

Abstract
The first stage of the development of a synthetic route for the total synthesis of laulimalide (1) is described. Our retrosynthetic analysis envisioned a novel macrocyclization route to the natural product using a Ru-catalyzed alkene-alkyne coupling. This would be preceded by an esterification of the C19 hydroxyl group, joining together two equally sized synthons, the northern fragment 7 and the southern fragment 8. Our first generation approach to the northern fragment entailed a key sequential Ru-Pd coupling sequence to assemble the dihydropyran. The key reactions proceeded smoothly, however, the inability to effect a key olefin migration led to the development of an alternative route based on an asymmetric dinuclear Zn-catalyzed aldol reaction of a hydroxyl acylpyrrole. This key reaction led to the desired diol adduct 66 with excellent syn:anti selectivity (10:1), and allowed for the successful completion of the northern fragment 7. The key step for the synthesis of the southern fragment was a chemoselective Rh-catalyzed cycloisomerization reaction to form the dihydropyran ring from a diyne precursor. This reaction proved to be selective for the formation of a six-membered ring, over a seven. The use of an electron-deficient bidentate phosphine allowed for the reaction to proceed with a reduced catalyst loading.
Keywords: Synthesis, Macrocyclic compd, Aldol react, Asymmetric synthesis, Chemoselectivity
Introduction
The focus of work in our laboratories has been the discovery of new and efficient methodologies for the synthesis of complex natural products. The development of these new techniques within the context of total synthesis presents a unique challenge and allows for in depth exploration of the scope and limitations of the methodology. Our ultimate goal is overcoming these limitations using complex natural product intermediates as substrates. In this and the following article, we wish to report the full account of our efforts that culminated in a successful total synthesis of the natural product laulimalide (1) and a biologically active analogue. A portion of this work has appeared in a communication.[1]
Laulimalide (1), also known as fijianolide B, is a structurally unique 20-membered marine macrolide that was isolated from two different marine sponges:[2] Cacospongia mycofijiensis and Hyattella sp simultaneously and independently by the Crews[2a] and Moore and Scheuer[2b] research groups. Two other closely related analogues have also been identified (Figure 1). Isolaulimalide (2) (fijianolide A) is an isomer of laulimalide where the C20 hydroxyl has opened the C16–17 epoxide resulting in a new tetrahydrofuran ring. This transformation readily occurs under mildly acidic conditions.[2a] A second regioisomer of laulimalide is neolaulimalide (3), which possesses an ester linkage at the C20 hydroxyl leading to a macrocycle that has been enlarged by one carbon (versus laulimalide).
Figure 1.

Laulimalide and Two Naturally Occurring Analogues
The structure and absolute stereochemical configuration of laulimalide was determined through X-ray crystallography.[3] Initially it was shown that laulimalide displays potent cytotoxicity towards numerous NCI cell lines,[2b] however, it did not attract the attention of synthetic chemists until Mooberry and coworkers discovered that laulimalide displays microtubule stabilizing activity similar to that of paclitaxel and the epothilones.[4] Additionally, laulimalide was found to bind to tubulin at a different site from paclitaxel, and was found to be active against multidrug-resistant cancer cell lines.[5] Recently, a distinct laulimalide microtubule binding site has been identified using mass shift perturbation mapping.[6] This discovery has the potential to help direct the synthesis of novel antimitotic laulimalide analogues. Due to both its unique pharmaceutical profile and challenging chemical architecture, laulimalide has attracted considerable interest from the synthetic chemistry community, leading to numerous attempts and several successful syntheses of both the naturally occurring compound and some analogues.[ 7 ] These approaches have underscored several unique structural features that can be addressed via the development of new efficient and atom economical transformations. Key features of laulimalide include nine stereogenic centers, two dihydropyran rings and a trans-disubstituted epoxide, which is susceptible to nucleophilic attack from the C20 hydroxyl group under mild acidic conditions to form a tetrahydrofuran regioisomer (isolaulimalide, 2) that is more stable but significantly less biologically active than the parent compound.
Synthetic Planning
Our main goal when designing a synthesis of laulimalide was to develop an efficient synthesis of this structurally challenging natural product that would serve as a springboard for the development of new synthetic methodologies. We were drawn to the use of the alkyne moiety as a synthetic handle for our key macrocyclization. As such, our retrosynthetic analysis for laulimalide was based on the assumption that the natural product could be formed from the 1,4-diene 5, which could be accessed via a key intramolecular ruthenium-catalyzed alkene-alkyne coupling of enyne 6 (Scheme 1). The resulting 1,4-diene 5 could be converted into compound 4 via a diastereoselective epoxidation of allylic alcohol 5 followed by an epoxide transposition (Payne rearrangement)[8] to give the correct oxidation pattern found in laulimalide. The enyne substrate 6 for the key macrocyclization would be accessed from an esterification between two dihydropyran-containing fragments of similar size and complexity: alcohol 7 (northern fragment) and acid 8 (southern fragment). Further retrosynthetic analysis revealed that the six-membered ring of the northern fragment 7 could be generated via sequential ruthenium-palladium catalysis between alkene 9 and alkyne 10.[9] We envisioned that alkene 9 could in turn be derived stereoselectively from the chiral pool using δ-gluconolactone 11. The dihydropyran of the southern fragment 8 was envisioned to be assembled with two key steps. A rhodium-catalyzed cycloisomerization of diyne 13 would provide dihydropyran 12,[10] and a Ferrier-type addition of an allenyl stannane to the resulting dihydropyran would complete the synthesis of the southern fragment 8.[11]
Scheme 1.

