

Abstract
Urotensin-II (U-II) has been shown to be the most potent mammalian vasoconstrictor known.[1, 2] Thus a U-II antagonist might be of therapeutic value in a number of cardiovascular disorders.[3] However, interspecies variability of several nonpeptidic ligands complicates the interpretation of in vivo studies of such antagonists in pre-clinical animal models of disease. Thus compound ACT058362 is a selective antagonist for human U-II receptor (hUT2R) with a reported Kd ~ 4 nM in a molecular binding assay, but it is reported to bind weakly to rat UT2R (rUT2R), with Kd ~ 1,500 nM.[4] In contrast, the arylsulphonamide SB706375 is a selective antagonist against both hUT2R (Kd: ~ 9 nM) and rUT2R (Kd: ~ 21 nM).[3] To understand the species selectivity of the UT2R, we investigated the binding site of ACT058362 and SB706375 complex with both hUT2R and rUT2R to explain the dramatic (~ 400-fold) lower affinity of ACT058362 for rUT2R and the similar (~10 nM) affinity of SB706375 for both UT2R. These studies.used MembStruk and MSCDock to predict the UT2R structure and the binding site for ACT058362 and SB706375. Based on binding energy, we found two binding modes each with D1303.32 as the crucial anchoring point. We predict that ACT058362 (an aryl-amine-aryl or ANA ligand) binds in the TM 3456 region while we predict that SB706375 (an aryl-aryl-amine or AAN ligand) binds in the TM 1237 region. These predicted sites explain the known differences in binding the ANA ligand to rat and human while explaining the similar binding of the AAN compound to rat and human. Moreover the predictions explain currently available SAR data. To further validate the predicted binding site of these ligands to hUT2R and rUT2R, we propose several mutations that would help define the structural origins of differential responses of UT2R among species potentially indicating novel UT2R antagonists with cross-species high affinity.
Keywords: docking, G protein-coupled receptors, Urotensin II, dynamics. MSCDock, MembStruk, MembScream
Introduction
Urotensin-II (U-II) is a C-terminal conserved disulfide bridged cyclic peptide (ETPDCFWKYCA). The human isoform of U-II, a undecapeptide (AGTADCFWKYCV), is the most potent mammalian vasoconstrictor known, 10 to 100 times more potent than Endothelin-1.[1, 2] Thus, an antagonist to human U-II receptor (hUT2R) could be of therapeutic value for cardiovascular disorders characterized by increased vasoconstriction, myocardial dysfunction, and even atherosclerosis.[3] The identification of U-II as the endogenous ligand for the GPR14[1] orphan G protein-coupled receptor (GPCR), mainly expressed in the cardiovascular system, has stimulated interest in developing novel peptidic and nonpeptidic UT2R agonists and antagonists.[3, 5, 6]
All non-peptidic reported UT2R antagonists contain a basic amino group and at least two aromatic moieties in one of two arrangements: Ar1-N-Ar2 (denoted as ANA) and Ar1-Ar2-N (denoted as AAN).[7] We will consider here prototypes for both types of UT2R antagonists (Figure 1):
Figure 1.
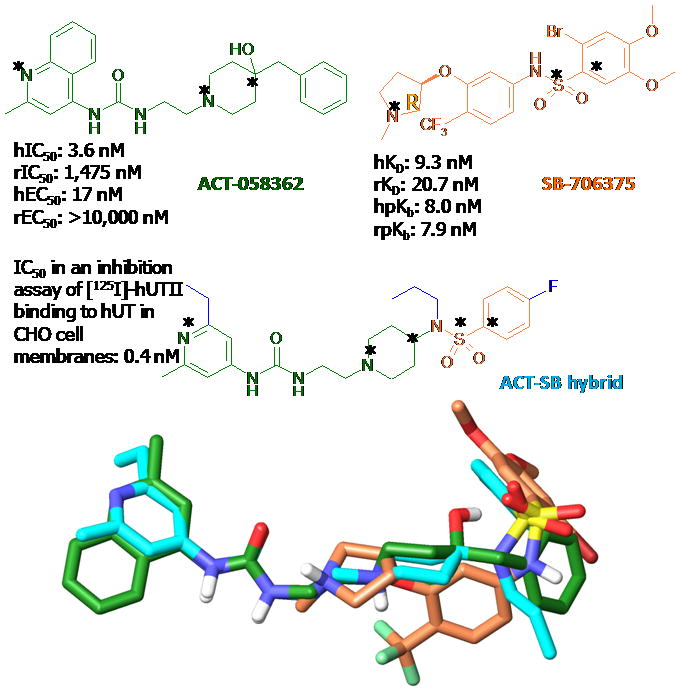
(Top) The chemical structures of ACT058362 in green and SB-706375 in orange with its hybrid compound in cyan. (Bottom) the superimposition of the lowest E conformers of these compounds through atom-by-atom fitting among three atoms marked with asterisks. The root mean square deviation (RMSD) are 0.63 (for N+, Nar, Cal of ACT-058362 to N+, Nar, Cal of ACT-SB hybrid) and 1.12 Å (for N+, S, Car of SB-706375 to N+, S, Car of ACT-SB hybrid).
ANA: Palosuran, ACT058362, 4-ureido-quinoline derivative, 1-[2-(4-benzyl-4-hydroxy-piperidin-1-yl)-ethyl]-3-(2-methyl-quinolin-4-yl)-urea sulfate salt), is a selective antagonist for hUT2R receptor (Kd ~ 4 nM from a molecular binding assay). However, it binds ~400 times more weakly to rat UT2R (rUT2R), with Kd ~ 1,500 nM.[4]
AAN: SB706375 is a arylsulphonamide (2-bromo-4,5-dimethoxy-N-[3-(R)-1-methyl-pyrrolidin-3-yloxy]-4-trifluro-methyl-phenyl)-benzenesulphonamide HCl) with high affinity to all five (mouse, rat, cat, monkey and human) of the mammalian UT2R tested (with Kd: ~ 9 nM to hUT2R and Kd: ~ 21 nM to rUT2R).[8]
This interspecies variability in which the ANA nonpeptidic ligands are very selective between human and rat while AAN ligands are not makes this system will be an excellent one for testing the validity of our structural models and for understanding species selectivity which may be crucial for developing useful preclinical drugs. Understanding the origin of these differences is useful for preclinical studies in delineating the (patho)physiological actions of U-II in animals and human. It may also hint at variations between individuals with differing SNP (single nucleotide polymorphism) patterns.
A major impediment to understanding such differences in the activity of ligands to GPCRs has been the lack of a 3D structure to use as a framework for comparing the binding of various ligands to the receptors of various species. As a result, few 3D experimental structures for a GPCR are available: bovine rhodopsin [bRho, PDB ID (resolution): 1f88 (2.8 Å) in 2000, 1hzx (2.8 Å) in 2001, 1l9h (2.6 Å) in 2002, 1gzm (2.65 Å) in 2003, and 1u19 (2.2 Å) in 2004], β2 adrenergic receptors [AR, PDB ID (resolution): 2r4s (3.4 Å) and 2rh1 (2.4 Å) in 2007], turkey β 1 adrenergic receptor [PDB ID (resolution): 2vt4 (2.7Å) in 2008], retinal-free native opsin [PDB ID (resolution): 3CAP (2.90Å) in 2008], active opsin bound with peptide derived from Gα subunit of transducin [PDB ID (resolution): 3dqb (3.2Å) in 2008], and human Adenosine A2A receptor [PDB ID (resolution): 3eml (2.6Å) in 2008]. Moreover, structural identity among different families of GPCR super family is generally low, 13% to 16%, which is too small for obtaining homology based structures sufficiently accurate for ligand binding predictions. Consequently, we developed the MembStruk method for predicting the 3D structures of GPCRs from sequence information alone without the use of homology,[9] with significant improvements in 2002 and 2004.[10, 11] These methods have been validated by a series of applications to human D2 dopamine receptor (DR),[12] human β2 AR,[13, 14] human M1 muscarinic receptor (M1MR),[15] human Chemokine (C-C) motif receptor 1 (CCR1),[16] the orphan GPCR mouse MrgC11 (Mas Related Genes) for the molluscan peptide FMRF-amide (FMRFa),[17, 18] human prostanoid DP receptor,[19] and human Serotonin 2C receptor.[20] To validate these 3D GPCR structures, we developed the HierDock methodology to predict ligand binding sites to the predicted GPCR structures and compared these to the results of mutation and binding experiments. The results were in excellent agreement with experiment, which for CCR1, MrgC11, and DP were carried out after the predictions. We consider that these studies validate that the 3D structures from MembStruk are sufficiently accurate for use in predicting ligand binding sites and that the predicted binding sites from HierDock are sufficiently accurate for interpreting subtype and species selectivity and for development of ligands with improved binding. More recently, we made what we consider to be dramatic improvements in MembStruk (the MembScream method) and in HierDock (the MSCDock method) which we use in this study.
We intend to report separately on the binding of agonists to hUT2R. In this paper our focus is on the binding of both ANA and AAN antagonists to both hUT2R and rat UT2R. We find that these structures provide an understanding of why the AAN antagonists bind equally well to hUT2R and rUT2R, whereas the ANA antagonist strongly prefers hUT2R to rUT2R.
The Methods section summarizes the MembStruk and MembScream methods to predict the 3D structure of UT2R and the MSCDock method to predict the binding sites. The Results section reports the details of the 3D structure of human and rat UT2R, with a focus on the differences between two structures and examines the binding of the AAN antagonist and the ANA antagonist to both structures, where we find a clear explanation for the differences.
Results and Discussion
1. GPCR structure and comparison of hUT2R and rUT2R
1.1 Alignments
The multiple alignments of a variety of receptors for the 23 GPCR sequences with 20 to 90% identity are shown in Figure A-3 in Supporting Information. The hydrophobicity plot from TMPred2nd is shown in Figure 2 which clearly displays the expected 7 hydrophobic TM domains of UT2R and their hydrophobic centers.
Figure 2.
