

Glutamate, the major excitatory neurotransmitter, functions in the brain via activation of ligand gated cation channels and also the eight subtypes of Class C G protein-coupled metabotropic glutamate receptors (mGlus).[1] Selective allosteric modulation of mGlu5 has been shown to have potential for treatment of a variety of neurological disorders[2,3] including anxiety disorders[4,5], Parkinson’s disease[6–8], Fragile X syndrome[9] and schizophrenia.[10–14] The majority of mGlu5 negative allosteric modulators (NAMs) developed to date either contain an alkyne moiety 1–4 or employ the alkyne topology as basis for ligand design,[15] as in 5–8 (Figure 1). Only recently have mGlu5 NAM chemotypes been identified, through high-throughput screening (HTS) campaigns, that are structurally unrelated to the classical acetylenic derivatives, such as 9–12 (Figure 1).[16] Due to the prevalence of ‘molecular switch‘phenomenon in MPEP related scaffolds, our interest focused on the discovery and development of novel mGlu5 NAM chemotypes, by both HTS and Artificial Neural Network (ANN) virtual screens.
Figure 1.

mGlu5 NAM chemotypes represented by alkyne chemotypes 1–4, chemotypes based on the alkyne topology 5–8 and novel chemotypes not based on an acetylenic, MPEP-like scaffold or topology 9–12.
High-throughput screens (HTS) leverage the often diverse chemical content found in screening collections of small molecules (105 – 106 compounds) to rapidly identify a subset of molecules with desired activity via automated plate based experimental assays.[17] A recent PubChem search[18] returned 120 HTS assays targeting G protein-coupled receptor (GPCR) signaling (pubchem.ncbi.nlm.nih.gov, accessed April 2011) that addressed targets like RGS16-Gαo (AID1441, a primary screen reporting 826 active compounds out of 218,535 tested), and 5-hydroxytryptamine (serotonin) receptor subtype 1a (5-HT1A) (AID567, a primary screen reporting 366 actives out of 64,907 compounds tested). The hit rates (0.4–0.6% active) in these examples are typical of HTS and highlight the frequently encountered limitation that approximately 99.5% of the screened compounds are routinely reported as inactive towards the desired screening target.[18,19] Theoretically, the increase of this sparse hit rate by an order of magnitude (5% actives) would enrich the screening results and require that only 10% of the original total amount of compounds would need to be tested in a second round to roughly double the amount of active compounds identified. A systematic method capable of producing this level of enrichment by utilizing virtual screens would significantly reduce iterative screening costs and accelerate experimental turnaround times to facilitate more rapid discovery of novel leads.
Rodriguez and co-workers recently screened approximately 160,000 small molecules to identify allosteric modulators of mGlu5 receptor signaling.[19] The primary assay revealed 624 potential antagonists by reduction of calcium flux at an 80% maximally effective concentration of glutamate (EC80) in an experimental screening protocol designed to detect various types of mGlu5 allosteric modulation. In confirmatory screens that employed 10-point concentration response curves, 345 mGlu5 antagonists were verified.[19] This experimental HTS data set formed the basis of model development for a virtual screen of the ChemDiv screening collection of over 700,000 compounds.[20] Results of that virtual screen and pharmacological characterization of a selected set of the predicted compounds are described herein. Medicinal chemistry optimization of a new chemotype and characterization of a novel tool compound is also described, including assessment of in vivo efficacy in two different behavioral assays.
Structure activity relationships are built on the paradigm that structurally similar compounds typically have similar biological activity. Quantitative structure activity relationships (QSARs) connect activity to structure by fitting a mathematical function through known structure activity relationship (SAR) data: f(structure) = activity.[21] Linear regression analysis is commonly used to establish QSAR models. However, this approach assumes that the biological activity can be described as a linear combination of all the structural descriptors which is not necessarily the case. Machine learning tools like artificial neural networks (ANNs) can alleviate this problem, since they employ non-linear functions to relate input (structure) and output (activity), as described by Winkler and co-workers.[22] The QSAR models reported here are neural networks employing one hidden layer with a variable number of inputs and hidden neurons trained with simple back-propagation of errors, or resilient propagation.[23]
Once this quantitative relationship is established in a model, it can be employed to predict the activity of molecules with known structure but unknown activity. Our approach for establishing QSARs from HTS experiments includes four steps: i) a dataset connecting molecules with known SAR needs to be obtained; ii) the structures of the molecules are described in a way that remains independent from spatial orientation (avoids a requirement to perform lengthy superimposition of the molecules); iii) training of models (ANNs) through supervised back-propagation of errors is performed; and iv) final prediction of molecular bioactivity at a given target without prior knowledge of activity for the query compounds is returned. Predicted active molecules from commercially available databases[20] (e.g. ChemDiv, ChemBridge, Enamine) are then ordered and tested to validate the model’s ability to enrich for compounds with mGlu5 pharmacologic activity. Noeske and co-workers[24,25] recently utilized virtual screens that employed a machine learning Self-Organizing Map (SOM) approach to search for mGlu1 NAMs and yielded one molecule showing potency below 1 µM, and five between 1 µM and 15 µM in a rat mGlu1 experimental assay. We also have recently applied an initial version of ANN-based virtual HTS to identify new mGlu5 positive allosteric modulators (PAMs).[26]
Six models with different subsets of ADRIANA descriptors were trained.[26–28] The ANN model trained on all 35 descriptor categories available in the ADRIANA[29] descriptor set achieved a root mean square deviation (rmsd) of the independent data set of 0.209, area under the curve (auc) of 0.83, and theoretical enrichment of 18.8. The descriptor subsets and model quality measures for auc, rmsd, and enrichment are shown in Table 2. For the first round of model optimization, the four least sensitive descriptor categories were removed leading to an rmsd of 0.199, auc of 0.87, and enrichment of 28.2. Removal of eight more descriptor categories led to the optimal model with 763 descriptors and to an rmsd of 0.201, auc of 0.86, and theoretical enrichment of 37.6. Further reduction of the number of descriptors led to slightly inferior models which were not considered for further predictions (Figure 2). While the rmsd and auc values represented the general quality of the trained ANNs, these metrics mainly captured the overall utility of the models to sort potentially active from likely inactive compounds. However, the success of a model is based primarily on the experimental enrichment of active compounds (true positives) for commercial orders and experimental testing. Therefore, the theoretical enrichment measure (see Supporting Information) was weighted more heavily in our final assessment of model metrics based on its closer correlation with the final experimental ratio of correctly identified compounds with mGlu5 activity.
