 |
3 (11) | 1.00E-07 |
| Heat shock protein HSP 90-alpha | P07901 | 84788 | 4.9 3 |
4 (12) | 2.00E-07 |
| Synaptosomal-associated protein 25-A | P60879 | 23315 | 4.6 6 |
2 (9) | 2.00E-06 |
| V-type proton ATPase subunit B, brain isoform | P62814 | 56516 | 5.4 8 |
2 (6) | 2.00E-07 |
| Glutamate dehydrogenase 1, mitochondrial | P26443 | 61299 | 7.9 6 |
7 (22) | 4.00E-06 |
The protein accession number found in the Swiss Prot (mouse) database.
The number of peptide sequences identified by ESI-MS/MS. The total number of peptide hits is indicated in ( ), including multiple hits across different charge states.
The probability associated with a false protein identification using the SEQUEST search algorithm.
Table 3.
Proteomic Identification of Differentially Expressed Brain Proteins in 7–12 mos. Treatment groups
| Protein Identified | Accession Numbera |
MW (kDa) |
pI | peptides (hits)b |
Probabilityc |
|---|---|---|---|---|---|
| Cofilin 1 | P18760 | 18548.7 | 8.1 9 |
4 (14) | 2.00E-09 |
| Cofilin 1 | P18760 | 29802.9 | 5.4 6 |
3 (9) | 6.00E-06 |
| ATP synthase subunit alpha, mitochondrial | Q03265 | 59716.6 | 9.5 3 |
5 (17) | 2.00E-06 |
| Cytochrome c oxidase, subunit 5A | P12787 | 16092.3 | 6.0 9 |
3 (11) | 1.00E-06 |
| Dynamin 1 | P39053 | 97742.3 | 7.6 2 |
9 (46) | 2.00E-09 |
| Glutamine synthetase | P15105 | 42119.65 | 6.6 4 |
2 (5) | 1.00E-03 |
| Hemoglobin subunit beta | P02088 | 15831.2 | 7.4 8 |
3 (13) | 5.00E-08 |
| Peptidyl-prolyl cis-trans isomerase A | P17742 | 17971.34 | 7.7 3 |
2 (5) | 2.00E-05 |
| Alpha-enolase | P17182 | 47112.2 | 6.3 8 |
7 (27) | 7.00E-08 |
| Pyruvate kinase isozymes M1/M2 | P52480 | 57809 | 7.2 | 12 (41) | 4.00E-08 |
The protein accession number found in the Swiss Prot (mouse) database.
The number of peptide sequences identified by ESI-MS/MS. The total number of peptide hits is indicated in ( ), including multiple hits across different charge states.
The probability associated with a false protein identification using the SEQUEST search algorithm.
(a) Effect of NAC on brain protein levels in WT mice
Following in vivo NAC treatment in 9-month-old WT mice, the proteins, fructose bisphosphate aldolase A (142.6 ↑, p-value <0.04) and superoxide dismutase 2 (6.75 ↑, p-value <0.04) were significantly increased in expression (Table 2). Prohibitin (2.95 ↑, p-value <0.01) was significantly increased, while cofilin 1 (0.56↓, p-value < 0.04) was significantly decreased in 12-month-old WT mice (Table 4).
Table 2.
Differentially Expressed Proteins in 4–9 mos. Treatment groups
| Protein Identified | WT H2O vs. WT NACa | WT H2O vs. APP/PS-1 H2Ob | APP/PS-1 H2O vs. APP/PS-1 NACc | |||
|---|---|---|---|---|---|---|
| Ratio | p-value | Ratio | p-value | Ratio | p-value | |
| Fructose-bisphosphate aldolase A | 142.59 | <0.04 | ||||
| Superoxide dismutase 2 | 6.75 | <0.04 | ||||
| Synaptosomal-associated protein 25-A | 20.52 | <0.0003 | ||||
| V-type proton ATPase subunit B, brain isoform | 16.20 | <0.03 | ||||
| Cytochrome c oxidase subunit 5A, mitochondrial | 4.19 | <0.02 | ||||
| ATP synthase subunit alpha, mitochondrial* | 0.35 | <0.02 | ||||
| ATP synthase subunit d, mitochondrial | 0.35 | <0.04 | ||||
| Heat shock protein HSP 90-alpha | 0.28 | <0.01 | ||||
| Fructose-bisphosphate aldolase C | 0.28 | <0.04 | ||||
| Aconitate hydratase, mitochondrial | 0.28 | <0.04 | ||||
| Glyceraldehyde-3-phosphate dehydrogenase | 0.28 | <0.02 | ||||
| Alpha-enolase | 0.02 | <0.02 | ||||
| ATP synthase subunit alpha, mitochondrial* | 0.01 | <0.02 | ||||
| Calbindin | 0.005 | <0.0003 | ||||
| Glutamate dehydrogenase 1, mitochondrial | 1.88 | <0.03 | ||||
See Figure 1 for the location of these spots
Ratio represents the average protein spot density WT NAC/ average protein spot density WT H2O. N=8 for WT H2O and N=9 for WT NAC.
Ratio represents the average protein spot density APP/PS-1 H2O/average protein spot density WT H2O. N=6 for for WT H2O and N=9 for APP/PS-1 H2O.
Ratio represents the average protein spot density APP/PS-1 NAC/average protein spot density APP/PS-1 H2O. N=9 for each group.
Listings in bold are statistically significantly different from the respective control.
Table 4.