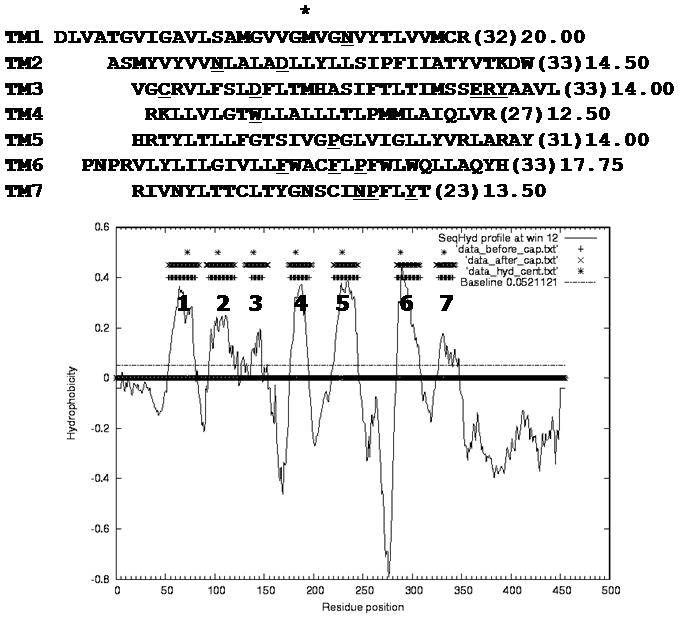
(Top) The predicted seven transmembrane (TM) regions and (Bottom) the hydropathy prediction from TMPred2nd for rat Urotensin II receptor. Hydrophobic centers marked with asterisks were calculated by the peak method. Highly conserved residues in each TM are underlined.
The pairwise alignment of rat and human UT2R in Figure 3 shows 74% sequence identity over the full protein and 89% sequence identity in the TM regions. In bold face, we marked the residues critical to binding of ACT058362 to hUT2R within 5 Å of the binding site. We see that rUT2R has mutations in several amino acids predicted to be important to the binding of ACT058362 to hUT2R (e.g., I1082.61, M1844.60, I1884.64).
Figure 3.

Pairwise alignment of rat and human Urotensin II receptor (GPCR14). Each transmembrane (TM) helix predicted by TMPredict program is shown with grey shading. Highly conserved residues in Family A receptors are displayed in boxes with Ballesteros numbers. The residues in bold face are important amino acids for UT-II binding. Length= 389 amino acids, Score= 489 bits (1125), Expect=1e-137, Method: Composition-based stats. Identities= 266/328 (74% in whole sequence, 89% in TM helix). Positives= 290/328 (88%), Gaps= 2/328 (0%).
GPCRs are partitioned into several families based on their sequences, including family A (the Rhodopsin-like family) to which UT2R belongs. Among all members of family A GPCRs there are characteristic conserved sequences. In the Ballesteros-Weinstein numbering, the most conserved residue in each of the 7 TM domains is taken as the reference and numbered as 50. This residue is designated x.50 where x is the number of the TM helix. All other residues on that helix are numbered relative to this conserved position. The conserved residues in family A GPCRs include: N1.50, N2.45, D2.50, C3.25, D/ERY in TM3, W4.50, C in the second extracellular loop (EL2), P5.50, FxxxW/FxP (P6.50) motif in TM6, and NP (P7.50)xxYx(5,6)F region in TM7, as shown in Figure 3. These highly conserved residues are expected to be important for the packing of the TM domains into a 7-helix bundle (through an H-bonding networks among 7 helices) and/or for binding or activation.[21] In particular, a disulfide bond between a cysteine at the top of TM3 with one in EL2 is conserved across the family A GPCRs.
1.2 MembStruk predictions
After the RotScan step of MembStruk (5° increments from 0 to 360° angle), we selected the best angles (η) based on a combination of the maximum number of total H-bonds, salt-bridges, and the total energy, leading to the angles for each TM indicated with the red circle in Figure A-4 in Supporting Information.
TM1: 30, −175°
TM2: −165, −5°
TM3: 50, −40, −10°
TM4: 60, −20°
TM5: −20°
TM6: 145, −85, −20°
TM7: 10°
This leads to 72 (2×2×3×2×1×3×1) different conformations, one of which is to be selected as the best packing structure. For each of these, we assigned the side chains using SCREAM and selected the one with the maximum number of H-bonds among the middle 15 residues. This led to a final best packing with helix angles of 30, −5, −10, −20, −20, −85, 10° for TMs 1 to 7.
The MembStruk procedure used SCRWL, which we found to give unreliable interhelical interactions. Consequently we used very small rotations (5°) and examined separately the number of hydrogen bonds in the central regions, the number of salt bridges, and the total energies in order to select the best angles. MembStruk led successfully to accurate structures but required a great deal of examination of the structures to judge which criterion was best for each case.
1.3 MembScream refinement
With SCREAM we found that it is sufficient to consider just 30° increments in the angles and that we could base the selection of the best angles on the total energies. Here we started with the optimum rotations from MembStruk, which we denote now as 0°.
MembScream also includes a measure of the energy penalty resulting from polar residues facing the hydrophobic central region of the membrane. This hydrophobic penalty is plotted radically in kcal/mol in Figure A-5 in Supporting Information. For example, for TM1, we consider η=0 to 90°, shown by the blue line, as the reasonable range of rotation angles for the bundle in the lipid bilayer environments. The range of angles allowed for the other TMs in hUT2R are:
0 ° for TMs 1, 2, and 7,
0, and −120° for TM3,
0 and 150° for TM4,
0 and 120° for TM5,
0, 30, −90, −30° for TM6,
as shown in Figure A-5 in Supporting Information. We found the case with all current 0° angles to be the lowest E. Within 30 kcal/mol from the lowest one, there are several minimums in TMs 3, 4, 5, and 6.
In rUT2R, the range of angles allowed for the other TMs are:
−90 to 0 ° for TM2,
0, 120, and −150° for TM3,
0 to 60° for TM4,
0 to 60° and −180 to −90° for TM5,
0 to −150° for TM6,
0 to 180° for TM7,
as shown in Figure A-5 in Supporting Information. Except for TMs 1 and 6, we found 0° angles to have the lowest penalty E. The appropriate rotational ranges of the hydrophobic scale within a 2 kcal/mol E difference were selected for further interhelical E scans.
The final energies from MembScream are shown in Figure 4. Thus we found that the energetically preferred angles of the rUT2R are 0° for TMs 2, 7, 1, and 3 (just as in MembStruk): 30° for TM4, 60° for TM5 and −30° for TM6. We then did a second round of rotations starting with η = 30° for TM4 and η = 60° for TM5. This led to an optimum with all other rotations at 0° including TM6. The interhelical energy of the new structure with 30° for TM4 and 60° for TM5 was re-examined twice for conformation. Finally, in the third round, all current 0 angles for all TM helixes were detected as an energetically favorable helix orientation by E-polar.
Figure 4.
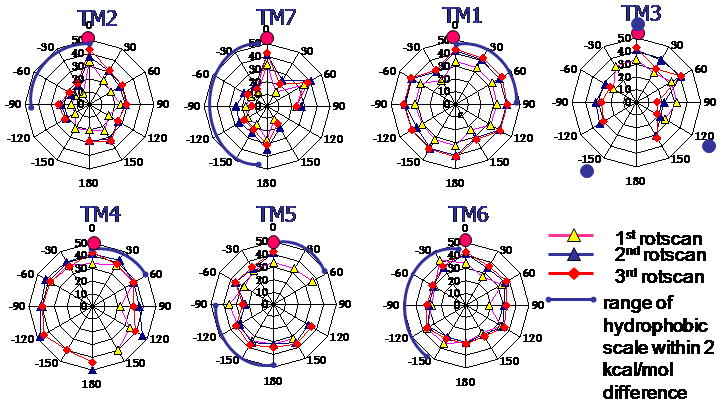
Interhelical interaction energies of MembScream. E-polar energy of each transmembrane (TM) was calculated and plotted radically outward in kcal/mol. In the plot of Scream E, 0 is the lowest Scream E, while the others are the relative E compared with the lowest one. Energetically preferred angles of the rUT2R were 30° of TM4, 60° of TM5, −30° for TM6 and 0° for other TMs 2, 7, 1, and 3 at the first round. Optimizing the rotations starting with TM4 at 30 ° and TM5 at 60° led to 0° preference for TM6 and other four TMs. The interhelical energy of the new structure with 30° for TM4 and 60° for TM5 was reexamined twice for conformation. Resetting TM4=30° and TM5=60° as 0, the third round let to 0 angles for all TM helixes (in pink circles) as the energetically favorable helix orientation by E-polar.
The final best structure from MembScream (0, 0, 0, 30, 60, 0, 0 ° angles for TMs 1–7) revealed improved H-bonding networks of TMs 2-3-4 (N2.45-T3.42-T4.49-W4.50), as shown in Figure A-6 in Supporting Information. Thus, the η=30 ° for TM4 facilitates the interaction between N2.45 and W4.50. In addition, important residues in TMs 4 and 5 known to be involved in ligand binding at rUTR[22] became directed more toward the binding site.
To compare the rUT2R and hUT2R structures, we recalculated the η angles with respect to the most conserved residue in each helix (N1.50, D2.50, D3.32, W4.50, S5.56, Q6.55, and N7.49) for both structures. Here η=0 is taken to point to the average value for the central residue in the central plane.
Comparing the η angles of N1.50, D2.50, D3.32, W4.50, S5.56, Q6.55, and N7.49 for rat and human UT2R structures we found all η angles to be within 10°. TMs 1 (N1.50), 5 (S5.46), and 6 (Q6.55) showed less than 2° difference. TMs 4 (W4.50) and 7 (N7.49) displayed 10.4 and 10.8 ° difference, respectively.
2. Ligand binding studies
The following predictions made use of the standard charge model for residues and ligands denoted Charge Residue Model (CRM) in which their charges are appropriate for pH 7.4 in aqueous solution (i.e. −1 for Asp and Glu and +1 for Lys and Arg). However, we found that the predicted binding energies were more consistent with each other and with experiment with the Neutral Residue Model (NRM) in which the proton is left on the Asp and Glu and removed from the Lys and Arg to yield neutral residues and for which corresponding changes are made in the ligands.
2.1 Conformations of the ANA (ACT058362) and AAN (SB706375) compounds
Using Jaguar we predict the pKa of ACT058362 to be 7.9 while the pka of SB706375 is predicted to be 5.6–7.3, depending on the torsional angle of C-S(O)-N(H)-C, as shown in Figure A-11 and 12 in Supporting Information. These pKa calculations show that the nitrogen in the quinoline ring of ACT is basic and will exist in a protonated state in neutral water, while the hydrogen of sulphonamide in SB is acidic indicating that SB will be a zwitterion in water.
In order to determine the most important conformations of SB706375, we rotated about the C-SO-NH-C bond and calculated the energy with B3LYP/LACVP*. Figure A-13 of Supporting Information showed that the two important angles are ± 60 and 180°. Consequently, we docked to all three of these to the protein.