Table 2.
The rmsd, auc and enrichment values for all mGlu5 NAMs QSAR models
| No. & Type of descriptor |
train | rmsd monitor (independ.) |
auc | Enrichment (at 0.3%) |
|---|---|---|---|---|
| 1252 1–35 | 0.184 | 0.203 (0.209) | 0.83 | 18.8 |
| 972 1–19, 21–26, 29–32, 34–35 | 0.168 | 0.202 (0.199) | 0.87 | 28.2 |
| 763 1–9, 11, 12, 14, 15, 17, 21–23, 25, 29–32, 35 | 0.157 | 0.201 (0.201) | 0.86 | 37.6 |
| 683 1–8, 14, 23, 25, 29–32, 35 | 0.178 | 0.204 (0.210) | 0.84 | 9.4 |
| 555 1–8, 14, 23, 25, 29–31, 35 | 0.204 (0.218) | 0.81 | 28.2 | |
| 416 1–8, 23, 25, 30, 31, 35 | 0.210 (0.215) | 0.82 | 9.4 |
Figure 2.
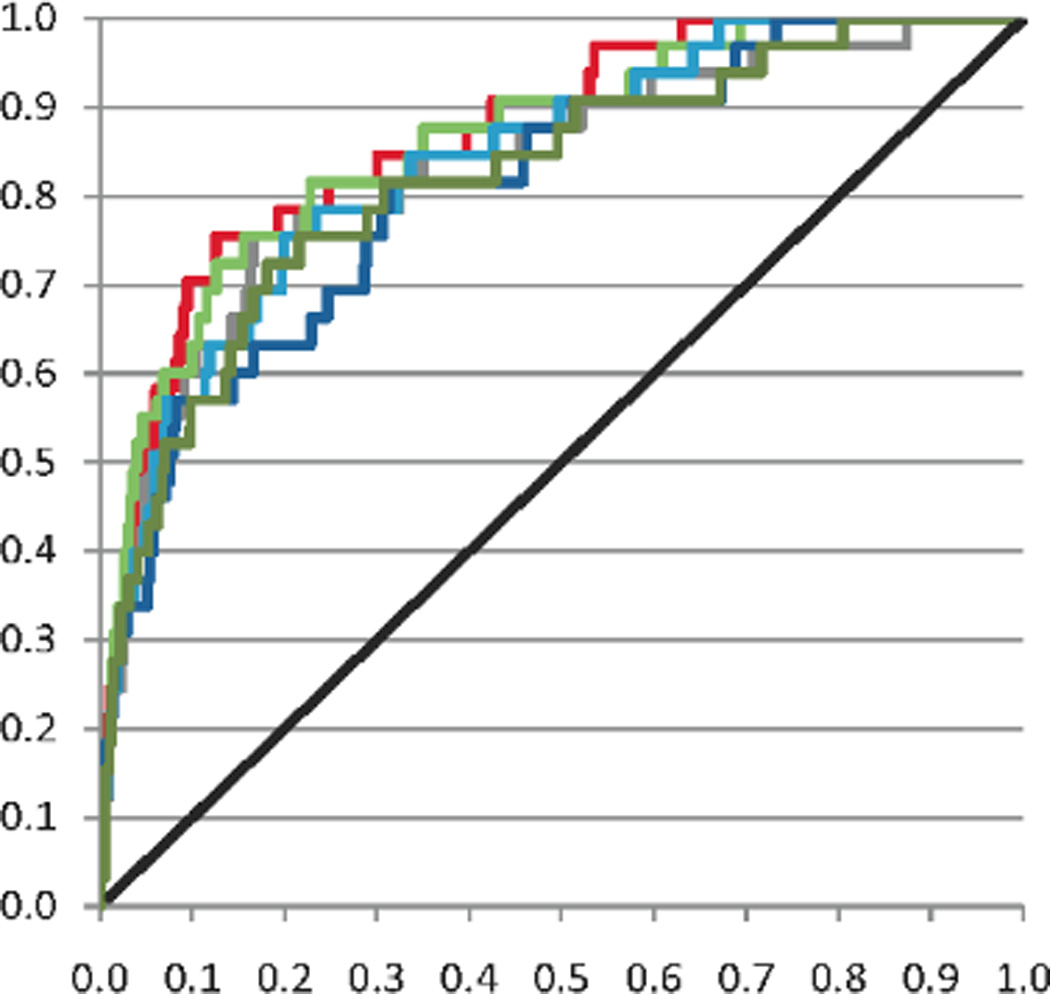
Receiver Operating Characteristic (ROC) curve plot for models using base (black), 416 (dark green), 555 (dark blue), 683 (light blue), 763 (light green), 972 (red), and 1252 (grey) descriptors: While the models with 972 (red) and 763 (light green) descriptors performed well over the entire ROC curve, the other models clearly show a reduced performance in the middle portion of the ROC curve. The steepness at beginning of the curve most effects theoretical enrichment, however, all of the models displayed similar performance for total area under the curve (AUC). Therefore, based on significantly better theoretical enrichment values, the model with 763 descriptors was selected for final prediction of active compounds to be ordered.