Differentially Expressed Proteins in 7–12 mos. Treatment groups
| Protein Identified | WT H2O vs. WT NACa | WT H2O vs. APP/PS-1 H2Ob | APP/PS-1 H2O vs. APP/PS-1 NACc | |||
|---|---|---|---|---|---|---|
| Ratio | p-value | Ratio | p-value | Ratio | p-value | |
| prohibitin | 2.95 | <0.01 | ||||
| cofilin 1 | 0.56 | <0.04 | ||||
| glutamine synthetase | 83.9 | <0.05 | ||||
| ATP synthase subunit alpha, mitochondrial | 4.18 | <0.03 | ||||
| cytochrome c oxidase, subunit 5A | 0.40 | <0.04 | ||||
| hemoglobin subunit beta | 0.33 | <0.02 | ||||
| Peptidyl-prolyl cis-trans isomerase A | 0.01 | <0.004 | ||||
| dynamin 1 | 0.009 | <0.04 | ||||
| alpha-enolase | 60.6 | <0.05 | ||||
| Pyruvate kinase isozymes M1/M2 | 4.60 | <0.005 | ||||
Ratio represents the average protein spot density WT NAC/average protein spot density WT H2O. N=6 for each group.
Ratio represents the average protein spot density APP/PS-1 H2O/average protein spot density WT H2O. N=6 for each group.
Ratio represents the average protein spot density APP/PS-1 NAC/ average protein spot density APP/PS-1 H2O. N=6 for each group.
(b) Comparison of WT vs. APP/PS-1 mice
Several brain proteins were differentially expressed in comparisons of WT and APP/PS-1 mice given drinking water at 9 months (Table 2). Synaptosomal-associated protein 25A [SNAP25 (20.5 ↑, p-value <0.0003)], V-type H+ ATPase subunit B (16.2 ↑, p-value <0.03), and cytochrome c oxidase subunit 5A [Cox5A (4.19 ↑, p-value <0.02)] were significantly increased in expression. The following brain proteins were significantly decreased in APP/PS-1 compared to WT mice: ATP synthase subunit alpha, mitochondrial (two isoforms: 0.35 ↓, p-value <0.02; 0.01↓, p-value <0.02), ATP synthase subunit d, mitochondrial (0.35 ↓, p-value <0.04), heat shock protein 90α [HSP90α (0.28 ↓, p-value <0.01)], fructose-bisphosphate aldolase C (0.28 ↓, p-value <0.04), aconitate hydratase (ACO2), mitochondrial (0.28 ↓, p-value <0.04), glyceraldehyde-3-phosphate dehydrogenase [GAPDH (0.28 ↓, p-value <0.02)], α-enolase (0.02 ↓, p-value <0.02), and calbindin (0.005↓, p-value <0.0003).
Glutamine synthetase (83.9 ↑, p-value < 0.05) and ATP synthase subunit alpha, mitochondrial (4.18 ↑, p-value <0.03) were significantly increased in expression in brains of 12-month-old APP/PS-1 H2O. Several brain proteins had decreased levels in APP/PS-1 H2O relative to WT H2O mice. These proteins are Cox5A (0.40 ↓, p-value <0.04), hemoglobin subunit β (0.33 ↓, p-value <0.02), peptidyl-prolyl cis-trans isomerase A [Pin1 (0.01 ↓, p-value <0.004)], and dynamin 1 (0.009 ↓, p-value <0.04).
(c) Effects of NAC treatment of APP/PS-1 mice
Glutamate dehydrogenase 1, mitochondrial (1.88 ↑, p-value <0.03), was significantly increased in 9-month-old APP/PS-1 NAC mice relative to APP/PS-1 H2O. The proteins α-enolase (60.6 ↑, p-value <0.05) and pyruvate kinase isozymes M1/M2 (4.60 ↑, p-value <0.005), had significantly increased levels of 12-month-old APP/PS-1 NAC mice (Table 4). Recent studies in our laboratory investigated the expression levels of Pin1 using Western blot analysis and determined that Pin1 was significantly decreased in the brains of APP/PS-1 mice relative to WT controls [21] at 12 months of age and Pin1 levels slightly increased after in vivo NAC treatment. These results are consistent with these observations that Pin1 is significantly decreased in the brains of 12-month-old APP/PS-1 H2O mice relative to WT H2O mice (see Table 4).
(c) Western blotting analysis
Western blotting analysis was performed to validate changes in protein expression for α-enolase in the 7–12 month treatment group. Figure 3a shows a Western image of three samples from each treatment group after being probed with primary enolase antibody (actin was used as a loading control). Figure 3b is a histogram plot representation of the results in which no significant changes are detected in normalized enolase expression levels across the treatment groups. However, there is a trend towards increased enolase expression in APP/PS-1 mice given drinking water treated with NAC relative to APP/PS-1 mice given drinking water. This result is generally consistent with the proteomics results reported above in Table 4. We note that the additional band most likely comes from a nonspecifically bound protein. A total N=3 used to measure the Western blots for enolase is small relative to the entire population of animals used in each group (i.e., N=8 or N=9) and could contribute to differences in the ratio differences measured by the two techniques. As noted above, one or two animals may have outlier ratios. We randomly chose a smaller subset of each group to carry out the validation study. Additionally, differences in the measured percent change in Western blots and the 2D gel blots could also be due to dynamic range limitations in colorometric staining as opposed to greater sensitivity with Sypro Ruby fluorescent staining, respectively. Finally, some of the largest changes measured in 2D gel analyses may be overestimated based on the software algorithm that estimates a threshold density value for missing or low-level spots.
Figure 3.