To find the lowest energy conformation of ACT058362, we rotated five angles (Cal-N+-C-C, N+-C-C-NH, C-C-NH-CO, Cal-Cal-C-Car, and Cal-C-Car-Car) randomly and selected the lowest energy structure for docking study.
Recently, a hybrid compound of ACT058362 and SB706375 has been developed by the same Swiss group. The ACT-SB hybrid 4-ureido-quinoline derivative displayed a high binding affinity at hUT2R of 0.4 nM. The lowest E conformers from the conformational search of three compounds were generated and superimposed through atom-by-atom fitting among heteroatoms, as shown in Figure 1. The RMSD of ACT058362 and SB706375 with their hybrid compound were 0.63 and 1.12 Å, respectively. Thus, the binding site of ACT058362 and SB706375 is expected to overlap at the common basic amino group. However, the binding site of the ureido in ACT058362 and the sulphonamide group in SB706375 should bind separately with different binding modes.
2.2 Docking study of non-selective AAN type SB706375 at rUT2R
SB706375 has the AAN type diarylsulphonamide scaffold. Here we docked both conformations to both binding modes 3456 and 1237.
-
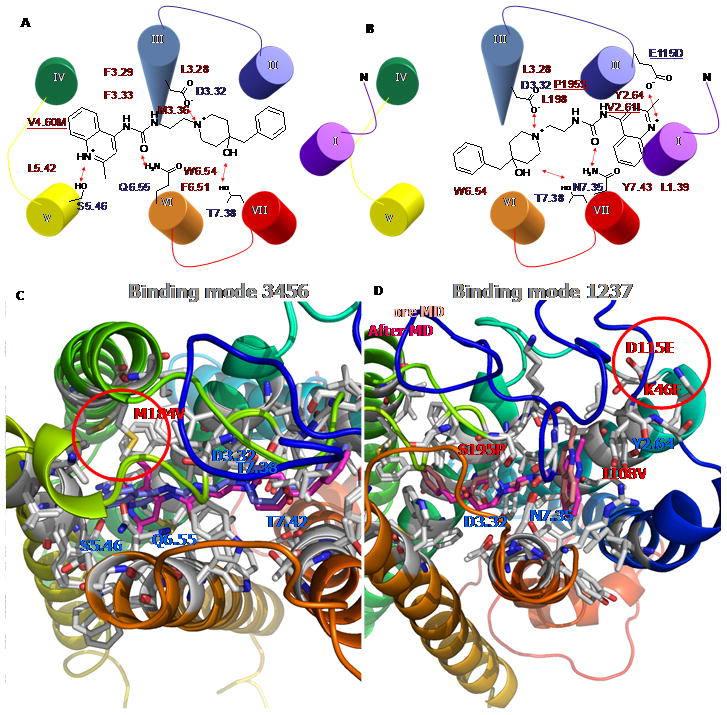
Binding mode 3456
For the binding mode 3456, Figure 5A/5C shows a major interaction of the basic amine group to D1303.32 plus important H-bonds of the sulphonamide oxygen atoms to S2195.46 and Q2796.55. The rat specific residue M1844.60 displays favorable nonbonding interactions with the phenyl ring of the ligand.
-
Binding mode 1237
For the binding mode 1237 of SB706375, Figure 5B/5D shows a strong bond at the anchor point at D1303.32 plus additional hydrogen bonds of the sulphonamide oxygen atoms to T3037.39 and Y3077.43 in TM7. The rat specific residue I1082.61 also displays energetically favorable nonbonding interactions with the phenyl ring.
Figure 5.
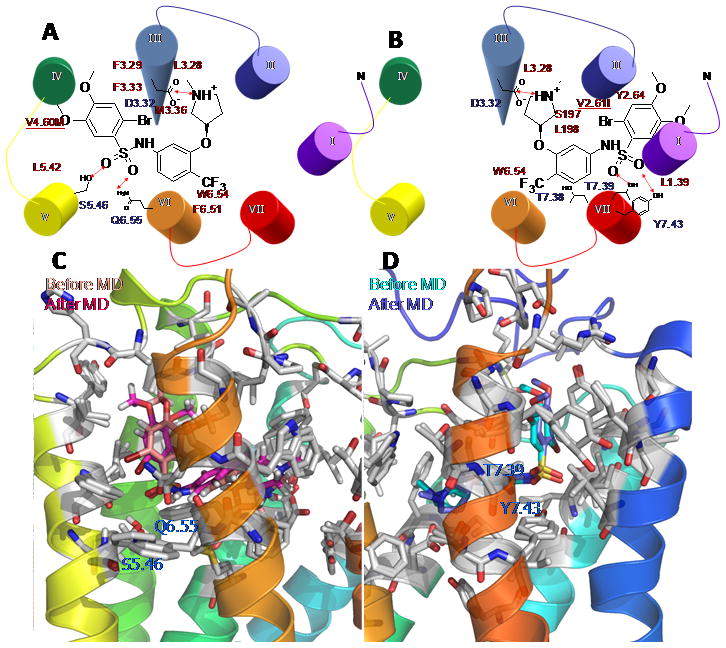
The two energetically favorable binding modes of SB706375 complex at rUT2R. A) the binding site of binding mode 3456 (TMs 4-5-6), B) the binding site of binding mode 1237 (TMs 1-2-7) Light color is the binding conformation before MD and dark color is the final configuration after MD for binding mode 3456 in C and binding mode 1237 in D. Hydrophilic residues in blue and hydrophobic residues in red interacting with SB706375 compound are labeled.
Both binding modes lead to strong binding for SB706375 to both human and rat UT2R, This is consistent with the similar binding observed experimentally. The only residues in the binding site that are different between rat and human are rM1844.60 vs hV1844.60 for binding mode 3456 and rI1082.61 vs hV1082.61 for binding mode 1237. In each case, the cavity analysis (Table 1) leads to similar energetics dominated by van der Waals (vdW) interactions. However, the binding site predicted for binding mode 1237 correlates better with the phylogenetically related opioid receptors, where the OH group of Y3077.43 contributes significantly to the binding of most ligands tested.[23] The following section 2.5 shows that binding mode1237 correlates better with available SAR data. The binding complex for the binding mode 1237 appears much more stable through the 10 ps quench annealing process, while the binding complex for the binding mode 3456 lead to significant fluctuation, as shown in Figure 5C.
Table 1.
The cavity analysis for SB706375/rat Urotensin II receptor complex in binding mode 3456 and 1237.
| Binding | mode | 3456 | Binding | Mode | 1237 |
|---|---|---|---|---|---|
| Res | # | NonBondE | Res | # | NonBondE |
| ASP | 130 | −37.79 | ASP | 130 | −27.11 |
| GLN | 279 | −8.81 | LEU | 198 | −7.21 |
| PHE | 275 | −4.77 | TYR | 307 | −3.52 |
| MET | 184 | −4.71 | TRP | 278 | −3.37 |
| LEU | 215 | −3.45 | THR | 303 | −3.14 |
| PHE | 131 | −3.00 | TYR | 111 | −2.95 |
| PHE | 127 | −2.64 | SER | 197 | −2.91 |
| TRP | 278 | −2.54 | LEU | 58 | −2.87 |
| LEU | 126 | −1.96 | THR | 302 | −2.78 |
| MET | 134 | −1.94 | LEU | 126 | −2.72 |
| TRP | 276 | −1.81 | ASN | 299 | −2.46 |
| LEU | 212 | −1.35 | THR | 306 | −2.43 |
| LEU | 200 | −1.30 | ILE | 108 | −2.41 |
| SER | 128 | −1.18 | PHE | 127 | −2.33 |
| ALA | 207 | −0.95 | TYR | 300 | −1.67 |
| SER | 219 | −0.94 | CYS | 123 | −1.53 |
| PHE | 216 | −0.92 | PHE | 275 | −1.18 |
| PRO | 274 | −0.49 | ILE | 104 | −1.18 |
| ILE | 188 | −0.49 | ILE | 107 | −0.99 |
| THR | 306 | −0.48 | SER | 195 | −0.95 |
| THR | 218 | −0.48 | ILE | 54 | −0.84 |
| ALA | 187 | −0.45 | GLY | 26 | −0.70 |
| HIS | 208 | −0.36 | CYS | 199 | −0.20 |
| TYR | 211 | −0.32 | PRO | 201 | 0.15 |
| LEU | 132 | 0.07 | TYR | 100 | 0.27 |
Rat specific residues in shading were shown in the cavity analysis, [b] The unit of energy is kcal/mol.
2.3 Binding of several SB706375 analogues into both binding modes
We also examined the binding of several SB706375 analogues shown in Figure 6 and Table 2 to validate the binding mode. The initial structures were obtained for both binding modes by matching to the predicted structure of SB706375. We then annealed the binding complex for all three SB706375 analogues. Here MD studies for all three cases led to stronger binding energy for binding mode 1237 (increased interactions) but weakened binding energy (decreased interactions) for binding mode 3456, as depicted in Figure A-7 in Supporting Information. This compares to structure-activity relationship (SAR) experiments that find tolerable binding at human and rat UT2R with the additional phenyl ring at the para position of the pyridine and the thiophene ring. The comparison of the experimental binding affinity vs. calculated binding energy, Unified cavity (UnifiedCav) and Partial Delphi (PartialDel), has a good correlation coefficient of r2= 0.76 in UnifiedCav E but not in PartialDel (r2= 0.19) for binding mode 1237 in CRM, while there is no correlation in binding mode 3456. In NRM, both of binding energies, UnifiedCav and PartialDel, revealed improved correlation of r2= 0.81 and r2=0.98, respectively. These results strongly support binding mode 1237 as the correct mode.
Figure 6.
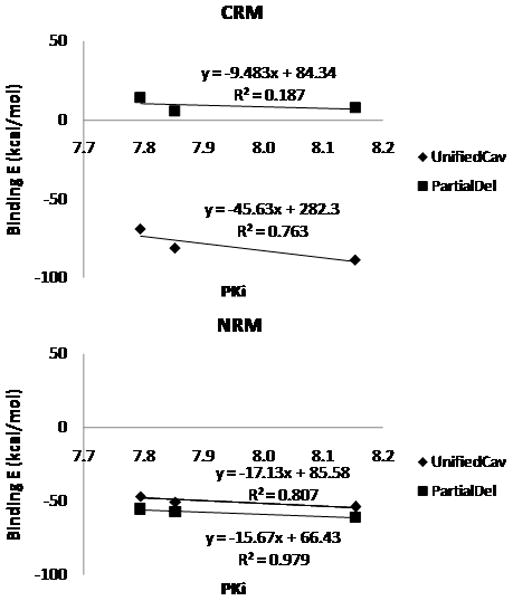
The comparison of binding energies of three SB706375 analogues in Charge Residue Model (CRM) and Neutral Residue Model (NRM). Unified cavity (UnifiedCav) in blue and Partial Delphi (PartialDel) in red are displayed with the linear equation and the r2 value on the chart.