First Generation Retrosynthesis
A Ru-Pd Coupling Sequence for the Synthesis of the Northern Fragment
The preparation of the northern fragment began with commercially available δ-gluconolactone 11 (Scheme 2). Following literature precedent, compound 11 was treated with 2,2-dimethoxypropane to form bis-acetonide 14,[12] which in turn was subjected to a Barton–McCombie deoxygenation affording the methyl ester 15 in high yield (94%). Next, methyl ester 15 was derivatized to alcohol 16 by reduction of the ester moiety followed by diastereoselective Barbier-type allylation of the resulting aldehyde,[13] resulting in an inseparable mixture of diastereoisomers favoring the desired anti product 16 derived from the Felkin-Anh mode of addition (d.r. = 6:1). Fortunately, the desired isomer was separable by flash column chromatography on silica gel after transforming the hydroxyl group of alcohol 16 to methyl carbonate 9.
Scheme 2.

A Ru-Pd Coupling Sequence
Having completed the preparation of the alkene partner 9, we began to explore the key Ru-catalyzed transformation.[ 14 ] Our group previously demonstrated that Ru-catalyzed alkene-alkyne coupling provides an atom-economical and highly functional group tolerant access to 1,4-dienes. Additionally, depending on the relative orientation of alkene and alkyne in the initial ruthenacycle formation, either “branched” or “linear” 1,4-diene products could arise. Earlier studies from our group showed that the branched/linear selectivity is influenced by steric modification on either substrate. In particular, silylation of the alkyne coupling partner has proven to be a reliable method to drive the reaction to afford high branched selectivity.[15] Based on these observations, we initiated the study of this reaction with a TMS-substituted alkyne as our substrate to attain high branched selectivity. However, we soon discovered that the unsubstituted terminal alkyne 3-butyn-1-ol 10 also gave the desired regioselectivity. Thus, when alkene 9 and alkyne 10 were exposed to 10 mol% of cationic ruthenium complex [CpRu(CH3CN)3]PF6 in acetone,[16] the desired branched diene 17 was obtained in good yield with no apparent formation of the undesired linear isomer as judged by 1H-NMR spectroscopy. An optimization study revealed that high yields were obtained when an excess of the alkene coupling partner 9 was employed. Most (>85%) of the unreacted alkene could be recovered via column chromatography. The requirement of excess alkene was presumably due to the stronger coordination ability of the alkyne to the metal, resulting in a catalytically inactive coordinatively saturated ruthenium-alkyne complex. Slow addition of the alkyne substrate to the reaction mixture was moderately effective in reducing the amount of alkene necessary. However, consistently better yields were obtained by simply using an excess of the alkene.
In the key ruthenium-catalyzed alkene-alkyne coupling step, the exclusive formation of a ‘branched’ 1,4-diene in the absence of the large molecular weight TMS group on the alkyne renders our approach to the northern fragment more atom-economical. Although the exact mechanism awaits further study, we believe the presence of a hydroxyl functionality at the homopropargylic position may play a role as illustrated in Scheme 3. In the initial complexation step, the head-to-head complexation (compound 19), which will ultimately lead to the ‘linear’ 1,4-diene product 20, is thought to be favored to avoid the steric encumbrance (compound 21) near the newly formed carbon-carbon bond. However, in the resulting ruthenacyclopentene 22, the pendant hydroxyl group is well positioned to provide ligation to the electrophilic Ru (III) metal center. It is expected that coordination of the hydroxyl group will slow the subsequent β-hydride elimination process by making the additional coordination site unavailable for the β-hydrogen.[17] This coordination has the overall effect of favoring the formation of the desired branched pathway (leading to compound 24), despite the greater hindrance of the head to tail coordination.
Scheme 3.