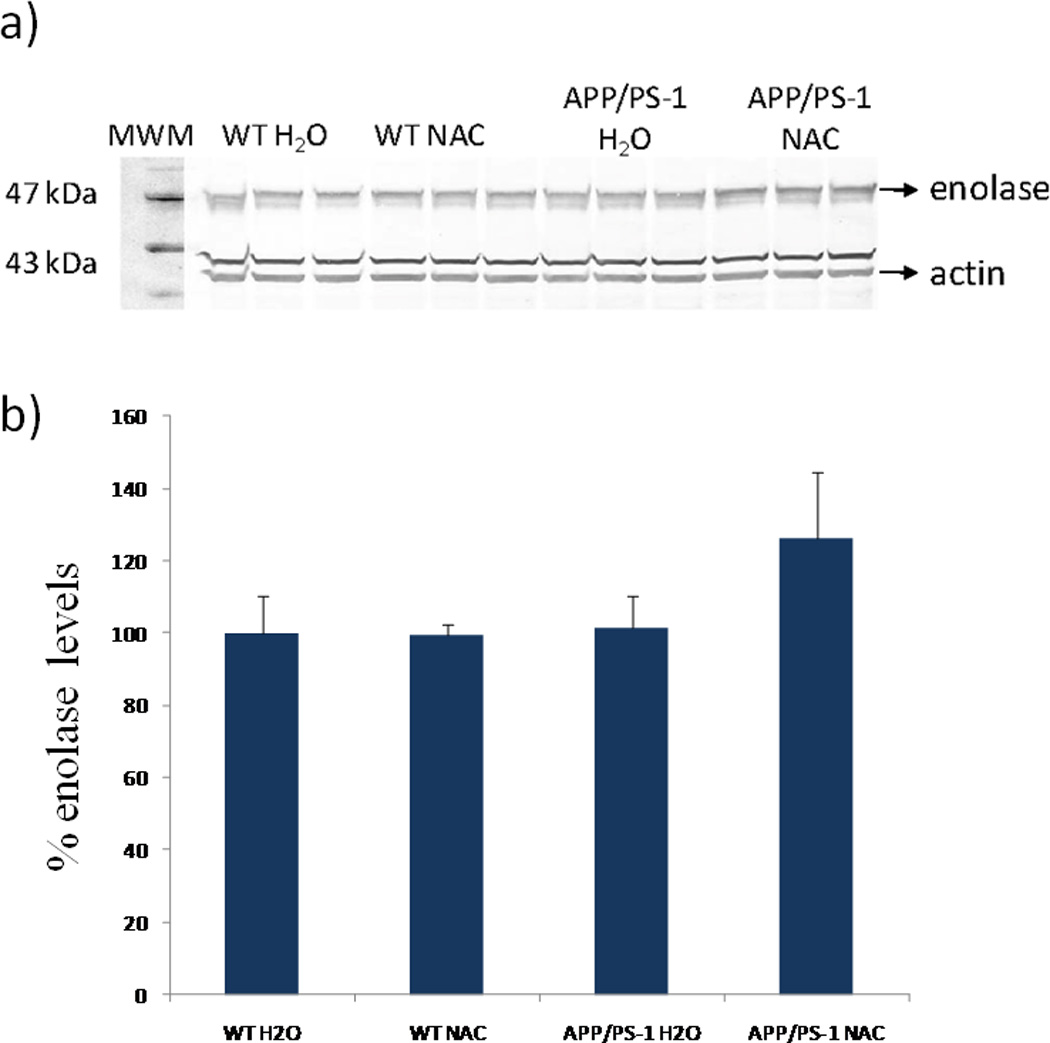
Western blot analysis of enolase expression in brain in the 7–12 month treatment groups. a) Western blot image corresponding to lane 1, MWM; lanes 2–4, WT mice given normal drinking water; lanes 5–7 WT mice given NAC supplemented water; lanes 8–10, APP/PS-1 mice given normal drinking water; and lanes 11–13, APP/PS-1 mice given NAC supplemented water. b) Histogram plot of enolase levels relative to percent controls (WT H2O). The percent values are based on normalized enolase densities (i.e., density of enolase band/density of actin band). Values shown are mean±SD for N = 3.
Discussion
Substantial evidence exists that Aβ(1–42) significantly contributes to oxidative stress in AD and in in vitro and in vivo model systems of AD [2, 13, 37]. As a part of this ongoing work, we have investigated the levels of oxidative stress in APP/PS-1 mice [20] at ages that correlate with the start of Aβ deposition and plaque formation in the brain[35]. The levels of PCO, 3-NT-modified proteins, and HNE-bound proteins are increased in APP/PS-1 mice relative to WT controls 21]. Simultaneously, the increase in oxidative stress levels correlate with an increase in soluble Aβ, Aβ load, and non-neuritic and neuritic plaques [38]. We note that while Aβ deposits are generally localized to the hippocampus and cortex regions, we examined entire brain homogenates as beneficial effects of NAC may appear in regions other than where plaques are localized. Key to the work reported herein is the ages that were used for investigation (i.e., 4–9 month- and 7–12 month-old mice) of the effects of Aβ deposition and effects of in vivo NAC treatment on the brain proteome of APP/PS-1 mice. These ages correspond to animals that have similar pathology to that observed in MCI and AD patients, respectively, and correlate with periods of NAC treatment given prior to and after amyloid deposition and plaque formation.
Based on the proteomics results obtained, we observe several proteins whose brain levels are altered in APP/PS-1 mice relative to WT controls in both the 4–9 month and 7–12 month groups given drinking water. Below we discuss biological processes of proteins that are substantially altered in APP/PS-1 mice and where applicable, the potential benefits of NAC in influencing protein levels in WT and APP/PS-1 mice. Some of the changes reported in Tables 2 and 4 are consistent with results obtained from studies in our laboratory that have previously investigated changes to the proteomes of oxidized proteins in AD and MCI [39–43].