2.4 Computational mutation study of human selective residues at both binding modes
To validate the binding model, we carried out mutation studies for both binding modes. Since two hydrophilic residues, S2195.46 and Q2796.55, were important for ligand binding in binding mode 3456, we mutated each of these sites to the other 19 amino acids and recalculated the binding E as shown in Figure A-8 in Supporting Information. Using charged residues, the results depended on the protonation state of sulphonamide group. In binding mode 3456, using the neutral NH for SB706375 (which has a net plus charge due to the protonated N) led to the most improved binding for the S219D (δE= −21.37 kcal/mol) and for S219E (δE= −22.13 kcal/mol) mutant receptor. Using neutral residues with the zwitterionic form of the ligand in which the sulphonamide N is deprotonated led to the most improved binding of SB706375 for the mutations S219R (δE= −11.24 kcal/mol) and Q279R (δE= −16.02 kcal/mol).
Based on these studies, we selected S219D and Q279R as the two most promising candidates for the CRM. We then carried out quench annealing for the NRM and found increased binding for both mutant receptors for both rat and human, as shown in Figure A-9 in Supporting Information. Here, we find that S210D is 17.4 kcal/mol more favorable compared with wild type (WT), while Q279R is more favorable by 6.4 kcal/mol.
The same procedure was carried out for binding mode 1237 with the results in Figure A-8 in Supporting Information. Depending on the protonation state, the mutations at T3037.39 and Y3077.43 gave opposite results. If SB706375 had a neutral NH, T303D (δE=−23.02 kcal/mol) and Y307E (δE= −16.05 kcal/mol) mutant receptors had favorable interactions. However, if SB706375 was in zwitterionic state, T303R (δE= −5.78 kcal/mol) and Y307K (δE= −6.26 kcal/mol) mutant receptors had more favorable interaction compared with the wild type. Two promising candidates, T303R and Y307E, were selected for neutralization. However, the neutral complex leads to slightly unfavorable interactions at both mutant receptors in Figure A-9 in Supporting Information.
The above calculations assumed that the protein packing was not affected by these mutations. To check this, we calculated the energies for various rotations of TMs 5, 6, and 7 for WT and both mutants with results as in Figure A-10 in Supporting Information. We found that the S219D (TM5) mutant prefers η = 0 and 30° for TM5, just like the wild type. However, the Q279R (TM6) mutant prefers η = 60° for TM6. Thus the Q279R (TM6) mutation is more likely to affect the helix packing than S219D (TM5). For T303R (TM7) and Y307E (TM7), we found that rUT2R preferred the same η = 0 of TM7. The Y307E rUT2R showed similar energy stability for specific angles as WT compared with the T303R mutant receptor.
These predictions of mutant receptors that would increase binding were done to provide a means of validating our predicted structures and hence determining the correct binding modes for SB706375. Based on the computational mutation study of NRM and the MD study, binding mode 1237 was more favorable through the docking study of SB706375.
2.5 Comparison of two binding modes of SB706375 at human UT2R with available SAR data
SB706375 compound has a similar (~10 nM) affinity for both hUT2R and rUT2R, and there are more binding affinity data available for hUT2R. In this section, we report the comparison of two binding modes (binding mode 1237 and binding mode 3456) of SB706375 at hUT2R. After comparing with binding affinity data, we reach the same conclusion as for rUT2R: binding mode 1237 is better than binding mode 3456 to correlate with binding affinity data.
Figure 7 shows the two binding modes we obtained for hUT2R with SB706375 compound, which are similar to mode 3456 and mode 1237 for rUT2R. For mode 3456, SB706375 compound not only interacts with D3.32, but also forms an H-bond between Q6.55 and its sulphonamide oxygen. For mode 1237, SB706375 compound forms a salt bridge with D3.32, and its sulphonamide has H-bonds with N283 and S183. In addition, its CF3 has favorable hydrophilic interactions with Y291 and Y86.
Figure 7.
Two energetically favorable binding modes of SB706375 complex at hUT2R. A) and B) are the binding site of binding mode 3456 (TMs 4-5-6) and 1237 (TMs 1-2-7), respectively.
In order to compare the correlation of the two binding modes with available binding affinities, we evaluated binding energies of mode 3456 and mode 1237 of 8 SB analogues as shown in Figure 8A. Figure 8A also shows the binding affinities of those 8 compounds. Figure 8B shows the correlation of binding energies of two binding modes with binding affinities. Binding energies were evaluated from CRM and NRM approaches. Mode 1237 shows stronger correlation than mode 3456 (R2 = 0.73 for NRM of mode 1237, R2 = 0.56 for CRM of mode 1237, R2 = 0.14 for NRM of mode 3456, R2 = 0.08 of mode 3456). Indeed, Figure 7 shows that mode 1237 has a cavity for bulky substitutions on the left side of SB compound, which is confirmed by binding energies in Figure 8B.
Figure 8.
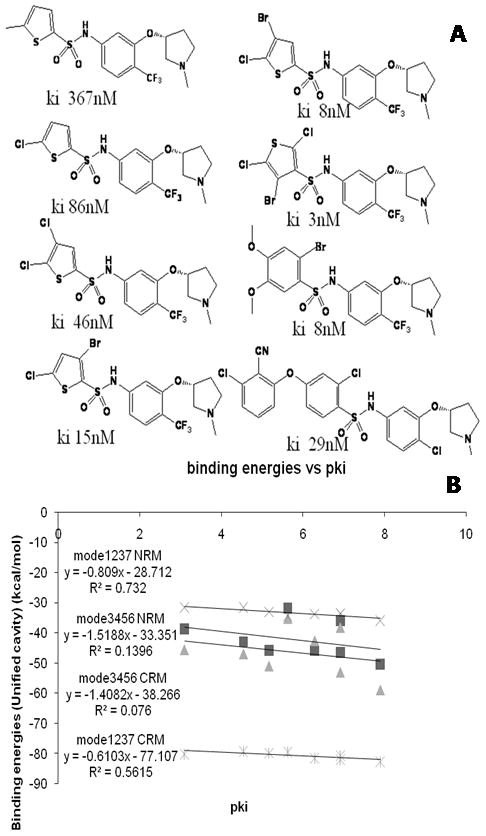
Comparison of the binding energies predicted for SB706375 analogues with hUT2R in the Charge Residue Model (CRM) and the Neutral Residue Model (NRM) based on the unified cavity analysis. A) Eight SB706375 analogues with binding affinities are used for binding energy analysis; B) Binding energy analysis of 8 SB analogues for both mode 1237 and mode 3456 with CRM or NRM. These results show that mode 1237 is better than mode 3456, leading to better correlation between binding affinities and binding energies.
In conclusion, our binding studies of SB analogues on hUT2R show that mode 1237 is better than mode 3456 in correlating with available binding affinities data, which is consistent with our results on rUT2R.
2.6 Docking study of human selective ANA type ACT058362 at the rUT2R
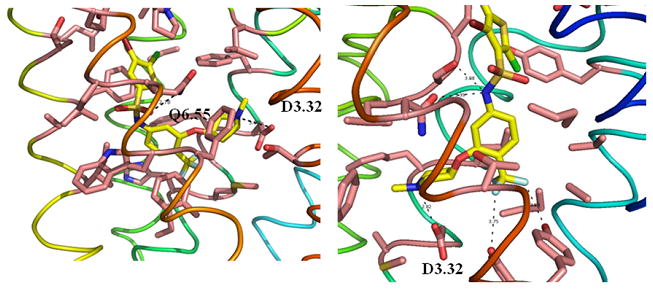
Our docking study of ACT058362 at hUT2R also led to two possible binding modes. Both modes showed the critical anchoring point at highly conserved D1303.32 in biogenic amine receptors with positively charged nitrogen in ligands.
As shown in Figure 9A/9C, binding mode 3456 displays hydrophilic interaction of S2195.46, Q2796.55, and T3027.38 interacting with the quinoline ring, the ureido group, and the hydroxyl group, respectively. However, binding mode 1237 from the hUT2R/ACT058362 complex leads to a salt-bridge interaction at E115 in EL1 with protonated N in the quinoline ring, and additional interactions at N2997.35 and T3037.39, as shown in Figure 9B/9D. Binding studies of phylogenetically related somatostatin receptors by site-directed mutagenesis show that Q6.55 and S7.35 determine the selectivity of the peptide agonist SMS 201-995 for the SSTR2 receptor.[24] Thus, both binding modes (Q6.55 in binding mode 3456 and N7.35 in binding mode 1237) could be explained the similar binding site between two homologous receptors.
Figure 9.
Two energetically favorable binding modes of ACT058362 complex at human UT2R. The region with in the red circle displays a major difference between human and rat. A) the binding site of binding mode 3456 (TMs 3-4-5-6) and B) the binding site of binding mode 1237 (TMs 1-2-3-7). Hydrophilic residues in blue and hydrophobic residues in red surrounded by ACT-058362 compound are shown. C) Final complexes followed by molecular dynamics (MD) exhibited different preferred binding conformation of the quinoline ring at hUT2R in magenta and at rUT2R in blue. D) Light color is the binding conformation before MD and dark color is the binding E after MD.
By matching to the predicted binding modes for hUT2R/ACT058362 complex we obtained the similar binding modes for rUT2R. In binding mode 3456, the amino acid corresponding to V1844.60 in human was Met in rUT2R. The side chain of M1844.60 in rUT2R has an unfavorable interaction with the quinoline ring, not present in hUT2R. The final rUT2R/ACT058362 complex followed by MDs exhibited a different preferred binding conformation of the quinoline ring at hUT2R in magenta and at rUT2R in blue in Figure 9C. Human preferred the horizontal orientation of the quinoline ring with favorable vdW interaction with V1844.60, while rat bound to the perpendicular conformation due to the bad contact at the bulky side chains of M1844.60 with the quinoline ring.
In binding mode 1237, there are four variable residues between human and rat: K46, I1082.61, D115, and S195 in Figure 9D. Interestingly, the EL1 and the N-terminal loops of rUT2R displayed highly constrained loop structure because of the salt-bridge between D115 and K46. In contrast, both of the corresponding amino acids of human are Glu, leading to different loop conformations due to unfavorable electrostatic interactions. The modeling suggested that the structural constraint at rUT2R blocked the direct interaction of D115 with a positively charged nitrogen, while the increased flexibility of E115 at hUT2R forms the optimum interaction with the ligand.