Mechanistic Rationale for the Formation of Branched Diene
With the desired 1,4-diene in hand, we now could form the six-membered ring via a palladium-catalyzed cyclization. When carbonate 17 was exposed to Pd2(dba)3·CHCl3 in the presence of diphenylphosphinoferrocene (dppf), clean cyclization occurred furnishing tetrahydropyran 18 as a single diastereomer in 87% isolated yield. This cyclization reaction is stereospecific as the stereochemistry at the C21 position of carbonate 17 was completely transferred to the C23 position of tetrahydropyran 18, in accordance with the well-known π-allylpalladium mechanism.[18]
With compound 18 in hand, an exo to endo olefin isomerization was necessary to provide the correct olefin regioisomer present in laulimalide (vide supra). It was initially expected that the propensity of the olefin to migrate to form a more stable isomer (exo to endo) and the steric bias from the C23-substituent would help to provide the correct regioselectivity; however, extensive experimentation utilizing various acids, bases, and transition metals led to either poor selectivity or low conversion.[19]
After screening a substantial set of conditions[20] to effect the double bond migration, the most promising proved to be the utilization of Pd(OAc)2 (20 mol%), in the presence of CSA (1.0 equiv.) in MeCN at room temperature which yielded the desired double bond isomer as the only product, albeit in poor yield (15%). However, satisfactory yields were only obtained when a stoichiometric amount of palladium was used (Scheme 4). Efforts to develop a catalytic system were unsuccessful.
Scheme 4.

Stoichiometic Pd Isomerization
The difficulty of achieving a direct, catalytic olefin migration led us to consider indirect methods. Ultimately, a successful route was developed that utilized a three-step sequence: (1) an oxidative cleavage of the exocyclic double bond, (2) a regioselective vinyltriflate formation, and (3) a cross-coupling reaction with a methyl donor (Scheme 5). The transformation of the exocyclic methylene group of compound 18 to ketone 26 could be achieved chemoselectively by an osmium tetroxide-catalyzed dihydroxylation, with the internal olefin remaining intact. This oxidation could be carried out either using a two-step (OsO4, NMO then NaIO4) or a one-step protocol (OsO4, NaIO4).[21] Although more convenient, the latter conditions gave somewhat lower yield than the stepwise procedure (70% vs 91% respectively). Regioselective enolate formation was achieved distal to C23 using bulky triphenylmethyllithium with good selectivity (9:1) furnishing enol triflate 27 with Comin’s reagent as the triflating agent in 66% yield.[22] More commonly used bulky amide bases, such as LDA or LTMP proved ineffective giving very poor regioselectivity even at −100 °C (< 2:1).
Scheme 5.

Synthesis of the Carbon Skeleton of the Northern Fragment
The conversion of 27 to the complete carbon skeleton of the northern fragment was achieved in 4 steps initiated by a Negishi coupling with dimethylzinc to provide dihydropyran 25.[23] A selective terminal acetonide deprotection, epoxide formation using catalytic tin oxide,[24] and finally a copper-catalyzed vinyl Grignard addition furnished dihydropyran 29 (Scheme 5).[25]
While the route described above provided an excellent example of the novel use of a Ru-Pd sequence for the construction of the enantiopure dihydropyran of the C14-C27 fragment of laulimalide, the lack of an efficient solution to the double bond migration problem prompted us to re-evaluate our synthetic strategy. In designing a second generation synthesis, our focus was on the potential implementation of a key zinc-catalyzed direct aldol addition to install the cis-diol at C19-C20.
A Dinuclear Zinc Aldol Reaction for the Synthesis of the Northern Fragment
Our new retrosynthetic approach envisioned a Julia- Kocieński olefination[ 26] to construct fragment 7 from phenyl tetrazole sulfone 30 and aldehyde 31 (Scheme 6). Aldehyde 31 would be assembled using a dinuclear zinc-catalyzed aldol reaction using (R,R)-ProPhenol[27] catalyst 32 with an aryl hydroxymethyl ketone 33 as the donor and aldehyde 34 as the acceptor. To assemble the dihydropyran of sulfone 30, we planned to utilize a ring-closing metathesis (RCM) approach using diene 35 which would be derived from (R)-glycidol (36). Several groups have used a RCM sequence for the construction of the exocyclic dihydropyran of laulimalide;[7o] however, we envisioned that we could improve upon these routes by utilizing a novel protecting group-free approach.
Scheme 6.