Energy-related enzymes
Alterations to proteins involved in various aspects of energy production through changes in expression levels or modifications could contribute to the overall dysregulation in glucose metabolism evidenced in AD [39–44]. Prior to Aβ(1–42) deposition (e.g., 4–9 months) the subunits of ATP synthase protein, α and d, have decreased expression in APP/PS-1 H2O mice relative to WT H2O mice. On the other hand, the V-type proton ATPase subunit B, which is the brain specific isoform, is increased in APP/PS-1 H2O mice relative to WT H2O mice. In the 7–12 month-old group, ATP synthase subunit α was observed to be increased in APP/PS-1 H2O mice relative to WT H2O mice. ATP synthase is involved in the synthesis of ATP and work by utilizing a proton gradient that is established through the ETC located in the inner mitochondrial membrane coupled to complex physical rotations of components of this complex [45].
Cox5A levels were increased in the brains of APP/PS-1 H2O mice relative to WT H2O in the 4–9 month treatment group and decreased in the 7–12 month treatment group. Cox5A is a mitochondrial enzyme involved in the final aspects of the ETC by providing reducing equivalents from cytochrome C to oxygen [46]. Elevated expression of Cox5A prior to substantial Aβ deposition could increase the amount of ROS present by leading to increased numbers of electrons coming out of the ETC. Alternatively, increased Cox5A expression could lead to better mitochondrial function to provide ATP. Decreased Cox5A expression however, may be an indirect or direct result of increased levels of Aβ deposition and correlate with increased levels of ROS and oxidative stress. It is possible that while we measure decreased Cox5A expression in the 7–12 month animals, there may be a concurrent increase in Cox5A oxidation due to increased oxidative stress. Additional experiments, such as redox proteomics, would be necessary however to support this hypothesis.
Alpha-enolase, fructose-bisphosphate aldolase C, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and pyruvate kinase are enzymes important in the glycolytic and TCA cycles and ATP production. Previous studies that investigated expression changes in the hippocampal region of brains from AD subjects observed significant increases in the levels of these proteins relative to age-matched controls [41]. Herein, we observed differing trends that may be a result of species differences and/or the lack of neuronal death and neurofibrillary tangles in APP/PS-1 mice. Decreased expression levels of α-enolase, fructose-bisphosphate aldolase C, and GAPDH in the brains of 4–9 month-old APP/PS-1 H2O mice may be directly reflective of lower glucose metabolism and ATP available in the brain. These mice have levels of Aβ deposition and plaque formation that are lower than that observed in 12 month-old animals whom better mimic the pathology observed in later stages of AD. Additionally, in the 7–12 month treatment group, NAC significantly increases α-enolase and PK expression, suggesting that NAC through downstream mechanisms may influence certain aspects of glucose metabolism, entry into the TCA cycle, and glutamate synthesis.
Excitotoxicity
Glutamine synthetase (GS) was reported as oxidatively modified in AD brain, [47]; thus, increased GS levels observed in 12-month-old APP/PS-1 H2O mice could represent a cellular response to elevated glutamate as a result of Aβ-induced oxidative stress. Glutamate dehydrogenase 1 (GDH) is a mitochondrial enzyme that catalyzes the oxidative deamination of glutamate to form α-ketoglutarate, thereby replenishing the TCA cycle. The levels of GDH do not change in APP/PS-1 H2O mice; however, NAC indirectly or directly causes an increase in GDH in APP/PS-1 NAC mice in the 9-month-old mice. Altered glutamate regulation is observed in AD [48], and we reported oxidative modification of the glutamate transporter in AD [8]. Calbindin, a calcium binding protein that buffers cytosolic Ca2+ levels, was significantly reduced in 9-month-old APP/PS-1 H2O mice. Reduced calbindin may contribute to Ca2+ dyshomeostasis through increasing the levels of intracellular Ca2+. Other Ca2+-binding proteins such as calcineurin have been linked to Ca2+ dyshomeostatis in aging [49] and AD [50]. These data are consistent with reports of altered Ca2+ levels in APP and other model systems of AD [51–53].
Cell cycle, tau phosphorylation, and Aβ production
Increased oxidation, decreased levels, and decreased activity of Pin1 in AD and MCI brain has been reported [54–55]. Similarly, decreased levels and activity of Pin1 exist in the brains of APP/PS-1 H2O mice relative to WT H2O mice in 9- and 12-month-old mice [21]. Pin1, by binding to p-Ser/p-Thr-Pro motifs and conversion of the Pro residue of the target protein from cis-to-trans conformation and vice versa, regulates the activity of target proteins [56]. In particular, Pin1 regulates target Tau-relevant kinases and protein phosphatase 2A, important for the phosphorylation/dephosphorylation of tau, and is necessary for cell growth and proper cell cycle functions [57–58]. Pin1 has also been implicated in Aβ production, in which Pin1 inhibition promotes APP processing in the amyloidogenic pathway and therefore causes elevated levels of Aβ [48, 59] and oxidative stress[13, 60]. This result is consistent with and may contribute to elevated numbers of senile plaques in AD and in neuritic plaques in this APP/PS-1 model [38].
Synaptic abnormalities
One pathological hallmark of AD is synapse loss [61]. SNAP-25 is a synaptosomal protein that is a part of the Q-SNARE complex that functions in neurotransmitter release at neuronal synapses. SNAP-25 is also involved in vesicular docking and associates with proteins such as syntaxin to anchor vesicles to their target membranes. This protein is oxidatively modified in AD brain [60] and has increased levels in brains from APP/PS-1 H2O mice relative to WT H2O mice. SNAP-25 elevation in APP/PS-1 mice brain conceivably may be a response to oxidative stress or decreased synaptic counts or an attempt to improve neurotransmission in this mouse model of AD.