The ANN model with the highest theoretical enrichment for mGlu5 NAMs was employed to virtually screen the ChemDiv (San Diego, CA) Discovery Chemistry database of 708,416 commercially available drug-like compounds.[20] At a 10 µM potency cutoff, the model predicted a set of 42,041 small molecules. Molecules with a weight above 600 Da, topological polar surface area greater than 130 Å2, a calculated XlogP[30] value of greater than 4.0, or compounds that contained labile or reactive fragments were removed via automated filtration.[31,32] Further removal of molecules with a Tannimoto substructure[33] similarity value above 0.6 reduced the predicted active compounds to a final order size of 749 compounds. These compounds were tested at the Vanderbilt Institute for Chemical Biology high-throughput screening center[34] to reveal a single point concentration (50% activation or inhibition @30 µM) experimental hit rate of 12% (88/749 compounds) that included a putative 51 negative allosteric modulators (NAMs), 18 positive allosteric modulators (PAMs) and 19 agonists. Concentration response curves (CRCs) at 10 concentrations (ranging 1 nM to 30 µM) confirmed the identification of 12 NAMs, 14 PAMs and 1 partial agonist compound (EC50/IC50 < 30 µM, a 3.6% overall hit rate) representing an enrichment factor of 15.7 for mGlu5 activity compared with the original mGlu5 experimental HTS data (a 0.22% original experimental hit rate). Compounds with confirmed antagonist activity (1.6% of predicted compounds) were enriched by a factor of 7.0 with novel mGlu5 NAM scaffolds. The optimized mGlu5 ANN models recognized substructure patterns related non-linearly to biological activity in chemical scaffolds that were not used in training the method and thus allowed for identification of previously unknown mGlu5 NAM compounds with novel chemotypes (Figure 3).[35] The discovery of two novel, potent mGlu5 NAMs 13 and 14, with IC50 values of 75 and 124 nM, respectively, possessing a previously unknown scaffold demonstrates the success of this approach.
Figure 3.

ANN pattern recognition of substructure fragments from known mGlu5 NAMs identified novel combinations of substructures with potent mGlu5 NAM activity. Highlighted substructures exist in several compounds in the training set from separate scaffolds. The optimized mGlu5 ANN model identified potent mGlu5 NAMs 13 and 14 that combined two or more of these features into the same scaffold.
Particularly interesting among the confirmed active compounds from the ANN virtual screen were the two potent 2-(2-benzoxazolylamino)-4-phenylpyrimidines 13 and 14, differing only in the substituent at the 5-position of the pyrimidine ring. Structurally, these compounds represent a significant departure from known mGlu5 NAM chemotypes. Whereas analog 13 contained a 5-cyano group, the corresponding group on compound 14 was an ethyl ester. Although esters are generally not suitable for use in many applications due to esterase mediated metabolism, cyano groups are often tolerated. The scaffold was quite attractive for a medicinal chemistry optimization program as there were multiple portions of the molecule amenable to modification and SAR development. Efforts were thus focused toward that objective.
Synthetic routes used to prepare the target compounds were quite straightforward (Scheme 1). In order to access 5-cyanopyrimidine targets with the generic structure 18, two different routes that both began with β-ketonitriles 15 were employed. Many β-ketonitriles are commercially available or can easily be prepared through additions of cyanomethanide anion to simple alkyl esters.[36] In equation I, 15 was treated with N,N-dimethylformamide-dimethyl acetal to afford intermediate 16, which was subsequently reacted with substituted guanidines 17 under basic conditions to afford 18. Alternatively in equation II, 15 could be reacted directly with 17 in the presence of triethyl orthoformate to produce 18, and this approach was generally preferred due to superior yields and fewer side reactions. For analogs lacking a 5-cyano substituent such as 21, palladium mediated coupling of amines 20 with 2-chloropyridine 19 were employed (Scheme 1, eq. III). Using the synthetic routes as described allowed for evaluation of three distinct areas of the chemotype: the necessity of the 5-cyano substituent, SAR at the 6-position of the pyrimidine ring (R1), and SAR around the 2-amino substituent (R2).
Scheme 1.

Reagents and conditions: a) Me2NCH(OMe)2, DMF ; b) Cs2CO3, DMF, µw, 200 °C, 10 min (5–40% over 2 steps); c) HC(OEt)3, 140 °C (50–75%); d) Pd2(dba)3, Cs2CO3, Xantphos, dioxane, µw, 140 °C, 20 min (10–50%).
Initial SAR exploration focused on substitution at the 4 and 5-position of the pyrimidine core as well as on the 2 and 3-position of the phenyl ring. The resynthesis of the hit compound 13 gave potency in good agreement with that observed with the original batch (IC50 = 89 nM). Removal of the 5-cyano substituent resulted in a dramatic loss of potency (IC50 >10 µM) as did the introduction of a 4-methyl substituent on the pyrimidine ring (IC50 >30 µM). Isosteric replacements for the 2-amino benzoxazole, such as benzothiazoles, benzimidazoles, quinolines and pyridines, also displayed diminished potency at mGlu5 (IC50 > 30 µM). In light of such data, the decision was made to focus SAR development on analogs containing the key 5-cyano substituent on the pyrimidine core and the 2-amino benzoxazole moiety at R2.
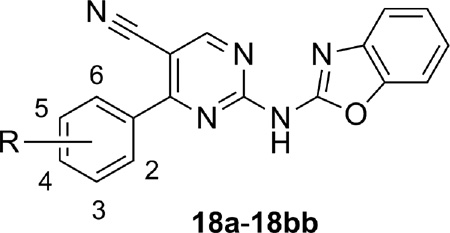
Further development of SAR around substitution of the phenyl ring was continued in the context of the 5-cyanopyrimidine core (Table 3). In order to evaluate the effect of substituents of varying electronic character, methyl, chloro, and methoxy analogs were prepared at each position (18a–18i). Both the 2-methoxyphenyl analog 18g and 4-methylphenyl analog 18c had similar activity to the hit compound 13. Other analogs demonstrated reduced potency relative to 13 with 3-chlorophenyl analog 18e being considerably less potent. Fluorophenyl analogs were more active than their chlorophenyl comparators (compare 18j to 18d and 18k to 18f), and analog 18k was equivalent in potency to 13. Several analogs with larger electron withdrawing groups were either inactive or weak antagonists (18l–18p). The size of the substituent on the phenyl ring was key to potency as evidenced by 4-ethylphenyl analog 18q (compare to 18c) and 2-ethoxyphenyl analog 18r (compare to 18g). A number of disubstituted analogs were prepared (18s–18bb) and compounds were generally weak antagonists. The exceptions were 4-chloro-2-fluorophenyl analog 18y, 4-fluoro-2-methoxyphenyl analog 18z, and 4-chloro-2-methoxyphenyl analog 18aa, which were only slightly less potent than 13. Interestingly, analog 18z was less potent than both 18g and 18k, which was somewhat surprising.