Second Generation Approach to the Northern Fragment
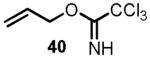
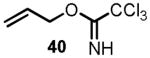
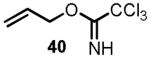
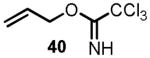
We began the synthesis of the sulfone coupling partner 30 via Mitsunobu coupling of (R)-glycidol 36 with 1-phenyl-1H-tetrazole-5-thiol to provide epoxysulfide 37 in good yield (Scheme 7). The epoxide underwent regioselective opening with isopropenylmagnesium bromide in the presence of a catalytic amount of copper iodide to provide alcohol 38 in near quantitative yield.[24] Allylation of alcohol 38 to provide diene 39 proved to be less straightforward than it initially appeared (see Scheme 7). Simple alkylations under acidic conditions using the trichloroimidate 40[28],[29] or under basic conditions with allyl bromide[30] failed.
Scheme 7.

Synthesis of the Sulfone Coupling Partner
| Entry | Conditions | Result |
|---|---|---|
| 1 |
 TfOH (50 mol %), CH2Cl2 |
Decomposition |
| 2 |
 TfOH (5 mol %), CH2Cl2-hexane |
Decomposition |
| 3 |
 Cu(OTf)2, PhMe |
No Reaction |
| 4 |
 BF3·OEt2 |
Decomposition |
| 5 | Allyl bromide, Ag2O, CaSO4, PhH | No Reaction |
| 6 | Et2Zn, Pd(OAc)2, PPh3, allyl acetate, THF | 61% of 39 |
Having been unsuccessful with several traditional allylation conditions, we investigated the possibility of a transition metal-catalyzed allylation reaction.[31] Aliphatic alcohols are poor nucleophiles and the corresponding alkoxide anions are hard nucleophiles making them unsuitable substrates for a metal-catalyzed reaction.[32] In order to overcome the reactivity mismatch between hard alkoxide anions and soft η3-allylmetal cations, Lee et al. have shown that the formation of a zinc alkoxide has the beneficial effect of softening the alkoxide anion (via the Zn (II) center), resulting in an attenuated basicity while retaining sufficient nucleophilicity toward a metal-bound allylic cation.[30]
Thus, addition of the zinc alkoxide of alcohol 38 to an in situ generated π-allylpalladium complex [Pd(OAc)2, PPh3, allylacetate] successfully resulted in the formation of the desired allylated compound 39 in 61% yield. Molybdenum-catalyzed oxidation of the sulfide into the corresponding sulfone 35 followed by a ring-closing metathesis using the second generation Grubbs catalyst (41)provided the dihydropyran 30 in excellent yield.[33]
With sulfone 30 in hand, our efforts shifted to the construction of the aldehyde partner for the Julia-Kocienski olefination. This piece was to be assembled using a dinuclear zinc aldol reaction using ProPhenol catalyst 32 to couple α-hydroxyacetophenone with a suitable aldehyde donor. The catalytic asymmetric aldol reaction is a powerful tool for the enantioselective generation of carbon-carbon bonds.[34] Previous studies in our group have demonstrated the feasibility of using dinuclear zinc ProPhenol catalyst 32 with α-hydroxyacetophenone as the donor to produce syn diols in up to 30:1 dr and 92% ee.[35] Shibasaki et al. has reported a similar reaction using α-hydroxyacetophenone donors with a dinuclear zinc BINOL-type catalyst.[36] However, the phenyl ketone product would not be synthetically useful in our synthesis of laulimalide. In order to overcome this obstacle, we looked into a strategy that Shibasaki had used for further functionalization of aryl ketone aldol products: a methoxy-substituted α-hydroxyacetophenone donor.[35b–d] It was shown that these electron-rich aryl ketone products could be either oxidized via a Baeyer-Villiger oxidation[37] to the corresponding phenyl ester, or subjected to a Beckmann rearrangement[38] to provide an aryl amide product. These precedents provided an attractive route to utilize the α-hydroxyacetophenone donor in our synthesis.
The synthesis of the aldehyde acceptor for the aldol reaction began with reduction of commercially available ester 42 to aldehyde 43 in excellent yield (Scheme 8). Brown allylation provided alcohol 44 in good yield and excellent stereoselectivity.[39] TBS-protection of the hydroxyl moiety and careful deprotection[40] of the diethyl acetal gave aldehyde 46. With a reliable and scalable route to the desired aldehyde 46 in hand, optimization studies of the aldol reaction were undertaken.
Scheme 8.