Defense systems
Heat shock protein 90α is a molecular chaperone protein that is induced in conditions of stress response. This elevated expression serves to protect cells from damaging effects caused by stress through maintaining proper folding/unfolding of proteins. HSP90α is significantly decreased in 9-month-old APP/PS-1 H2O mice suggesting a lack of sufficient brain defenses in place to combat oxidative stress.
Structural proteins
Dynamin 1 was significantly decreased in APP/PS-1 H2O mice in the 7–12 month group. Among its functions, dynamin is an inhibitor of phosphatidylinisotol 3-kinase and has been reported to be elevated in brains of aged mice, an elevation that leads to cell death [62]. However, in AD, dynamin mRNA and protein levels are significantly decreased and may be associated with altered synaptic vesicle recycling and endocytosis [63]. Additionally, dynamin was also observed to be decreased in the olfactory bulbs of old mice relative to young mice which may be a regional specific expression [64]. Lower levels of dynamin in 12-month-old APP/PS-1 H2O mice conceivably could be related to lessened synaptic vesicle recycling due to lower numbers of synapses.
Mitochondrial proteins
With aging, the activity of ACO2, mitochondrial matrix enzyme, is altered [65–66], and has decreased expression in aged rats [67]. ACO2 in brain is oxidatively modified in AD by the lipid peroxidation product, HNE [68]. ACO2 was significantly decreased in the 4–9 month treatment group In APP/PS-1 H2O mice.
Effects of NAC on the proteome of WT mice
Fructose bisphosphate aldolase A and superoxide dismutase 2 (SOD2) were significantly increased in 9 month-old WT mice treated with NAC relative to WT H2O. Fructose bisphosphate aldolase A is an enzyme involved in the glycolytic pathway. SOD2, also known as MnSOD, is an antioxidant enzyme localized in the mitochondrion that protects cells from toxic superoxide anion. Elevated SOD2 upon in vivo NAC treatment in WT mice may be an early protective mechanism against augmented oxidative stress which is known to increase with aging.
In the 12-month-old age group, NAC treatment of WT mice led to prohibitin being significantly increased and cofilin 1 significantly decreased in brain. Prohibitin is localized to the inner mitochondrial membrane and is known to inhibit DNA synthesis, regulate the cell cycle, and be involved in apopotosis and aging [69]. Prohibitin has shown cellular defense against oxidative stress in epithelial cells [70–71] and decreases in expression as a function of cellular senescence in human and chicken fibroblasts [69]. Because prohibitin also has roles as a chaperone and participates in the assembly of subunits in the mitochondrial respiratory chain complex, this protein regulates mitochondrial respiratory activity and aging [72–73]. Elevated prohibitin expression following NAC may be helpful in delaying detrimental changes associated with mitochondrial respiration in aging by increasing the availability of components involved in the electron transport chain (ETC). Cofilin 1 is the non-muscle isoform of cofilins that constitute a major portion of actin rods and plays roles in actin depolymerization and polymerization. Taken together, the results suggest that NAC, which many persons use as an over-the-counter dietary supplement, can affect levels of brain proteins involved in metabolism, prevention of oxidative stress, mitochondrial function, and cytoskeletal integrity. Each of these processes could be important in slowing mitochondrial alterations associated with aging.
Examining the proteomes of WT and APP/PS-1 mice given drinking water prior to (i.e., 9 months) and after (i.e., 12 months) periods of significant Aβ(1–42) deposition revealed several altered brain proteins involved in energy production, cell signaling, defense systems, excitotoxicity, synapse-related, cellular structure, and mitochondria in APP/PS-1 mice. In vivo NAC treatment for 5 months has been reported to provide protection against oxidative stress in brain of APP/PS-1 human double mutant knock-in mice at 9 and 12 months of age, consistent with the notion that NAC has potential to be a therapeutic approach for both MCI and AD [21]. NAC provides protection against oxidative stress because it acts as a free radical scavenger directly and increases endogenous GSH levels. Our results show that NAC alters the expression levels of some proteins in the brain proteomes of WT and APP/PS-1 mice. It is possible larger numbers of proteins affected by NAC may be detectable in specific cell types or tissues (e.g., hippocampus and/or cortex) and global effects in brain are minimized in these studies. Studies of this nature and others that investigate the possibility for NAC as a potential therapeutic approach in MCI and AD are strongly suggested and are currently ongoing in our laboratory.
Supplementary Material
Acknowledgements
This research was supported in part by grants from NIH to D.A.B. [AG-10836; AG-05119]. R.A.S. was supported by the University of Kentucky Lyman T. Johnson priority Postdoctoral Fellowship and a UNCF-Merck Science Initiative Postdoctoral Fellowship. She is now an Assistant Professor of Chemistry at the University of Pittsburgh.