Table 3.
Structures and activities of 5-Cyano pyrimidine analogues 18.
 | |||
|---|---|---|---|
| Compd | R | IC50 (nM)[a] | %Glu Max[b] |
| 13 | H | 89 ± 32 | 1.9 ± 3 |
| 18a | 2-CH3 | 403 ± 47 | 1.9 ± 0.4 |
| 18b | 3-CH3 | 138 ± 47 | 3.6 ± 1.1 |
| 18c | 4-CH3 | 90 ± 32 | 2.6 ± 0.7 |
| 18d | 2-Cl | 516 ± 54 | 2.2 ± 0.5 |
| 18e | 3-Cl | 2830 ± 1320 | 3.2 ± 0.8 |
| 18f | 4-Cl | 315 ± 110 | 2.5 ± 0.5 |
| 18g | 2-OCH3 | 62 ± 10 | 0.9 ± 0.4 |
| 18h | 3-OCH3 | 266 ± 47 | 2.4 ± 0.3 |
| 18i | 4-OCH3 | 223 ± 74 | 2.1 ± 0.3 |
| 18j | 2-F | 258 ± 51 | 1.8 ± 0.4 |
| 18k | 4-F | 91 ± 22 | 1.7 ± 0.1 |
| 18l | 3-CF3 | >30,000 | — |
| 18m | 4-CF3 | >30,000 | — |
| 18n | 3-Br | >30,000 | — |
| 18o | 4-Br | 4120 ± 1860 | 3.8 ± 1.2 |
| 18p | 4-OCF3 | >30,000 | — |
| 18q | 4-CH2CH3 | 1530 ± 358 | 2.8 ± 0.5 |
| 18r | 2-OCH2CH3 | 6580 ± 1670 | 4.5 ± 0.9 |
| 18s | 2,3-di-F | 1910 ± 773 | 2.5 ± 0.3 |
| 18t | 3,4-di-F | 2580 ± 1170 | 9.6 ± 0.7 |
| 18u | 2,4-di-Cl | 666 ± 238 | 3.0 ± 0.4 |
| 18v | 3,4-di-Cl | >10,000[c] | 46 ± 13 |
| 18w | 3,5-di-Cl | >30,000 | — |
| 18x | 3,5-di-OCH3 | >10,000[c] | 24 ± 10 |
| 18y | 2-F, 4-Cl | 375 ± 90 | 1.8 ± 0.5 |
| 18z | 2-OCH3, 4-F | 217 ± 10 | 1.5 ± 0.4 |
| 18aa | 2-OCH3, 4-Cl | 148 ± 42 | 2.0 ± 0.2 |
| 18bb | 3-F, 4-OCH3 | 1060 ± 696 | 2.9 ± 0.6 |
IC50 values are the average of at least three determinations,
% Glu Max is the maximum response of compounds relative to the maximal glutamate response – at least three determinations,
CRC does not plateau.35
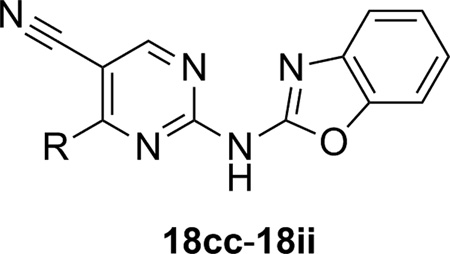
Having identified several similarly potent mGlu5 NAMs in this manner, subsequent effort focused on the replacement of the phenyl ring (R1) altogether. A variety of 5- and 6-membered heterocycles were evaluated with limited benefit (Table 4). The evaluation of pyridine (18cc and 18dd) and thiophene (18ee and 18ff) isosteres revealed that these were not well tolerated, although 3-pyridyl analog 18dd remained moderately potent. Similarly, napthyl analogs such as 18gg and 18hh were only weak antagonists. Finally, introduction of a cyclohexyl group (18ii) resulted in only an approximately 3-fold drop in potency relative to 13. The tolerance for the cyclohexyl ring is a bit surprising given the sensitivity demonstrated with other analogs (such as 18ee and 18ff) that are much more similar in size to 13. Based on the SAR described in the analogs presented thus far, it was determined that potency was substantially influenced by relatively minor changes in this portion of the chemotype. Such observations are quite common with mGlu5 NAMs from other chemotypes and have been noted in the literature.[37–40] A final effort was made to introduce substituents onto various positions of the benzoxazole ring; however, even small groups such as methyl and fluoro were not generally tolerated.
Table 4.
Structures and activities of 5-Cyano pyrimidine analogues 18.
 | |||
|---|---|---|---|
| Compd | R | IC50 (nM) [a] | %Glu Max[b] |
| 18cc | pyridin-2-yl | >10,000c | 22 ± 2 |
| 18dd | pyridin-3-yl | 500 ± 23 | 2.1 ± 0.7 |
| 18ee | thiophen-2-yl | >10,000[c] | 35 ± 10 |
| 18ff | thiophen-3-yl | 4740 ± 1530 | 13 ± 5 |
| 18gg | naphthalen-2-yl | >10,000[c] | 47 ± 11 |
| 18hh | naphthalen-1-yl | >10,000[c] | 40 ± 12 |
| 18ii | cyclohexyl | 216 ± 71 | 3.8 ± 0.8 |
IC50 values are the average of at least three determinations,
% Glu Max is the maximum response of compounds relative to the maximal glutamate response – at least three determinations,
CRC does not plateau.35
Having a number of compounds with good functional potency in hand, an evaluation of the metabolic stability of several compounds was in order. Compounds of interest were examined with regard to their stability in human and mouse liver microsomes following 15 minutes of incubation.[41] A general trend of greater stability in human compared to mouse liver microsomes was noted. Enhanced stability relative to 13 was observed with the 4-substituted phenyl analogs 18c, 18i, and 18k.[35] The nature of the 4-substituent does not appear to be a determining factor as these compounds have similar stability in both species. Although there was little difference between their respective metabolic profiles in microsomes, the methyl group of 18c was still viewed as a risk for in vivo oxidative metabolism. The fluorophenyl analog 18k offered a convenient and equipotent alternative without such a concern.