Both binding modes in the rat model were energetically favorable. Binding mode 1237 gave better SolvE, with 9.5 kcal/mol (SolvE-Delphi), while other binding energies supported binding mode 3456 with more favorable total complex E (6.1 kcal/mol), ligand strain E (4.7 kcal/mol), and Cavity E (15.9 kcal/mol). When compared to human model, the Total complex E (Total), Partial salvation E (PartialSolv), Unified Cavity E (UnifiedCavity), and Interaction E (Interaction) were calculated and compared in Table 3. In the charge system, only UnifiedCavity showed the human case to be energetically favorable. However, using neutral scoring E all showed that the human model was energetically favorable, which is consistent with experimental binding affinities.
Table 3.
Calculated binding energies (kcals/mols) of binding mode 3456 for ACT058362 complex
| Charge
| ||||
|---|---|---|---|---|
| Receptor | Total | PartialSolv | UnifiedCavity | Interaction |
|
| ||||
| human | 2465.21 | 257.96 | −110.91 | 220.12 |
| rat | 1495.64 | 145.21 | −108.44 | 107.40 |
|
| ||||
| Neutral | ||||
|
| ||||
| Receptor | Total | PartialSolv | UnifiedCavity | Interaction |
|
| ||||
| human | −2775.48 | −55.91 | −54.13 | −53.60 |
| rat | −1856.93 | −54.16 | −50.17 | −52.01 |
Total: Total E, [b] PartialSolv: Partial solvation E (Delphi method), [c] UnifiedCavity: Unified cavity E, [d] Interaction: Complex E – (Protein E – Ligand E), [e) Energetically favorable E was in bold face.
2.7 Computational mutation study of human selective residues for each binding mode
We found that mutations of such variable amino acids as human, M184V, K46E, I108V, D115E, S195P, led to binding E suggesting that binding mode 1237 would be the more favorable. Using the neutral cavity analysis, Table 4 shows that binding mode 3456 has M184 in rat 1.25 kcal less favorable compared with the corresponding amino acid of V184 in human. The total cavity energy shows that human is 3.96 kcal/mol more favorable. Figure 10A shows that mutation at M184 to V increases the binding for mode 3456 by ~ 4 kcal/mol, while the binding mode 1237 displays a more dramatically increased interaction by ~20 to 50 kcal/mol. Double mutation of K46E and D115E showed a 185 kcal/mol more favorable interaction. Figure 10B shows that the set of mutations, K46E, I108V, D115E, and S195P, were also 193 kcal/mol more favorable. Thus, K46 and D115 are involved in major contributions of species selectivity.
Table 4.
The cavity analysis for ACT058362 at human and rat Urotensin II receptor in binding mode 34567.
| hIC50: 3.6 nM | rIC50: 1,475 nM | ||||
|---|---|---|---|---|---|
|
| |||||
| Res | # | NonBond | Res | # | NonBond |
|
| |||||
| TYR | 100 | −0.56 | TYR | 100 | −0.59 |
| SER | 103 | −0.23 | SER | 103 | −0.18 |
| ILE | 104 | −1.12 | ILE | 104 | −1.24 |
| ILE | 107 | −1.38 | ILE | 107 | −1.40 |
| VAL | 108 | −1.15 | ILE | 108 | −1.60 |
| LEU | 126 | −2.19 | LEU | 126 | −1.93 |
| PHE | 127 | −3.44 | PHE | 127 | −2.91 |
| APP | 130 | −3.06 | APP | 130 | −3.01 |
| PHE | 131 | −3.86 | PHE | 131 | −4.03 |
| MET | 134 | −2.62 | MET | 134 | −2.72 |
| HIS | 135 | −0.32 | HIS | 135 | −0.28 |
| LEU | 180 | −0.26 | LEU | 180 | −0.24 |
| VAL | 184 | −0.18 | MET | 184 | 1.07 |
| MET | 188 | −0.74 | ILE | 188 | −0.26 |
| SER | 197 | −0.85 | SER | 197 | −0.85 |
| LEU | 198 | −3.51 | LEU | 198 | −3.44 |
| LEU | 200 | −0.99 | LEU | 200 | −1.05 |
| PRO | 201 | −0.30 | PRO | 201 | −0.33 |
| LEU | 215 | −2.14 | LEU | 215 | −2.16 |
| PHE | 216 | −1.26 | PHE | 216 | −1.30 |
| THR | 218 | −0.50 | THR | 218 | −0.41 |
| SER | 219 | −0.48 | SER | 219 | −0.52 |
| PHE | 272 | −0.77 | PHE | 272 | −0.74 |
| PHE | 275 | −2.54 | PHE | 275 | −3.08 |
| TRP | 276 | −0.89 | TRP | 276 | −0.71 |
| TRP | 278 | −6.02 | TRP | 278 | −5.90 |
| GLN | 279 | −4.75 | GLN | 279 | −3.09 |
| ASN | 299 | −0.42 | ASN | 299 | −0.43 |
| THR | 302 | −2.28 | THR | 302 | −2.39 |
| THR | 303 | −1.55 | THR | 303 | −1.12 |
| THR | 306 | −1.23 | THR | 306 | −1.20 |
| TYR | 307 | −2.55 | TYR | 307 | −2.14 |
|
| |||||
| SUM | −54.13 | SUM | −50.17 | ||
Rat specific residues in shading are shown in the cavity analysis. The unit of energy is kcal/mol.
Figure 10.
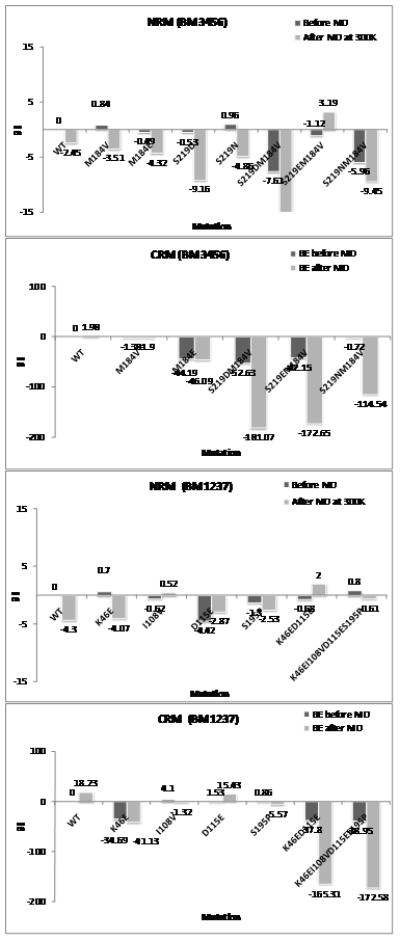
The relative binding energies from Delphi method among mutant receptors of rat Uroteinsin II receptor complex with ACT058362 in binding mode 3456 and binding mode 1237. Relative binding energies before and after 10 ps quench annealing for the Charged Residue Model (CRM) (50 to 600 K) and the Neutral Residue Model (NRM) (50 to 300 K) were calculated and compared with the binding energy of wild type set to 0.
Since the charged model displayed ~20 to 200 kcal energy differences among mutant receptors, we did not trust the energy differences. To reduce the effect of long-range charge interactions, we neutralized all charged residues and ligands through protonation or deprotonation and calculated the binding E of neutral complex for each mutation at species-specific residues before and after the quench annealing procedure. We found that using neutral binding energies leads to variations within 1 to 12 kcal/mol for the various mutations, which we consider more reliable. Similar results were found by Bray and Goddard.[20]
Using the neutral system we found that binding mode 1237 does not increase interactions for any mutant receptors in Figure 10B. For binding mode 3456, only a single mutation of M184V gave a slight increase in ligand interaction. However, other mutations of hydrophilic residues in TM5 displayed the dramatic increases of interaction due to the proximity of a protonated nitrogen in the quinoline ring. The mutation results suggest position dependence since the basic N was directed toward TM 5. Mutation at the 5.46 residue was better than mutation at the 4.60 residue because of an optimum interaction of the salt-bridge as well as the H-bonding between basic nitrogen and hydrophilic residue. A short MD study reported in Figure 10A suggests stabilization by hydrophilic interactions for the M184E, S219D, and S219N mutants. Protonated D forms H-bonding with the N in the quinoline ring. The S219D mutation leads to ~4.3 kcal/mol increased favorability compared to the S219N mutation. We found that double mutation of S219D/M184V and S219N/M184N are more favorable by 13.1 and 7 kcal/mol than WT. However, the S219E/M184V mutant receptor is less favorable by ~5.5 kcal/mol because of bad contacts with the quinoline ring.
Thus, we expect that the following mutations might increase the binding affinity of ACT058362 compound compared to WT (100%) by the order of the following priority. 1) M184V/S219D (127%), 2) S219D (116%), 3) M184V/S219N (111%), 4) S219N (103%). Such mutation experiments could also validate the binding mode of ACT058362. Based on the mutation study of the neutral system and the MD study, we find that ACT058362 binds in binding mode 3456, unlike the SB706375/rUT2R complex.
Thus, our docking studies suggested that the ANA compound ACT058362 prefers to bind at TMs 3-4-5-6 region (binding mode 3456), while the AAN compound SB706375 prefers to bind at TMs 1-2-3-7 region (binding mode 1237). Common to both binding sites is at D1303.32 with the basic amine in the ligand. However, the binding site of ACT058362 has favorable interaction with S2195.46 and Q2796.55 in the TMs 3-4-5-6 region, while the sulphonamide group in SB706375 binds at T3037.39 and Y3077.43 in TMs 1-2-3-7. Thus the computational model explains ACT058362 is selective for human over rat while SB706375 is non-selective between human and rat UT2R. This model provides an understanding of the species selective residues in the binding site, but further experimental studies will be useful in validating these predictions.
3. Full membrane full solvent simulations
As described in Methods, we inserted the apo hUT2R/rUT2R and the ligand-bound forms into a full periodically infinite membrane-water box and carried out 1 ns MD. These results support the stability of the predicted protein structures, with no loss of significant interactions. This suggests that the predicted structures are stable (Details in Supporting Information).
Conclusion
We report the 3D structure for UT2R predicted using the MembStruk method as improved using the MembScream method and we compare the structure of hUT2R and rUT2R. We used MSCDock to predict the binding sites for both cases.