References
- 1.Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA. Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–1122. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 2.Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer's disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–1060. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- 3.Lovell MA, Gabbita SP, Markesbery WR. Increased DNA oxidation and decreased levels of repair products in Alzheimer's disease ventricular CSF. J Neurochem. 1999;72:771–776. doi: 10.1046/j.1471-4159.1999.0720771.x. [DOI] [PubMed] [Google Scholar]
- 4.Mecocci P, MacGarvey U, Kaufman AE, Koontz D, et al. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- 5.Nunomura A, Perry G, Pappolla MA, Wade R, et al. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer's disease. J Neurochem. 2005;93:953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 7.Halliwell B. Role of free radicals in the neurodegenerative diseases: therapeutic implications for antioxidant treatment. Drugs Aging. 2001;18:685–716. doi: 10.2165/00002512-200118090-00004. [DOI] [PubMed] [Google Scholar]
- 8.Lauderback CM, Hackett JM, Huang FF, Keller JN, et al. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer's disease brain: the role of Abeta1-42. J Neurochem. 2001;78:413–416. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- 9.Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging. 1998;19:33–36. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- 10.Beckman KB, Ames BN. The free radical theory of aging matures. Physiol Rev. 1998;78:547–581. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 11.Markesbery WR. Oxidative stress hypothesis in Alzheimer's disease. Free Radic Biol Med. 1997;23:134–147. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- 12.Sano M, Ernesto C, Thomas RG, Klauber MR, et al. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer's disease. The Alzheimer's Disease Cooperative Study. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 13.Butterfield DA, Drake J, Pocernich C, Castegna A. Evidence of oxidative damage in Alzheimer's disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–554. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- 14.Casserly I, Topol E. Convergence of atherosclerosis and Alzheimer's disease: inflammation, cholesterol, and misfolded proteins. Lancet. 2004;363:1139–1146. doi: 10.1016/S0140-6736(04)15900-X. [DOI] [PubMed] [Google Scholar]
- 15.Borchelt DR, Ratovitski T, van Lare J, Lee MK, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- 16.Mohmmad Abdul H, Sultana R, Keller JN, St Clair DK, et al. Mutations in amyloid precursor protein and presenilin-1 genes increase the basal oxidative stress in murine neuronal cells and lead to increased sensitivity to oxidative stress mediated by amyloid beta-peptide (1–42), HO and kainic acid: implications for Alzheimer's disease. J Neurochem. 2006;96:1322–1335. doi: 10.1111/j.1471-4159.2005.03647.x. [DOI] [PubMed] [Google Scholar]
- 17.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, et al. Familial Alzheimer's disease-linked presenilin 1 variants elevate Abeta1-42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 18.van Groen T, Kiliaan AJ, Kadish I. Deposition of mouse amyloid beta in human APP/PS1 double and single AD model transgenic mice. Neurobiol Dis. 2006;23:653–662. doi: 10.1016/j.nbd.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 19.van Groen T, Liu L, Ikonen S, Kadish I. Diffuse amyloid deposition, but not plaque number, is reduced in amyloid precursor protein/presenilin 1 double-transgenic mice by pathway lesions. Neuroscience. 2003;119:1185–1197. doi: 10.1016/s0306-4522(03)00215-x. [DOI] [PubMed] [Google Scholar]
- 20.Abdul HM, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Oxidative damage in brain from human mutant APP/PS-1 double knock-in mice as a function of age. Free Radic Biol Med. 2008;45:1420–1425. doi: 10.1016/j.freeradbiomed.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Q, Aluise CD, Joshi G, Sultana R, et al. Potential in vivo amelioration by N-acetyl-L-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-in mice: toward therapeutic modulation of mild cognitive impairment. J Neurosci Res. 2010;88:2618–2629. doi: 10.1002/jnr.22422. [DOI] [PubMed] [Google Scholar]
- 22.Prescott LF, Park J, Ballantyne A, Adriaenssens P, Proudfoot AT. Treatment of paracetamol (acetaminophen) poisoning with N-acetylcysteine. Lancet. 1977;2:432–434. doi: 10.1016/s0140-6736(77)90612-2. [DOI] [PubMed] [Google Scholar]
- 23.Benzi G, Moretti A. Are reactive oxygen species involved in Alzheimer's disease? Neurobiol Aging. 1995;16:661–674. doi: 10.1016/0197-4580(95)00066-n. [DOI] [PubMed] [Google Scholar]
- 24.Butterfield D, Castegna A, Pocernich C, Drake J, et al. Nutritional approaches to combat oxidative stress in Alzheimer's disease. J Nutr Biochem. 2002;13:444. doi: 10.1016/s0955-2863(02)00205-x. [DOI] [PubMed] [Google Scholar]
- 25.Anderson ME, Luo JL. Glutathione therapy: from prodrugs to genes. Semin Liver Dis. 1998;18:415–424. doi: 10.1055/s-2007-1007174. [DOI] [PubMed] [Google Scholar]
- 26.Pocernich CB, La Fontaine M, Butterfield DA. In-vivo glutathione elevation protects against hydroxyl free radical-induced protein oxidation in rat brain. Neurochem Int. 2000;36:185–191. doi: 10.1016/s0197-0186(99)00126-6. [DOI] [PubMed] [Google Scholar]
- 27.Koppal T, Drake J, Butterfield DA. In vivo modulation of rodent glutathione and its role in peroxynitrite-induced neocortical synaptosomal membrane protein damage. Biochim Biophys Acta. 1999;1453:407–411. doi: 10.1016/s0925-4439(99)00014-9. [DOI] [PubMed] [Google Scholar]