Consideration of the SAR and the stability data in mouse liver microsomes indicated that compound 18k would be a potentially interesting compound for evaluation in an in vivo pharmacokinetics study. Prior to initiation of that study, more detailed molecular pharmacology studies with 18k were conducted. A radioligand binding study (Figure 4B) measuring the ability of the compound to compete with the equilibrium of [3H]-3-methoxy-5-(pyridin-2-ylethynyl)pyridine,[42] a close structural analog of MPEP, confirmed the interaction of 18k with the known mGlu5 allosteric binding site (mGlu5 Ki = 84 ± 14 nM). We evaluated the effects of 18k in a native system, rat cortical astrocytes, where mGlu5-mediated responses have previously been studied.[43,44] 18k induced a rightward shift in the glutamate concentration response curve (CRC) and reduced the maximal effect of glutamate (Figure 4C), confirming activity in the native system and suggesting a non-competitive inhibition of the glutamate response. It was also important to examine the effects of 18k in multiple signalling pathways as it has been shown that mGlu5 allosteric modulators can have an effect through some pathways and not others.[43,44] To this end, we determined the effects of 18k on the ERK1/2 phosphorylation response to glutamate in HEK293 cells expressing rat mGlu5. We found 18k blocked the effect of glutamate in a concentration dependent manner (Figure 4D), confirming antagonist activity in the ERK pathway. Examination of the activity of compound 18k against other mGlu receptors35 revealed no observable activity at 10 µM against mGlu1–4 and mGlu6–8. Compound 18k was also run at 10 µM in Ricerca Bioscience’s LeadProfilingScreen®,[45] which consists of radioligand binding assays in 68 primary molecular targets from classes such as GPCRs, kinases, and ion channels, including many targets relevant to CNS drug discovery. In this screen, an inhibition of more than 50% of radioligand binding was observed only in the adenosine A3 receptor assay (73% inhibition of binding), indicating that 18k has a remarkably clean pharmacology profile.
Figure 4.

In Vitro Characterization of 18k. a) Concentration response curve (CRC) in mGlu5 calcium mobilization assay (IC50 = 91 ± 22 nM, % Glu Max = 1.7 ± 0.1); b) Competition binding CRC between 18k and [3H]MeOPEPy, affording a Ki of 84 ± 14 nM, suggesting 18k is fully competitive with the classical MPEP allosteric site in mGlu5; c) Effect of 18k on glutamate CRC in rat cortical astrocytes (● = glutamate + vehicle, ■ = glutamate + 300 nM 18k, ▲= glutamate + 1 µM 18k, ▼ = glutamate + 30 µM 18k); d) Effect of 18k on glutamate-induced ERK phosphorylation in mGlu5 expressing HEK293 cells (● = glutamate + vehicle, ■ = glutamate + 10 nM 18k, ▲= glutamate + 30 nM 18k, ▼= glutamate + 100 nM 18k, ♦ = glutamate + 1 µM 18k). Data represent the mean ± S.E.M. of at least three independent experiments.
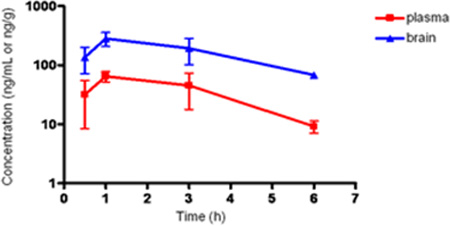
Confident of the molecular pharmacology profile, a study of the exposure of 18k following oral dosing in mice was conducted.[35] Unfortunately, the results revealed no exposure above detectable limits in either plasma or brain. As such, the compound was subsequently studied using intraperitoneal dosing (Table 5). Although exposure of 18k was moderate in plasma (AUC = 224 ng·h/mL), because of a more than 4 to 1 brain to plasma ratio, the brain exposure (AUC = 1006 ng·h/g) was quite good.[46] Protein binding studies revealed that compound 18k was highly bound to both mouse plasma (99.4%) and mouse brain homogenates (99.5%).[47] While limited free fraction represented a concern for the ability of 18k to function in an in vivo behavior study, the brain exposure data argued for its evaluation in spite of this fact.
Table 5.
Plasma and brain exposure of 18k in mice.
 | |||
|---|---|---|---|
| Time (h) |
Plasma [ ] (ng/mL) |
Brain [ ] (ng/mL) |
Brain:Plasma ratio |
| 0.5 | 31.8 | 136 | 4.3 |
| 1 | 64.7 | 283 | 4.4 |
| 3 | 45.4 | 193 | 4.3 |
| 6 | 9.2 | 68 | 7.4 |
Ideally, evaluation of a new tool compound such as 18k in a behavioral assay would initially take place in an assay known to be sensitive to other mGlu5 antagonists. It has been established that mice will bury foreign objects such as glass marbles in deep bedding.[48] Furthermore, studies have shown that low doses of anxiolytic benzodiazepines inhibit this behavior.[49] Additionally, the well known mGlu5 NAM tool compounds MPEP and fenobam are effective in this model.[50–52] Marble burying is also an operationally convenient assay, making it useful in an in vivo screening paradigm. We examined compound 18k as well as MTEP (positive control) in this assay using a 15 minute pretreatment with both compounds (Figure 5). Robust inhibition of marble burying with the 15 mg/kg dose of MTEP was observed as expected. Significant inhibition of marble burying was also observed with 18k at the highest dose of 56.6 mg/kg. Consideration of the results in the context of the pharmacokinetic study provided some insight. In mice, the average brain concentration of 18k at 30 minutes was 0.41 µM while the brain concentration at 1 hour post dose was 0.85 µM. The marble burying assay was conducted between the 15 and 45 minute time points post dose so the brain concentrations at these time points are relevant. If a dose linear increase in exposure is assumed, a 56.6 mg/kg dose should lead to brain exposures between 2.3 and 4.8 µM. It is certainly possible that the highly protein bound nature of 18k is restricting the availability of free drug to interact the receptor, thus necessitating relatively high brain concentrations in order to achieve efficacy.