These structures were validated by using them to predict the binding sites and energies for two types of antagonist structures:
The aryl-aryl-amine (AAN) ligand typified by SB706375 which binds similarly to hUT2R and rUT2R.
The aryl -amine-aryl (ANA) ligand typified by ACT058362 which binds strongly to hUT2R but 400 times weaker to rUT2R.
For the AAN case, we found that the preferred binding site is in the TM 1237 pocket with the most important contacts involving D1303.32, T3037.39, and Y3077.43. We found similar binding to hUT2R and rUT2R, and we found good agreement with the experimental SAR data for rUT2R cases. This structure was further validated by MD with full membrane and water solvent. We found no substantial changes in the binding site.
For the ANA case, ACT058362, we found that the preferred binding is the TM 3456 region pocket. Here we found that the critical contacts for hUT2R involve D1303.32, S2195.46, and Q2796.55. In contrast, for rUT2R we find reduced binding because the M1844.60 is mutated from Val in human to Met in rat. This explains the observed difference in the binding. This structure was further validated for rUT2R by MD with full membrane and water solvent. We found no substantial changes in the binding site, but we did find an alternative H-bonding of the quinoline NH and the backbone CO atom of L2155.42 because of water mediation.
There is good agreement between the predicted ligand-protein structures and available SAR and mutation data. There is also the good agreement with cross species comparisons (h to r).
We found that the Neutral Residue Model (NRM) leads to predicted binding energies much more consistent with each other and with experiment than the standard Charged Residue Model (CRM).
Experimental and Methods Section
We used the MembStruk method,[10, 11] which is based on the primary sequence of UT2R plus Monte Carlo, energy minimization, and dynamics simulations using a force field. The 3D structure of known ligands to the binding site was predicted using the HierDock or MSCDock hierarchical docking methods, leading to predicted structures in excellent agreement with experimental mutation and ligand binding experiments. These methods had been used previously for several cases (CCR1, MrgC11, and DP), in which experimental validation was carried out after the predictions, indicating that such predicted structures are accurate enough for use in drug design developments.
The MembStruk methods have been described previously,[10, 11] but many improvements have been, leading to the MembScream method used in these studies. Similarly the HierDock and MSCDock methods have been described previously,[25] but many improvements have also been made. Consequently, we will review the elements of the methods while describing the specifics for our applications and any changes in the methods.
Force fields and structures
Protein
For the structure prediction methods (MembStruk, HierDock, and MSCDock), all energy and force calculations used the DREIDING force field23 (DFF) with CHARMM22 charges for the protein.[26, 27] These calculations used the MPSim molecular dynamics (MD) program.
Ligands
The ligands used in this study are shown in Fig. 1.
The charges for the ligands came from quantum mechanics (QM) using the B3LYP flavor of density function theory with the 6-311** basis set (using Jaguar). The DFF for the sulfonyl group was modified slightly to fit the quantum mechanics (QM).
To determine the state of protonation of the ligands, we used the Jaguar pKa module with B3LYP/LACVP*.
Prediction of the UT2R structure
The first step is to predict the three-dimensional structure of UT2R. We used the MembScream method, which builds on the lessons learned with MembStruk. MembScream involves the following steps as depicted in Figure A-1 in Supporting Information.
Predict transmembrane (TM) domains and hydrophobic center
We did a blast search on the query sequence of hUT2R (Q9UKP6, UR2R_HUMAN, with 376 amino acids) or rUT2R (P49684, UR2R_RAT, with 386 amino acids) to obtain 226 protein sequences for GPCRs, with sequence identities ranging from 13% to 90% (E threshold of 10). Here we used the ExPASy (Expert Protein Analysis System) home page (us.expacy.org) with NCBI BlastP 2.2.15 from UniProtKB Swiss-Prot DB (options; mammalian and no fragment).
We found that multiple sequence analysis using the full 226 sequences led to gaps within the predicted TM domains. We considered that this might be caused by redundancy in gene sequences; hence, we selected just the 120 sequences corresponding to human, rat, and mouse species. We still found some gaps within predicted TM regions and carried out three passes eliminating sequences leading finally to 23 sequences with 20% to 90% identity that led to seven TM domains without gaps. These sequences are listed in Figure A-3 of Supporting Information.
These calculations used the clustalW program (v.1.8.3) for the multiple sequence alignments. The seven TM regions of the UT2R were predicted by hydropathicity analysis [“TM2ndS”][11] with the Eisenberg hydrophobicity scale.[28] Using window sizes (WS) ranging from 12 to 30 the average hydrophobicity was calculated in each TM to obtain the consensus hydrophobicity for every residue position in the alignment.
The hydrophobic center of each TM was determined by the maximum peak in the hydrophobicity[29] after averaging over the stable windows that deviate ≤5 from the value at 20 WS. Then each TM domain was positioned to have all hydrophobic centers in the same xy plane (defining z=0).
For each TM domain, we generated the canonical helix (with extended side chains) and minimized its energy. Using standard CHARMM charges, we subjected each isolated helix to 200 ps Cartesian dynamics at 300K (NVT) and selected the lowest potential energy conformation. We found that NEIMO torsional dynamics[30, 31] produced severe unrealistic kinking and unraveling of TMs 4 and 7, as did Cartesian MD with neutral QEq charges.[32] Thus, we eliminated them as feasible options. Individual helix dynamics of TM 4 and 7 structures was feasible because there was little unraveling and minor kinking caused by the presence of Pro. Thus, the structures of TMs 4 and 7 were merged into the result of the helix dynamics.
Create PDB template
To build the starting structure for the 7-helix bundle, we needed the x and y position for each TM domain (within the z=0 plane) and three orientations θ (the tilt of the helix axis with respect to the z axis), φ (the azimuthal position for the project of the axis on the xy plane), and η (the rotation of the helix about it axis). The rotation angle η is critical for obtaining an accurate 3D structure, and our method focused on optimizing this angle. The initial values for the other four numbers (x, y, θ, φ) were taken from the 7.5 Å electron density map of frog rhodopsin,[33] which was successful in our previous predictions. We considered that x, y, θ, φ can relax under minimization and dynamics, whereas z and η are likely to get trapped into wells.
Rotation by phobic face
To choose an initial value of η, we associated the Eisenberg hydrophobicity with the C-alpha carbon positions for the middle 15 residues around the hydrophobic centers. This was projected along the helix axis onto the xy plane to obtain a net hydrophobic value on this plane. Then we considered just the sector or projected values that are exposed to the membrane alkyl chains and chose the η so that summing over the exposed residues would yield the minimum value (most hydrophobic).
RotMin, RotScan, and CombiRot
After packing the 7 helices into a bundle as described above, the RotMin procedure was used to refine the initial orientation of the TM helices. The procedure was to rotate one of the helices by ±5° increments and reassign the side chains for all 7 TMs using SCWRL (Side Chain With Rotamer Library)[34] in order to minimize all atoms of the bindle. Then the best η was selected. This was followed by ±10°, ±15°, ±20°, ±25° increments. This was done in the order of TMs 3->2->1->7->6->5->4. The final rotation from the original values was given: 15°, −5°, 5°, 55°, −25°, −60°, and 5° angles for 7 TMs sequentially.
This was then followed by the RotScan procedure to find the optimum packing of the helices. Here each specific helix was considered one-by-one and rotated through the full 0 to 360° range in 5° increments. Again, for each case we used SCWRL to re-assign all side chain conformations, followed by a minimization of the atoms for all 7 helices. To choose the optimum η for each helix, we considered separately which value led to the maximum number of total H-bonds, the maximum number of salt-bridges, and the total energy. This led to several good choices for each TM and a combinatorial set of 2×2×3×2×1×3×1= 72 conformations input into the CombiRot step.
Based on the 72 combinatorial combinations, we selected the best cases with middle H-bond analysis (total inter H-bonds: 7) and classical H-bonding networks among TMs 1-2-7 (1.50-2.50-7.49) and TMs 2-3-4 (2.45-3.42-4.50) in family A GPCRs. The final rotation from the original values was given: 30°, −5°, −10°, −20°, −20°, −85°, and 10° angles for 7 TMs sequentially.
RotScream
We found that SCWRL[34] did not lead to accurate interhelix hydrogen bonds and consequently switched to the SCREAM (Side Chain Rotamer Energy Analysis Method).[35] SCREAM uses a library of residue conformations for each amino acid (it allows a range of diversity of 0.2 to 5.0 Å; we used 1.0 Å, leading to 1478 rotamers) in conjunction with a Monte Carlo sampling based on the sum over four energy terms: full valence, hydrogen bond, electrostatic, and van der Waals (vdW). A special aspect is the use of optimized flat bottom vdW potentials that reduce the penalty for contacts that are slightly too short while retaining the normal attractive interactions at full strength. Thus, starting with the best structure from MembStruk, we rotated each of the 7 TM by 30° increments while reassigning the side chain conformation by SCREAM method. The MembScream rotational scan of all TMs was performed for the rUR2R in the sequence of TM 2 ->7 -> 1 -> 3 -> 4 -> 5 -> 6.
In addition to the FF energies, we estimated solvation energy in the membrane as follows. The solvent accessible surface area (SASA) was determined for every residue in the middle of the membrane. Then, we calculated a Hydrophobicity penalty E over all residues having a SASA ≤45% using the following equation (1).
| (1) |
We used this Hydrophobicity penalty to determine the allowed rotational range for each helix. Thus, we allowed only cases in which the hydrophobic penalty was less than 2 kcal/mol. At each allowed angle, the side chains of all residues were optimized using SCREAM.
SCREAM does not change Ala, Gly, Pro, or Cys that are involved in disulfide bonds. In selecting the optimum η, we considered the total energy (denoted as Totfm), the total energy in which the vdW terms are deleted (denoted as Totfm-vdW), and polar energy (Epolar). The reason for doing so is that even with the flat bottom vdW, there can be slightly bad contacts for what should be a good configuration.
For self-consistency, the hydrophobicity penalty and interhelical energy scans were calculated again for the best packing structure.
Generation of loops
The N-terminals, three intracellular loops, three extracellular loops, and C-terminals of the hUT2R were generated using homology modeling based on the structure of bRho (PDB ID: 1f88). The same loops were copied into the rUT2R, and we refined the side chain through SCREAM.
The disulfide bond between C1233.25 and C199EC2 was constructed. Loops and N/C terminals were relaxed through 500 ps MD with backbone constraints in the α–helix, followed by minimization without any constraints.