- 28.Fontaine MA, Geddes JW, Banks A, Butterfield DA. Effect of exogenous and endogenous antioxidants on 3-nitropionic acid-induced in vivo oxidative stress and striatal lesions: insights into Huntington's disease. J Neurochem. 2000;75:1709–1715. doi: 10.1046/j.1471-4159.2000.0751709.x. [DOI] [PubMed] [Google Scholar]
- 29.Pocernich CB, Cardin AL, Racine CL, Lauderback CM, Butterfield DA. Glutathione elevation and its protective role in acrolein-induced protein damage in synaptosomal membranes: relevance to brain lipid peroxidation in neurodegenerative disease. Neurochem Int. 2001;39:141–149. doi: 10.1016/s0197-0186(01)00012-2. [DOI] [PubMed] [Google Scholar]
- 30.Farr SA, Poon HF, Dogrukol-Ak D, Drake J, et al. The antioxidants alpha-lipoic acid and N-acetylcysteine reverse memory impairment and brain oxidative stress in aged SAMP8 mice. J Neurochem. 2003;84:1173–1183. doi: 10.1046/j.1471-4159.2003.01580.x. [DOI] [PubMed] [Google Scholar]
- 31.Chetelat G, Desgranges B, de la Sayette V, Viader F, Eustache F. At the boundary between normal aging and Alzheimer disease. Rev Neurol (Paris) 2004;160:S55–S63. doi: 10.1016/s0035-3787(04)70944-3. [DOI] [PubMed] [Google Scholar]
- 32.Petersen RC. Mild cognitive impairment: transition between aging and Alzheimer's disease. Neurologia. 2000;15:93–101. [PubMed] [Google Scholar]
- 33.Reaume AG, Howland DS, Trusko SP, Savage MJ, et al. Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer's disease mutations and a "humanized" Abeta sequence. J Biol Chem. 1996;271:23380–23388. doi: 10.1074/jbc.271.38.23380. [DOI] [PubMed] [Google Scholar]
- 34.Siman R, Reaume AG, Savage MJ, Trusko S, et al. Presenilin-1 P264L knock-in mutation: differential effects on abeta production, amyloid deposition, and neuronal vulnerability. J Neurosci. 2000;20:8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anantharaman M, Tangpong J, Keller JN, Murphy MP, et al. Beta-amyloid mediated nitration of manganese superoxide dismutase: implication for oxidative stress in a APPNLH/NLH X PS-1P264L/P264L double knock-in mouse model of Alzheimer's disease. Am J Pathol. 2006;168:1608–1618. doi: 10.2353/ajpath.2006.051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andreassen OA, Dedeoglu A, Klivenyi P, Beal MF, Bush AI. N-acetyl-L-cysteine improves survival and preserves motor performance in an animal model of familial amyotrophic lateral sclerosis. Neuroreport. 2000;11:2491–2493. doi: 10.1097/00001756-200008030-00029. [DOI] [PubMed] [Google Scholar]
- 37.Varadarajan S, Yatin S, Aksenova M, Butterfield DA. Review: Alzheimer's amyloid beta-peptide-associated free radical oxidative stress and neurotoxicity. J Struct Biol. 2000;130:184–208. doi: 10.1006/jsbi.2000.4274. [DOI] [PubMed] [Google Scholar]
- 38.Murphy MP, Beckett TL, Ding Q, Patel E, et al. Abeta solubility and deposition during AD progression and in APPxPS-1 knock-in mice. Neurobiol Dis. 2007;27:301–311. doi: 10.1016/j.nbd.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 39.Butterfield DA, Gnjec A, Poon HF, Castegna A, et al. Redox proteomics identification of oxidatively modified brain proteins in inherited Alzheimer's disease: an initial assessment. J Alzheimers Dis. 2006;10:391–397. doi: 10.3233/jad-2006-10407. [DOI] [PubMed] [Google Scholar]
- 40.Butterfield DA, Sultana R. Redox proteomics identification of oxidatively modified brain proteins in Alzheimer's disease and mild cognitive impairment: insights into the progression of this dementing disorder. J Alzheimers Dis. 2007;12:61–72. doi: 10.3233/jad-2007-12107. [DOI] [PubMed] [Google Scholar]
- 41.Sultana R, Boyd-Kimball D, Cai J, Pierce WM, et al. Proteomics analysis of the Alzheimer's disease hippocampal proteome. J Alzheimers Dis. 2007;11:153–164. doi: 10.3233/jad-2007-11203. [DOI] [PubMed] [Google Scholar]
- 42.Reed TT, Pierce WM, Jr, Turner DM, Markesbery WR, Butterfield DA. Proteomic identification of nitrated brain proteins in early Alzheimer's disease inferior parietal lobule. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sultana R, Perluigi M, Butterfield DA. Oxidatively modified proteins in Alzheimer's disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis. Acta Neuropathol. 2009;118:131–150. doi: 10.1007/s00401-009-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pettegrew JW, Panchalingam K, Klunk WE, McClure RJ, Muenz LR. Alterations of cerebral metabolism in probable Alzheimer's disease: a preliminary study. Neurobiol Aging. 1994;15:117–132. doi: 10.1016/0197-4580(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 45.Leyva JA, Bianchet MA, Amzel LM. Understanding ATP synthesis: structure and mechanism of the F1-ATPase (Review) Mol Membr Biol. 2003;20:27–33. doi: 10.1080/0968768031000066532. [DOI] [PubMed] [Google Scholar]
- 46.Harris LK, Black RT, Golden KM, Reeves TM, et al. Traumatic brain injury-induced changes in gene expression and functional activity of mitochondrial cytochrome C oxidase. J Neurotrauma. 2001;18:993–1009. doi: 10.1089/08977150152693692. [DOI] [PubMed] [Google Scholar]
- 47.Castegna A, Aksenov M, Aksenova M, Thongboonkerd V, et al. Proteomic identification of oxidatively modified proteins in Alzheimer's disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002;33:562–571. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- 48.Lee HG, Zhu X, Ghanbari HA, Ogawa O, et al. Differential regulation of glutamate receptors in Alzheimer's disease. Neurosignals. 2002;11:282–292. doi: 10.1159/000067427. [DOI] [PubMed] [Google Scholar]
- 49.Foster TC, Sharrow KM, Masse JR, Norris CM, Kumar A. Calcineurin links Ca2+ dysregulation with brain aging. J Neurosci. 2001;21:4066–4073. doi: 10.1523/JNEUROSCI.21-11-04066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sultana R, Butterfield DA. Alterations of some membrane transport proteins in Alzheimer's disease: role of amyloid beta-peptide. Mol Biosyst. 2008;4:36–41. doi: 10.1039/b715278g. [DOI] [PubMed] [Google Scholar]