Figure 5.

In Vivo Characterization of 18k. Inhibition of marble burying with 18k (VU0366058) relative to MTEP (2). Dosing (i.p.) vehicle was 10% tween 80. No sedation was observed in any groups. N=12 per dosing group. Significance determined as P < 0.05 vs. vehicle control group (Dunnett’s test).
Seeking to examine compound 18k in a behavioral model of addiction, focus turned to a relatively new model. C57Bl/6J mice readily acquired operant responding to varied visual and auditory stimuli without prior training, a phenomenon termed operant sensation seeking (OSS).[53,54] During this assay mice will ‘self-administer’ visual cues consisting of flashing lights of random duration in combination with an auditory stimulus. Recent studies using low doses of the dopamine antagonist cis-flupenthixol to disrupt dopamine signaling demonstrated increased responding for OSS stimuli, similar to effects seen with cocaine self-administration.[55] Furthermore, mice lacking mGlu5 also fail to acquire OSS despite having normal acquisition of food self-administration,[56] suggesting that OSS would represent another interesting assay for assessment of novel mGlu5 NAMs with therapeutic potential for addiction. Since it has also been established that mGlu5 knockout mice do not self-administer cocaine,[57] the reinforcing effects of OSS may in fact be more similar to psychostimulants than food. Recently, the results of studies with both MTEP and the first novel small molecule mGlu5 NAM were reported in the OSS model.[58] Gratifyingly, compound 18k also dose dependently reduced progressive ratio responding for OSS stimuli with no significant effects observed with a food reinforcer (Figure 6).[53,54,56,58] Unlike the marble burying assay, here significant efficacy was observed at both 30 and 56.6 mg/kg doses of 18k.
Figure 6.

Efficacy of compound 18k in the OSS model of addiction. a) OSS, b) food. Dosing (i.p.) vehicle was 10% tween 80. OSS: n=8. Food: n=6. Significance determined as P < 0.05 vs. vehicle control group (Dunnett’s test).
In summary, we have developed a novel mGlu5 NAM tool compound from a completely new 4-aryl-5-cyanopyrimidine chemotype, identified through an ANN virtual screening exercise. Although SAR in this series was somewhat flat, some compounds with excellent potency were identified. Compound 18k is a potent non-competitive antagonist in vitro that interacts with the known allosteric binding site. It is also selective against other mGlu receptors and demonstrated excellent selectivity in a large, diverse panel of other drug targets. When examined in both the marble burying model of anxiety and the OSS model of addiction, compound 18k demonstrated efficacy. Although the molecule lacks oral bioavailability in mice and is highly protein bound, these features are overcome by intraperitoneal dosing and the excellent brain to plasma ratio of 18k. Of particular value, is the demonstration that the artificial neural network screening approach employed here can be used as a viable method for the identification of fundamentally new chemotypes. Further studies including additional in vivo profiling of compound 18k are planned and will be reported in due course.
Supplementary Material
Table 1.
Summary of 1,252 molecular descriptors in 35 categories computed with ADRIANA
| Description Method |
Description Property | Abbreviation | Number | |
|---|---|---|---|---|
| 1 | Scalar | MW of comound | Weight | 1 |
| 2 | Desciptors | No. H-Bond donors | HDon | 1 |
| 3 | No. H-Bond acceptors | HAcc | 1 | |
| 4 | Oct/water part. coeffic. | XlogP | 1 | |
| 5 | Polar surface area | TPSA | 1 | |
| 6 | Molecular polarizability | Polariz | 1 | |
| 7 | Dipole moment | Dipol | 1 | |
| 8 | Solubility (water) | LogS | 1 | |
| 9 | 2D | Atom identities | 2DA_Ident | 11 |
| 10 | Autocorrelation | σ atom charges | 2DA_SigChg | 11 |
| 11 | π atom charges | 2DA_PiChg | 11 | |
| 12 | Total charges | 2DA_TotChg | 11 | |
| 13 | σ atom electroneg. | 2DA_SigEN | 11 | |
| 14 | π atom electroneg. | 2DA_PiEN | 11 | |
| 15 | Lone pair electroneg. | 2DA_LpEN | 11 | |
| 16 | Effective atom polariz. | 2DA_Polariz | 11 | |
| 17 | 3D | Atom identities | 3DA_Ident | 12 |
| 18 | Autocorrelation | σ atom charges | 3DA_SigChg | 12 |
| 19 | π atom charges | 3DA_PiChg | 12 | |
| 20 | Total charges | 3DA_TotChg | 12 | |
| 21 | σ atom electroneg. | 3DA_SigEN | 12 | |
| 22 | π atom electroneg. | 3DA_PiEN | 12 | |
| 23 | Lone pair electroneg. | 3DA_LpEN | 12 | |
| 24 | Effective atom polariz. | 3DA_Polariz | 12 | |
| 25 | Radical | Atom identities | RDF_Ident | 128 |
| 26 | Distribution | σ atom charges | RDF_SigChg | 128 |
| 27 | Function | π atom charges | RDF_PiChg | 128 |
| 28 | Total charges | RDF_TotChg | 128 | |
| 29 | σ atom electroneg. | RDF_SigEN | 128 | |
| 30 | π atom electroneg. | RDF_PiEN | 128 | |
| 31 | Lone pair electroneg. | RDF_LpEN | 128 | |
| 32 | Effective atom polariz. | RDF_Polariz | 128 | |
| 33 | Surface | Mol. Electrostat. pot. | Surf_ESP | 12 |
| 34 | Autocorrelation | H-Bonding pot. | Surf_HBP | 12 |
| 35 | Hydrophobicity pot. | Surf_HPP | 12 | |
| Total 1252 | ||||
Acknowledgements
The authors warmly thank the NIH and NIDA for support of our programs in drug discovery. We specifically acknowledge NIDA (R01DA023947-01) ad NIMH (R01090192-01). We thank Daryl F. Venable for technical assistance with calcium flux and radioligand binding assays.