Route to the Aldehyde Substrate for the Aldol Reaction
In the event, aldehyde 46 was coupled with α-hydroxyacetophenone (33) using 2.5 mol% of (R,R)-ProPhenol 32 and 5 mol % Et2Zn in THF at room temperature to produce the desired aldol adduct 47 with a 4.8:1 syn:anti diol ratio (Table 1, entry 1). The desired diastereomer was separated via flash column chromatography on silica gel and obtained in a 52% isolated yield. It was found that switching the protecting group on the hydroxyl moiety of the aldehyde from a TBS to a p-methoxybenzyl group did not significantly alter the syn:anti diol ratio, but the isolated yield of the desired diastereomer 48 increased. The syn configuration of the major product was confirmed via coupling constants of the C20-C19 hydrogens (laulimalide numbering) after conversion to the corresponding carbonate 49 (vida infra, Scheme 9) (J = 3.8 Hz for the syn diol carbonate, J = 6.1 Hz for anti diol carbonate). Switching the solvent from THF to toluene, acetonitrile or dichloromethane led to dramatic improvements in the selectivity for the reaction with toluene proving optimal, providing the desired product with a 9:1 syn:anti selectivity and an 83% isolated yield (entry 3). In addition to a less favorable syn:anti ratio, the use of acetonitrile and dichloromethane led to a decrease in the isolated yield of the desired product when compared to toluene.
Table 1.
Optimization of the α-Hydroxyl Acetophenone Aldol Reaction
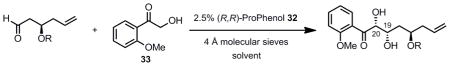
| |||||
|---|---|---|---|---|---|
| Entry | R | Solvent | syn: anti | de of syn (%) | Isolated yield (%), (cmpd) |
| 1 | TBS | THF | 4.8: 1 | 91 | 52 (47) |
| 2 | PMB | THF | 5: 1 | 92 | 69 (48) |
| 3 | PMB | Toluene | 9: 1 | 94 | 83 (48) |
| 4 | PMB | MeCN | 7.5: 1 | 90 | 70 (48) |
| 5 | PMB | DCM | 6: 1 | 88 | 51 (48) |
Scheme 9.