Prediction of the binding site for antagonists to UT2R
We used the MSCDock methodology[36] to predict the putative binding for the ACT058362 and SB706375 compounds to UT2R. Earlier predictions on binding of ligands to GPCRs had used HierDock.[25] MSCDock is an ensemble approach designed to ensure that a complete set of configurations is sampled for each conformation of the ligand. This leads to a hierarchy of families with a specified range of diversities, allowing the best to be selected at a coarse level by evaluating only energies of family heads. This leads to an ensemble of the best diversity families that are used for higher-level calculations. MSCDock was applied as described previously in Heo et al 2007, Bray et al 2008, and Goddard et al 2010.[18–20] Some of the key steps in the MSCDock procedure are outlined here (see Figure A-2 in Supporting Information)
-
FindBindSite. We first identify 2 or 3 putative binding regions of the protein using FindBindSite. In this procedure we replace the bulky hydrophobic residues (Phe, Trp, Tyr, Val, Ile, and Leu) with Ala prior to the completeness and enrichment steps of ligand docking. After bulky residue alanization, the grids and the spheres were created to find all possible binding regions. For rUT2R, 46, 294, 416 grid points were generated to encompass the whole protein, with box dimensions of 100×86×83 Å. This was subdivided into 39 boxes with box dimensions of 10×10×10 Å i Then we do extremely crude docking (using Dock4)[37] to find 1000 structures within each cube with a bump max of MaxBump = Max (2, 0.1 N) where N is the number of atoms in the ligand. Then, we selected 10 ligand positions and evaluated the energy using the Dreiding FF (with MPSim). These cubes are then combined into distinct binding regions, which are filled with spheres using Sphgen of Dock4 for subsequent high quality docking. This led to two distinct binding regions
Binding mode 3456: in which TMs 3, 4, 5, and 6 were involved in ligand binding. This involved grid box numbers 23, 35, and 46 with 432 spheres. These spheres were reduced to 208 spheres with 0.7 Å radius.
Binding mode 1237: in which TMs 1, 2, 3, and 7 were involved in ligand binding. This involved grid box numbers 13, 17, and 44 with 448 spheres. These spheres were reduced to 208 spheres with 0.7 Å radius.
Completeness and enrichment: To ensure that the complete binding site is sampled (completeness), we aim to generate a complete set of poses to span the binding region and then to select the best by energy. Here, we sampled binding configurations having no more than “MaxBump” bad contacts until we had generated ~2,500 families with a 1.2 Å diversity. Here, MaxBump = Max (2, 0.1 N) where N is the number of atoms in the ligand. We then calculated the Dock score for each of these and kept the best 10% (250). Then we generated additional configurations and kept the ones overlapping any one of these 250 families until we had an average of 6 children in each family. These 1500 structures were then partitioned into families with 0.6 Å diversity, leading to 750 families. For each family we evaluated the Dreiding energy using MPSim and selected the best 150 families (10%) for further consideration. For each of these 150, we evaluated the MPSim energy for each structure and kept the best for each family.
MSC-Dock-Selection of the optimum protein side chains from each of the best 150 ligand poses. Here we use SCREAM[38] to dealanize the protein back to the original residue, finding the optimum side chains for each of the 150 ligand poses. SCREAM (Side Chain Rotamer Energy Analysis Method) SCREAM uses a library of residue conformations for each amino acid (allowing a range of diversity of 0.2 to 5.0 Å; we used 1.0 Å, leading to 1478 rotamers) in conjunction with a Monte Carlo sampling based on the sum over four energy terms: full valence, hydrogen bond, electrostatic, and vdW. A special aspect of SCREAM is the use of optimized flat bottom vdW potentials that reduce the penalty for contacts that are slightly too short while retaining the normal attractive interactions at full strength. Dreiding3 was used to obtain accurate HB and vdw energies. We then picked the best 50 and carried out 50 steps of minimization for the unified binding site and selected a subset (~5). Then for this subset, we minimized the structure for the full ligand-protein complex and picked one or two for subsequent analysis.
This procedure solved the problem of allowing bulky residues and bulky ligands to accommodate each other. This procedure has been used successfully for serotonin 2C,[20] MrgC11,[18] and DP receptors.[19]
Rescoring using neutral residues and ligands
Traditionally, the charges on each Asp and Glu add up to −1 while the charges on each Lys and Arg add up to +1. Although this Charge Residue Model (CRM) seems to be a reasonable description in aqueous media, we have found that it can lead to increased uncertainties in the binding energies. The origin of the problem is that our calculations use the common approach in which the charges are fixed onto atoms with no polarization of the protein, ligand, of solvent molecules. In the real systems, such effects dampen long-range effects. We have redeveloped a Dreiding-like FF in which the hydrogen bond terms are adjusted so that protonated Asp and Glu and deprotonated Lys and Arg are described consistently. Similarly, in this scheme, an amine will be deprotonated so that it is neutral. Then, after calculating binding energies, we correct the final states of the ligands that should be charged in aqueous media based on their pKa. We refer to this as the Neutral Residue Model (NRM).
Solvation
SCDock allows, at each step, for a percentage of the protein+ligand complexes to be eliminated based on energy criteria, including continuum AVGB (Analytical Volume Generalized Born) continuum salvation and Delphi-based solvation method.[39, 40] Several best structures were scored using the following energy criteria.
- Solvation E: the binding energy with solvation without ligand minimization.
- Solvation Emin: the binding energy with solvation with ligand minimization.
Ligand strain E: ligand strain energy in the binding site, ligandcom − ligandmin
Cavity E: the cavity analysis where only residues within the local 5.0 Å binding cavity contribute to the energy.
Validation: To validate the model, we neutralized the system, re-docked, and re-scored using the NRM. This removes the sensitivity to the distances to charged residues (and counter ions) remote from the active site, which sometimes can change binding energy by over 10 kcal/mol. The NRM leads to a much smoother electrostatic potential in the binding region of the GPCRs and leads to much smaller solvation energies, both of which change much less for small changes in ligands.
Validation
To validate the model, we neutralized the system, re-docked, and re-scored using the NRM. This removes the sensitivity to the distances to charged residues (and counter ions) remote from the active site, which sometimes can change binding energy by over 10 kcal/mol. The NRM leads to a much smoother electrostatic potential in the binding region of the GPCRs and leads to much smaller solvation energies, both of which change much less for small changes in ligands.
Full lipid, full solvent MD
After predicting the protein-ligand complex, this structure was inserted into a fully equilibrated hydrated palmitoyl-oleyl-phosphatidylcholine (POPC) lipid bilayer having cell size in the XY plane of 75 Å × 75 Å (172 POPC molecules) and solvated with 6,689 water molecules using Membrane builder in the VMD program. The lipid tails were almost fully extended, allowing for easy insertion of the protein into the membrane, reducing the required equilibration time. The distance between the layers was set at c=72.3 Å to fit the actual membrane thickness plus the solvent thickness. The lateral supercell was set to fit the actual surface density of lipid molecules, a=69.9 and b=68.4 Å. We introduced disorder into the lipid bilayer patches to better resemble a membrane at 300K by allowing random orientation of each lipid about its axis, with a truncated obtained with a short (1ps) equilibration at 300K in vacuum. This eliminated steric contacts between the lipid atoms but left the lipid tails mostly extended. These steps in our procedure reduce the time required for equilibration of the lipid/protein complex. After inserting the protein into the lipid-water cell, lipids overlapping within 1 Å of protein and waters overlapping within 5 Å were removed.
For the particle mesh Ewald (PME) in the electrostatics calculation,[41, 42] the charge of system was balanced through replacing waters into 15, 17, 16, and 30 Cl- ions to the system of the apo protein rUT2R, the ACT058362-bound rUT2R, the SB706375-bound rUT2R, and the ACT058362-bound hUT2R, respectively. These systems contain 36,612 atoms (apoprotein-rUT2R), 36,056 atoms (nonselective antagonist SB706375-rUT2R), 36,224 atoms (human selective antagonist ACT058362-rUT2R, and 36,032 atoms (human selective antagonist ACT058362-hUT2R). This structure was then equilibrated at 300K for 1 ns using the NAMD 2.5 (NAnoscale Molecular Dynamics) program.[43] We used the CHARMM22 force field parameters for the protein, the TIP3 model for water,[27] and the CHARMM27 force field parameters for the lipids.[44] Minimization of both ligands in vacuum for these ligands led to bond lengths, angles, and dihedrals similar to the quantum mechanical results and the previous structures determined with DREIDING.[26] Quantum charges from DFT/6311G** method were used for these ligands.
The process was first to minimize the water, ion, and lipid bilayer while keeping the protein and ligand fixed. This was followed by an all-atom conjugate gradient minimization of the entire system for 1,000 steps. After this minimization, we carried out 500 ps of MD simulations for equilibration with 1-fs time steps, followed by 1,000 steps of minimization of the full system. Langevin dynamics was used for temperature control with the thermostat set at 310 K. The Nosé–Hoover Langevin piston pressure control was used to control fluctuations in the barostat, which was set at a pressure of 1 bar. Here the periodic cell was constrained to remain orthorhombic, but the cell parameters were allowed to vary. A dielectric constant of 1 was used for the electrostatic interactions, which were calculated by using the PME method.[41, 42] The grid in the x, y, and z directions used for the PME method was set at 96, 80, and 108 points, respectively. The van der Waals interactions were described using a Lennard–Jones function multiplied by a cubic spline switching function starting at 8 Å and stopping at 12 Å. The cut off radius for including atoms in the nearest-neighbor list was 13.5 Å. All 1–2 and 1–3 interactions were excluded, and 1–4 interactions were scaled by multiplication with a predefined factor. The bonded interactions were calculated every time step, the nonbonded interactions were calculated every other time step, and the electrostatic interactions were calculated every fourth time step. The nearest-neighbor list was updated every 20 time steps. Every 10 ps a snapshot was written to the trajectory file for subsequent analysis.
The hardware configuration used for these simulations was a Linux-based Beowulf cluster running on RedHat Linux 7 at the Materials Process and Simulation Center at the California Institute of Technology. Each central processing unit in either of the two clusters was an Intel P4 2.2-GHz processor with 1 GB of memory.
Supplementary Material
Table 2.
Calculated binding energies (kcals/mol) of three SB analogues in binding mode 1237
| Charge
| |||||
|---|---|---|---|---|---|
| Compound | Ki (nM) | Pki | UnifiedCav | PartialDel | TotalE |
|
| |||||
| SBphenoxy | 7 | 8.15 | −88.97 | 7.68 | −1255.35 |
| SB706375 | 14 | 7.85 | −81.35 | 5.77 | −1224.86 |
| SBthiophen | 16 | 7.80 | −69.03 | 13.85 | −1264.38 |
|
| |||||
| Neutral | |||||
|
| |||||
| Compound | Ki (nM) | Pki | UnifiedCav | PartialDel | TotalE |
|
| |||||
| SBphenoxy | 7 | 8.15 | −53.88 | −61.28 | −1803.06 |
| SB706375 | 14 | 7.85 | −50.74 | −57.11 | −1765.64 |
| SBthiophen | 16 | 7.80 | −46.55 | −55.33 | −1812.32 |
The unit of energy is kcal/mol.