Footnotes
Supporting information for this article is available on the WWW under http://www.chemmedchem.org or from the author.
Contributor Information
Jens Meiler, Email: jens.meiler@vanderbilt.edu.
Craig W. Lindsley, Email: craig.lindsley@vanderbilt.edu.
References
- 1.Niswender CM, Conn PJ. Annu. Rev. Pharmacol. Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gasparini F, Bilbe G, Gomez-Mancilla B, Spooren W. Curr. Opin. Drug Discov. Devel. 2008;11:655–665. [PubMed] [Google Scholar]
- 3.Conn PJ, Christopoulos A, Lindsley CW. Nat. Rev. Drug Discov. 2009;8:41–54. doi: 10.1038/nrd2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter RH, Jaeschke G, Spooren W, Ballard TM, Buttelmann B, Kolczewski J-U, Peters S, Prinssen E, Wichmann J, Viera E, Muhlemann A, Gatti S, Mutel V, Malherbe P. J. Pharmacol. Exp. Ther. 2005;315:711–721. doi: 10.1124/jpet.105.089839. [DOI] [PubMed] [Google Scholar]
- 5.Swanson CJ, Bures M, Johnson MP, Linden AM, Monn JA, Schoepp DD. Nat. Rev. Drug Discov. 2005;4:131–144. doi: 10.1038/nrd1630. [DOI] [PubMed] [Google Scholar]
- 6.Marino MJ, Conn PJ. Curr. Drug Target CNS Neurol. Disord. 2002;1:239–250. doi: 10.2174/1568007023339319. [DOI] [PubMed] [Google Scholar]
- 7.Marino MJ, Awad H, Poisik O, Wittmann M, Conn PJ. Amino Acids. 2002;23:185–191. doi: 10.1007/s00726-001-0127-1. [DOI] [PubMed] [Google Scholar]
- 8.Marino M, Valenti O, Conn PJ. Drugs Aging. 2003;20:377–397. doi: 10.2165/00002512-200320050-00006. [DOI] [PubMed] [Google Scholar]
- 9.Bear MF, Huber KM, Warren ST. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Marino MJ, Conn PJ. Curr. Drug Target CNS Neurol. Disord. 2002;1:1–16. doi: 10.2174/1568007023339544. [DOI] [PubMed] [Google Scholar]
- 11.Moghaddam B. Psychopharmacology. 2004;174:39–44. doi: 10.1007/s00213-004-1792-z. [DOI] [PubMed] [Google Scholar]
- 12.Kinney GG, Burno M, Campbell UC, Hernandez LM, Rodriguez D, Bristow LJ, Conn PJ. J. Pharmacol. and Exper. Ther. 2003;306:116–123. doi: 10.1124/jpet.103.048702. [DOI] [PubMed] [Google Scholar]
- 13.Conn PJ, Lindsley CW, Jones CK. Trends in Pharmacol. Sci. 2009;30:25–31. doi: 10.1016/j.tips.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindsley CW, Emmitte KA. Curr. Opin. Drug Discov. & Dev. 2009;12:446–457. [PubMed] [Google Scholar]
- 16.Emmitte KA. ACS Chem. Neurosci. 2011;2:411–432. doi: 10.1021/cn2000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bleicher KH, Bohm H-J, Muller K, Alanine AI. Nat. Rev. Drug Discov. 2003;2:369–378. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 18.See: http://pubchem.ncbi.nlm.nih.gov
- 19.Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, Blatt TN, Jadhav S, Menon U, Vinson PN, Rook JM, Stauffer SR, Niswender CM, Lindsley CW, Weaver CD, Conn, P.J. PJ. Mol. Pharmacol. 2010;78:1105–1123. doi: 10.1124/mol.110.067207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.See: www.chemdiv.com
- 21.Hansch C, Hoekman D, Leo A, Weininger D, Selassie CD. Chem. Rev. 2002;102:783–812. doi: 10.1021/cr0102009. [DOI] [PubMed] [Google Scholar]
- 22.Winkler D. Mol. Biotechnol. 2004;27 doi: 10.1385/MB:27:2:139. 139-167-167. [DOI] [PubMed] [Google Scholar]
- 23.Riedmiller M, Braun H. Proc. Int. Conf. Neural Networks. 1993:586–591. [Google Scholar]
- 24.Noeske T, Jirgensons A, Starchenkovs I, Renner S, Jaunzeme I, Trifanova D, Hechenberger M, Bauer T, Kauss V, Parsons CG, Schneider G, Weil T. ChemMedChem. 2007;2:1763–1773. doi: 10.1002/cmdc.200700151. [DOI] [PubMed] [Google Scholar]
- 25.Noeske T, Trifanova D, Kauss V, Renner S, Parsons CG, Schneider G, Weil, T. T. Bioorg. Med. Chem. 2009;17:5708–5715. doi: 10.1016/j.bmc.2009.05.072. [DOI] [PubMed] [Google Scholar]
- 26.Mueller R, Rodriguez AL, Dawson ES, Butkiewicz M, Oleskiewicz S, Bleckmann A, Weaver CD, Lindsley CW, Conn PJ, Meiler J. ACS Chem. Neurosci. 2010;1:288–305. doi: 10.1021/cn9000389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zupan J, Gasteiger J. Neural Networks for Chemists. Weinheim: VCH; 1993. [Google Scholar]
- 28.Gasteiger J, Rudolph C, Sadowski J. Tetrahedron Computer Methodology. 1990;3:537–547. [Google Scholar]
- 29.Computerchemie MNG, Schwab CH, Gasteiger J. J. ADRIANA.Code; Algorithms for the Encoding of Molecular Structures, version 2.0, program description. Erlangen, Germany: Molecular Networks GmbH; 2006. [Google Scholar]
- 30.Wang R, Gao Y, Lai L. Drug Discov. Res. 2000;19:47–66. [Google Scholar]