Attempted Baeyer-Villiger Reaction
| entry | oxidant | Additive | Temperature | Solvent | Result |
|---|---|---|---|---|---|
| 1 | TMSO-OTMS | SnCl4 | −20 °C | DCM | 80% PMB deprotection |
| 2 | TMSO-OTMS | CeCl3 | −20 °C to rt | DCM | No reaction |
| 3 | TMSO-OTMS | Toluene | −20 °C to rt | DCM | No reaction |
| 4 | mCPBA | TBAF | 0 °C | DCM | No reaction |
| 5 | mCPBA | None | rt | DCM | 70% epoxidation |
| 6 | Peroxyacetic acid | None | 0 °C | CHCl3 | 78% epimerization |
| 7 | Peroxytrifluoroacetic acid | None | 0 °C | CHCl3 | 62% epimerization |
| 8 | Mono peroxyphthalate (Mg salt) | None | rt | CHCl3 | 30% epimerization |
After optimization of the aldol reaction, the next step was a Baeyer-Villiger oxidation to convert the ketone moiety to the corresponding phenyl ester to complete the synthesis of fragment 50 (Scheme 9).[35b–d] Carbonate formation proceeded smoothly to give the protected triol 49. However, several difficulties were encountered during the course of the oxidation (Scheme 9).
Using bis(trimethylsilyl)peroxide and SnCl4 as the Lewis acid for the oxidation led only to deprotection of the PMB group (entry 1).[35c] Switching the PMB group to a silyl ether (TBS) led to no reaction. The use of different additives (entries 2 and 3) was ineffective. mCPBA was an effective oxidant for the substrates employed by Shibasaki.[35b–d] In our example it proved to be a poor oxidant for the reaction, either leading to no reaction at low temperature or undesired epoxidation of the terminal olefin (entries 4 and 5). The use of other peroxyacids (entries 6–8) led to extensive epimerization of the C20 hydroxyl group (laulimalide numbering) with no detectable oxidation product. Several additional reaction conditions were tested, including different oxidants and solvents, all to no avail (data not shown). At this point, it was decided that even though the dinuclear zinc aldol reaction had proceeded smoothly, we had hit an impassable roadblock in the inability to affect a Baeyer-Villiger type process with our substrate. We therefore decided to change the donor for the aldol reaction from α-hydroxyacetophenone to α-hydroxyacylpyrrole.
In contrast to the use of α-hydroxyacetophenone as the donor for the aldol reaction, the use of α-hydroxyacylpyrrole should lead to aldol products that can readily be transformed without having to resort to oxidative rearrangements since the products are already at the carboxylic acid oxidation state.[41] Acylpyrroles previously have been used in several types of asymmetric transformations. For example, N-acylpyrroles have been utilized as ester surrogates in asymmetric conjugate addition reactions.[ 42 ] Additionally, acylpyrrole enolsilanes have been employed in enantioselective aminations,[43] and diastereoselective Mukaiyama-Michael reactions.[44] α-Hydroxyacylpyrrole itself has been employed in asymmetric Mannich-type reactions.[45]
Our initial investigations using a TBS protected β-hydroxyaldehyde revealed that the reaction proceeds in high yield, but with a low diastereoselectivity (diol syn:anti = 1.5:1) (Table 2, entry 1). The focus of the reaction optimization would be on the improvement of the syn:anti ratio. Additional experiments indicated that the syn:anti dr for the newly formed 1,2-diol was somewhat dependent on the nature of the protecting group P of the β-hydroxyaldehyde (entries 1–4). The largest effect arose by the use of a strong zinc-chelating group, methylthiomethyl (MTM, entry 4) which resulted in a slight improvement in the syn:anti ratio, but a drastic reduction in the yield. Turning to modifications of the donor, it was thought that increasing the steric bulk at the 2-position of the acylpyrrole may improve the observed diastereoselectivity. Gratifyingly, 2-ethyl-N-acylpyrrole significantly improved the syn:anti ratio to 4:1 when P was a TBS group (entry 5).
Table 2.
Initial Optimization of the Acyl Pyrrole Aldol Reaction
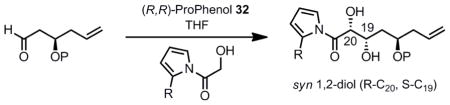
| |||||
|---|---|---|---|---|---|
| Entry | P | R | Yield (%) | syn: anti | de (syn) |
| 1 | TBS | H | 80 | 1.5: 1 | 86 |
| 2 | TBDPS | H | 85 | 1.1: 1 | 68 |
| 3 | PMB | H | 70 | 1.1: 1 | 41 |
| 4 | MTM | H | 25 | 1.9: 1 | 60 |
| 5 | TBS | Et | 80 | 4: 1 | ND |
At this point, we wanted to demonstrate that the observed selectivity of the aldol reaction was due to catalyst control and not substrate control (due to the presence of the existing stereocenter). To this end, the opposite enantiomer of the catalyst (S,S) was used in place of the (R,R)-ProPhenol that had provided the aldol products with the desired configuration. When the reaction was conducted with the (S,S)-catalyst, the opposite configuration of the diol was achieved (S-C20, R-C19) in a similar syn:anti ratio (2:1) to that of the desired configuration (R-C20, S-C19) shown in Table 2. This result confirmed that it was indeed catalyst and not substrate control of the stereoselectivity that provided the desired aldol products.
Considering the results in Table 2, it appeared that the use of a non-chelating protecting group was necessary to achieve good chemical yield and an acceptable syn:anti ratio, and the size of the hydroxyl protecting group should ideally be as small as possible. In addition, the use of 2-ethylpyrrole instead of pyrrole led to a significant syn:anti improvement. With this information in hand, we envisioned that switching from a TBS group to a smaller TES protecting group may furnish the desired product with improved diastereoselectivity. The synthesis of this new substrate also provided an opportunity to revisit our synthetic route to the β-hydroxyaldehyde.
In order to circumvent the use of a stoichiometric amount of a chiral reagent (Brown allylation) and the large amount of waste generated in that reaction, a new route to the aldehyde acceptor was adopted (Scheme 10).[46] The required β-triethylsiloxyaldehyde 51 was prepared from the commercially available (S)-glycidyl tosylate 52 which was treated with lithiated 1,3-dithiane to give dithiane 53. Subsequent copper-catalyzed vinyl Grignard addition followed by protection of the resulting homoallyic alcohol as its triethylsilyl ether (TES) gave compound 54. The desired aldehyde 51 was ultimately obtained upon treatment of dithiane 54 with MeI in the presence of CaCO3 (74% yield).
Scheme 10.