Acknowledgments
This work was funded partially by Boehringer-Ingelheim and by NIH. In addition, the computational resources used here were provided by grants from ARO-DURIP and ONR-DURIP. We thank MS. Lindsay Riley from Univ. of California, Los Angeles, for helpful discussion.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Ames RS, Sarau HM, Chambers JK, Willette RN, Aiyar RV, Romanic AM, Louden CS, Foley JJ, Sauermelch CF, Coatney RW, Ao Z, Disa J, Holmes SD, Stadel JM, Martin JD, Liu WS, Glover GI, Wilson S, McNutty DE, Ellis CE, Eishourbagy NA, Shabon U, Trill JJ, Hay DVP, Ohlstein EH, Bergsma DJ, Douglas SA. Nature. 1999;401:282–286. doi: 10.1038/45809. [DOI] [PubMed] [Google Scholar]
- 2.Douglas SA. Curr Opin Pharmacol. 2003;3:159–167. doi: 10.1016/s1471-4892(03)00012-2. [DOI] [PubMed] [Google Scholar]
- 3.Carotenuto A, Grieco P, Campiglia P, Novellino E, Rovero P. J Med Chem. 2004;47:1652–1661. doi: 10.1021/jm0309912. [DOI] [PubMed] [Google Scholar]
- 4.Clozel M, Binkert C, Birker-Robaczewska M, Boukhadra C, Ding S-S, Fischli W, Hess P, Mathys B, Morrison K, Müller C, Müller C, Nayler O, Qiu C, Rey M, Scherz MW, Velker J, Weller T, Zi J-F, Ziltenerm P. J Pharmacol Exp Ther. 2004;311:204–212. doi: 10.1124/jpet.104.068320. [DOI] [PubMed] [Google Scholar]
- 5.Flohr S, Kurtz M, Kostenis E, Brkovich A, Fournier A, Klabunde T. J Med Chem. 2002;45:1799–1805. doi: 10.1021/jm0111043. [DOI] [PubMed] [Google Scholar]
- 6.Grieco P, Carotenuto A, Campiglia P, Marinelli L, Lama T, Patacchini R, Santicioli P, Maggi CA, Rovero P, Novellino E. J Med Chem. 2005;48:7290–7297. doi: 10.1021/jm058043j. [DOI] [PubMed] [Google Scholar]
- 7.Carotenuto A, Grieco P, Rovero P, Novellino E. Curr Med Chem. 2006;13:267–275. doi: 10.2174/092986706775476061. [DOI] [PubMed] [Google Scholar]
- 8.Douglas SA, Behm DJ, Aiyar NV, Naselsky D, Disa J, Brooks DP, Ohlstein EH, Gleason JG, Sarau HM, Foley JJ, Buckley PT, Schmidt DB, Wixted WE, Widdowson K, Riley G, Jin J, Gallagher TF, Schmidt SJ, Ridgers L, Chirstmann LT, Keenan RM, Knight SD, Dhanak D. Br J Pharm. 2005;145:620–635. doi: 10.1038/sj.bjp.0706229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Floriano WB, Vaidehi N, Goddard WA, III, Singer MS, Shepherd GM. Proc Natl Acad Sci USA. 2000;97:10712–10716. doi: 10.1073/pnas.97.20.10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaidehi N, Floriano WB, Trabanino R, Hall SE, Freddolino P, Choi EJ, Goddard WA., III Proc Natl Acad Sci USA. 2002;99:12622–12627. doi: 10.1073/pnas.122357199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trabanino RJ, Hall SE, Vaidehi N, Floriano WB, Kam V, Goddard WA., III Biophys J. 2004;86:1904–1921. doi: 10.1016/S0006-3495(04)74256-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kalani Y, Vaidehi N, Hall SE, Floriano WB, Trabanino RJ, Freddolino PL, Kam V, Goddard WA., III Proc Natl Acad Sci USA. 2004;101:3815–3820. doi: 10.1073/pnas.0400100101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Freddolino PL, Kalani MY, Vaidehi N, Floriano WB, Trabanino RJ, Freddolino PL, Kam V, Goddard WA., III Proc Natl Acad Sci USA. 2004;101:2736–2741. doi: 10.1073/pnas.0308751101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Spijker P, Vaidehi N, Freddolino PL, Hilbers PA, Goddard WA., III Proc Natl Acad Sci, USA. 2006;103:4882–4887. doi: 10.1073/pnas.0511329103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peng JY, Vaidehi N, Hall SE, Goddard WA., III Chem Med Chem. 2006;1:878–890. doi: 10.1002/cmdc.200600047. [DOI] [PubMed] [Google Scholar]
- 16.Vaidehi N, Schlyer S, Trabanino RJ, Floriano WB, Abrol R, Sharma S, Kochanny M, Koovakat S, Dunning L, Liang M, Fox JM, de Mendonca FL, Pease JE, Goddard WA., III J Biol Chem. 2006;281:27613–27620. doi: 10.1074/jbc.M601389200. [DOI] [PubMed] [Google Scholar]
- 17.Heo J, Han S-K, Vaidehi N, Wendel J, Kekenes-Huskey P, Goddard WA., III Chem Bio Chem. 2007;8:1527–1539. doi: 10.1002/cbic.200700188. [DOI] [PubMed] [Google Scholar]
- 18.Heo J, Vaidehi N, Wendel J, Goddard WA., III J Mol Graph Model. 2007;26:800–812. doi: 10.1016/j.jmgm.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li Y, Zhu F, Vaidehi N, Goddard WA, III, Sheinerman F, Reiling S, Morize I, Mu L, Harris K, Ardati A, Laoui A. J Am Chem Soc. 2007;129:10720–10731. doi: 10.1021/ja070865d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bray JK, Goddard WA., III J Mol Graph Model. 2008;27:66–81. doi: 10.1016/j.jmgm.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 21.Gao ZG, Kim SK, Gross AS, Chen A, Blaustein JB, Jacobson KA. Mol Pharm. 2003;63:1021–1031. doi: 10.1124/mol.63.5.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boucard AA, Sauvé SS, Guillemette G, Escher E, Leduc R. Biochem J. 2003;370:829–838. doi: 10.1042/BJ20021566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Befort K, Tabbara L, Kling D, Maigret B, Kieffer BL. J Biol Chem. 1996;271:10161–10168. doi: 10.1074/jbc.271.17.10161. [DOI] [PubMed] [Google Scholar]
- 24.Kaupmann K, Bruns C, Raulf F, Weber HP, Mattes H, Lübbert H. EMBO J. 1995;14:727–735. doi: 10.1002/j.1460-2075.1995.tb07051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Floriano WB, Vaidehi N, Zamanakos G, Goddard WA., III J Med Chem. 2004;47:56–71. doi: 10.1021/jm030271v. [DOI] [PubMed] [Google Scholar]
- 26.Mayo SL, Olafson BD, Goddard WA., III J Phys Chem. 1990;94:8897–8909. [Google Scholar]
- 27.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 28.Eisenberg D, Weiss RM, Terwilliger TC. Proc Natl Acad Sci USA. 1984;8:140–144. doi: 10.1073/pnas.81.1.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jayasinghe S, Hristova K, White SH. J Mol Biol. 2001;312:927–934. doi: 10.1006/jmbi.2001.5008. [DOI] [PubMed] [Google Scholar]
- 30.Jain A, Vaidehi N, Rodriguez G. J Comp Phys. 1993;106:258–268. [Google Scholar]
- 31.Vaidehi N, Jain A, Goddard WA., III J Phys Chem. 1996;100:10508. [Google Scholar]
- 32.Rappé AK, Goddard WA., 3rd J Phys Chem. 1991;95:3358–3363. [Google Scholar]
- 33.Schertler GFX, Villa C, Henderson R. Eye. 1998;12:504–510. [Google Scholar]
- 34.Bower MJ, Cohen FE, Dunbrack RL., Jr J Mol Biol. 1997;267:1268–1282. doi: 10.1006/jmbi.1997.0926. [DOI] [PubMed] [Google Scholar]
- 35.McClendon CL, Vaidehi N, Kam VWTK, Zhang D, Goddard WA., 3rd Protein Eng Design & Selection. 2006;19:195–203. doi: 10.1093/protein/gzl001. [DOI] [PubMed] [Google Scholar]
- 36.Cho AW, JA, Vaidehi N, Kekenes-Huskey PM, Floriano WB, Maiti PK, Goddard WA., III J Comp Chem. 2005;26:48–71. doi: 10.1002/jcc.20118. [DOI] [PubMed] [Google Scholar]
- 37.Kuntz ID, Blaney JM, Oatley SJ, Langridge R, Ferrin TE. J Mol Biol. 1982;161:269–288. doi: 10.1016/0022-2836(82)90153-x. [DOI] [PubMed] [Google Scholar]
- 38.Kam VWT, Goddard WA., III J Chem Theo & Comp. 2008;4:2160–2169. doi: 10.1021/ct800196k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zamanakos G. Thesis (PhD) PQ#30454682002etd-04062005-082441. available at http://resolver.caltech.edu/caltechETD:etd-04062005-04082441.
- 40.Liu HY, Zou X. J Phys Chem B. 2006;110:9304–9313. doi: 10.1021/jp060334w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Darden T, Perera L, Li L, Pedersen L. Structure. 1999;7:R55–R60. doi: 10.1016/s0969-2126(99)80033-1. [DOI] [PubMed] [Google Scholar]
- 42.Bhandarkar M, Brunner R, Chipot C, Dalke A, Dixit S, Grayson P, Gullingsrud J, Gursoy A, Hardy D, Hénin J, Humphrey W, Hurwitz D, Krawetz N, Kumar S, Nelson M, Phillips J, Schinozaki A, Zheng G, Zhu F. NAMD User’s Guide. Theoretical Biophysics Group, University of Illinois at Urbana-Champaign and Beckman Institute; Urbana: 2008. Version 2.6. [Google Scholar]
- 43.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feller SE, Yin D, Pastor RW, MacKerell AD., Jr Biophys J. 1997;73:2269–2279. doi: 10.1016/S0006-3495(97)78259-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.