- 31.Bauknecht H, Zell A, Bayer H, Levi P, Wagener M, Sadowski J, Gasteiger J. J. Chem. Inf. Comput. Sci. 1996;36:1205–1213. doi: 10.1021/ci960346m. [DOI] [PubMed] [Google Scholar]
- 32.Wagener M, Sadowski J, Gasteiger J. Angew. Chem, Int. Ed. Engl. 1995;34:2674–2677. [Google Scholar]
- 33.Dominguez AR, Nandi AK. Med. Phys. 2007;34:4256–4269. doi: 10.1118/1.2791034. [DOI] [PubMed] [Google Scholar]
- 34.See: www.vanderbilt.edu/vicb/
- 35.For complete details, see Experimental Section
- 36.Ji Y, Trenkle WC, Vowels JV. Org. Lett. 2008;8:1161–1163. doi: 10.1021/ol053164z. [DOI] [PubMed] [Google Scholar]
- 37.Felts AS, Lindsley SR, Lamb JP, Rodriguez AL, Menon UN, Jadhav S, Jones CK, Conn PJ, Lindsley CW, Emmitte KA. Bioorg. Med. Chem. Lett. 2010;20:4390–4394. doi: 10.1016/j.bmcl.2010.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma S, Rodriguez AL, Conn PJ, Lindsley CW. Bioorg. Med. Chem. Lett. 2008;18:4098–4101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. Mol. Pharmacol. 2005;68:1793–1802. doi: 10.1124/mol.105.016139. [DOI] [PubMed] [Google Scholar]
- 40.Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. Biochemistry. 2011;50:2403–2410. doi: 10.1021/bi200129s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.The test compounds (1 µM) were incubated for 15 min at 37 °C with shaking, in medium containing human/mouse liver microsomes, phosphate buffer, and the cofactor NADPH.
- 42.Cosford NDP, Roppe J, Tehrani L, Schweiger EJ, Seiders TJ, Chaudary A, Rao S, Varney MA. Bioorg. Med. Chem. Lett. 2003;13:351–354. doi: 10.1016/s0960-894x(02)00997-6. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Y, Rodriguez AL, Conn PJ. J. Pharmacol. Exp. Ther. 2005;315:1212–1219. doi: 10.1124/jpet.105.090308. [DOI] [PubMed] [Google Scholar]
- 44.Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, Xiang Z, Daniels JS, Lindsley CW, Weaver CD, Conn PJ. Mol. Pharm. doi: 10.1124/mol.111.075184. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.See, https://pharmacology.ricerca.com/Catalog
- 46.Compound 18k was dosed at 10 mg/kg intraperitoneally as a microsuspension in 10% tween 80 in male CD-1 mice using two mice per time point. Brain and plasma samples were collected at 0.5, 1, 3, and 6 hours post dose. IP dosing was chosen as a convenient route to help maximize exposure.
- 47.A 96-well rapid equilibrium dialysis (RED) apparatus (Thermo Scientific) was used to determine the free fraction of 25 in mouse plasma and brain homogenates.
- 48.Deacon RMJ. Nature Protocols. 2006;1:122–124. doi: 10.1038/nprot.2006.20. [DOI] [PubMed] [Google Scholar]
- 49.Njung’e K, Handley SL. Brit. J. Pharmacol. 1991;104:105–112. doi: 10.1111/j.1476-5381.1991.tb12392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Broekkamp CL, Rijk HW, Joly-Gelouin D, Lloyd KL. Eur. J. Pharmacol. 1986;126:223–229. doi: 10.1016/0014-2999(86)90051-8. [DOI] [PubMed] [Google Scholar]
- 51.Spooren WP, Vassout A, Neijt HC, Kuhn R, Gasparini F, Roux S, Porsolt RD, Gentsch C. J. Pharmacol. Exp. Ther. 2000;295:1267–1275. [PubMed] [Google Scholar]
- 52.Nicolas LB, Kolb Y, Prinssen EPM. Eur. J. Pharmacol. 2006;547:106–115. doi: 10.1016/j.ejphar.2006.07.015. [DOI] [PubMed] [Google Scholar]
- 53.Olsen CM, Winder DG. Neuropsychopharmacology. 2009;34:1685–1694. doi: 10.1038/npp.2008.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olsen CM, Winder, D.G. DG. J. Visualized Exp. 2010;45:e2292. doi: 10.3791/2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ettenberg A, Pettit HO, Bloom FE, Koob GF. Psychopharmacology. 1982;78:204–209. doi: 10.1007/BF00428151. [DOI] [PubMed] [Google Scholar]
- 56.Olsen CM, Childs DS, Stanwood GD, Winder DG. PLoS ONE. 2010;5:e15085. doi: 10.1371/journal.pone.0015085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chiamulera C, Epping-Jordan MP, Zocchi A, Marcon C, Cottiny C, Tacconi S, Corsi M, Orzi F, Conquet F. Nat. Neurosci. 2001;9:873–874. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- 58.Lindsley CW, Bates BS, Menon UN, Jadhav S, Kane AS, Jones CK, Rodriguez AL, Conn PJ, Olsen CM, Winder DG. ACS Chem.Neurosci. 2011;2:471–482. doi: 10.1021/cn100099n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Niswender CM, Myers-Johnson KA, Weaver CD, Jones CK, Luo Q, Rodriguez AL, Marlo JE, de Paulis T, Thompson A, Days E, Nalywajko NT, Austin C, Williams M, Ayala JE, Williams R, Lindsley CW, Conn PJ. Mol. Pharm. 2008;74:1345–1358. doi: 10.1124/mol.108.049551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.