- 51.Haughey NJ, Nath A, Chan SL, Borchard AC, et al. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem. 2002;83:1509–1524. doi: 10.1046/j.1471-4159.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- 52.Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer's disease. Cell Calcium. 2003;34:385–397. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- 53.Sama MA, Mathis DM, Furman JL, Abdul HM, et al. Interleukin-1beta-dependent signaling between astrocytes and neurons depends critically on astrocytic calcineurin/NFAT activity. J Biol Chem. 2008;283:21953–21964. doi: 10.1074/jbc.M800148200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Butterfield DA, Poon HF, St Clair D, Keller JN, et al. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer's disease. Neurobiol Dis. 2006;22:223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 55.Sultana R, Boyd-Kimball D, Poon HF, Cai J, et al. Oxidative modification and down-regulation of Pin1 in Alzheimer's disease hippocampus: A redox proteomics analysis. Neurobiol Aging. 2006;27:918–925. doi: 10.1016/j.neurobiolaging.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 56.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3:619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 57.Butterfield DA, Abdul HM, Opii W, Newman SF, et al. Pin1 in Alzheimer's disease. J Neurochem. 2006;98:1697–1706. doi: 10.1111/j.1471-4159.2006.03995.x. [DOI] [PubMed] [Google Scholar]
- 58.Lu KP. Phosphorylation-dependent prolyl isomerization: a novel cell cycle regulatory mechanism. Prog Cell Cycle Res. 2000;4:83–96. doi: 10.1007/978-1-4615-4253-7_8. [DOI] [PubMed] [Google Scholar]
- 59.Pastorino L, Sun A, Lu PJ, Zhou XZ, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440:528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 60.Butterfield DA, Reed T, Newman SF, Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer's disease and mild cognitive impairment. Free Radic Biol Med. 2007;43:658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scheff SW, Price DA. Synaptic pathology in Alzheimer's disease: a review of ultrastructural studies. Neurobiol Aging. 2003;24:1029–1046. doi: 10.1016/j.neurobiolaging.2003.08.002. [DOI] [PubMed] [Google Scholar]
- 62.Poon HF, Vaishnav RA, Getchell TV, Getchell ML, Butterfield DA. Quantitative proteomics analysis of differential protein expression and oxidative modification of specific proteins in the brains of old mice. Neurobiol Aging. 2006;27:1010–1019. doi: 10.1016/j.neurobiolaging.2005.05.006. [DOI] [PubMed] [Google Scholar]
- 63.Yao PJ, Zhu M, Pyun EI, Brooks AI, et al. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer's disease. Neurobiol Dis. 2003;12:97–109. doi: 10.1016/s0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
- 64.Poon HF, Vaishnav RA, Butterfield DA, Getchell ML, Getchell TV. Proteomic identification of differentially expressed proteins in the aging murine olfactory system and transcriptional analysis of the associated genes. J Neurochem. 2005;94:380–392. doi: 10.1111/j.1471-4159.2005.03215.x. [DOI] [PubMed] [Google Scholar]
- 65.Curti D, Benzi G. Age-related modification of enzyme activities in synaptosomes isolated from rat cerebral cortex. J Neurosci Res. 1989;22:346–350. doi: 10.1002/jnr.490220315. [DOI] [PubMed] [Google Scholar]
- 66.Curti D, Benzi G. Role of synaptosomal enzymatic alterations and drug treatment in brain aging. Clin Neuropharmacol. 1990;13(Suppl 3):S59–S72. doi: 10.1097/00002826-199013003-00007. [DOI] [PubMed] [Google Scholar]
- 67.Poon HF, Shepherd HM, Reed TT, Calabrese V, et al. Proteomics analysis provides insight into caloric restriction mediated oxidation and expression of brain proteins associated with age-related impaired cellular processes: Mitochondrial dysfunction, glutamate dysregulation and impaired protein synthesis. Neurobiol Aging. 2006;27:1020–1034. doi: 10.1016/j.neurobiolaging.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 68.Perluigi M, Sultana R, Cenini G, Di Domenico F, et al. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer's disease: Role of lipid peroxidation in Alzheimer's disease pathogenesis. Proteomics Clin Appl. 2009;3:682–693. doi: 10.1002/prca.200800161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coates PJ, Nenutil R, McGregor A, Picksley SM, et al. Mammalian prohibitin proteins respond to mitochondrial stress and decrease during cellular senescence. Exp Cell Res. 2001;265:262–273. doi: 10.1006/excr.2001.5166. [DOI] [PubMed] [Google Scholar]
- 70.Theiss AL, Idell RD, Srinivasan S, Klapproth JM, et al. Prohibitin protects against oxidative stress in intestinal epithelial cells. FASEB J. 2007;21:197–206. doi: 10.1096/fj.06-6801com. [DOI] [PubMed] [Google Scholar]
- 71.Theiss AL, Vijay-Kumar M, Obertone TS, Jones DP, et al. Prohibitin is a novel regulator of antioxidant response that attenuates colonic inflammation in mice. Gastroenterology. 2009;137:199–208. 208, e191–e196. doi: 10.1053/j.gastro.2009.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nijtmans LG, Artal SM, Grivell LA, Coates PJ. The mitochondrial PHB complex: roles in mitochondrial respiratory complex assembly, ageing and degenerative disease. Cell Mol Life Sci. 2002;59:143–155. doi: 10.1007/s00018-002-8411-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nijtmans LG, de Jong L, Artal Sanz M, Coates PJ, et al. Prohibitins act as a membrane-bound chaperone for the stabilization of mitochondrial proteins. EMBO J. 2000;19:2444–2451. doi: 10.1093/emboj/19.11.2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.