Modified Route to the Aldehyde Substrate for the Aldol Reaction
We now had the required intermediates for the crucial zinc aldol reaction. The reaction proceeded as anticipated using 15 mol% of (R,R)- Prophenol 32 and 2-ethyl acylpyrrole 55 as donor furnishing the desired syn 1,2-diol 56 with a 9:1 dr, in 54% isolated yield (Scheme 11).
Scheme 11.

Aldol Reaction Using a TES Protecting Group and 2-Ethyl Acylpyrrole
In order to unequivocally prove the absolute stereochemistry of the N-acylpyrrole aldol product, it was converted to a substrate that could be intercepted from our previous synthetic route using gluconolactone 11 (vide supra, Scheme 2). Deprotection of the terminal acetonide of ester 15 gave diol 57 in good yield (Scheme 12). Tosylate formation mediated by dibutyltin oxide and base induced[47] cyclization led to epoxide 58 in good yield. Epoxide opening with higher order lithium divinyl cyanocuprate in the presence of BF•OEt2 led to the formation of the corresponding homoallylic alcohol which was subsequently protected with a TBS group to provide ester 59. This intermediate was intercepted by the acyl pyrrole aldol adduct 60 (from Table 2) by installation of the acetonide, followed by methanolysis of the pyrrole. Both the sugar derived ester 59 and the aldol derived adduct were identical by 1H and 13C NMR spectroscopy and optical rotation, thus confirming our stereochemical assignment.
Scheme 12.

Confirmation of the Absolute Stereochemistry for the Aldol Reaction Product
Having the desired syn 1,2-diol 56 in hand, we needed to orthogonally protect the two hydroxyl groups, since the northern fragment will later need to be selectively attached to the southern fragment via an esterifcation of the alcohol at the β-position (C19 laulimalide numbering) of the carbonyl group (vide supra). Taking advantage of the enhanced acidity of the alcohol α to the carbonyl group of diol 56 (C20), we intended to protect it selectively as a TBS-ether (Scheme 13). To this end, diol 56 was treated with TBSCl, in the presence of imidazole in DMF which furnished the desired protected alcohol 61 along with compound 62 (inseparable from 61) resulting from the migration of the TES group.
Scheme 13.

Attempted Protection Strategy for the syn Diol
To circumvent this silyl migration issue, we planned to protect the 1,2-diol of compound 56 as its p-methoxyphenyl (PMP) acetal, then replace the TES ether with a MOM ether. Additionally, replacement of the TES protecting group by a chelating MOM group should in principle allow for a selective opening of the PMP acetal with DIBAL-H via chelation, to afford the desired C20 PMB ether. It was found that the protection of 1,2-diol 56 as a PMP-acetal was best performed by employing a large excess of the dimethylacetal of p-anisaldehyde (10 equiv) in the presence of CSA (64% yield). Indeed, when only one equivalent is used the reaction is slow and significant deprotection of the secondary TES-ether occurs. The excess of p-anisaldehyde dimethylacetal can be separated from the desired product 63 by simply stirring the crude residue with silica in chloroform. These mildly acidic conditions allowed the conversion of p-anisaldehyde dimethylacetal into p-anisaldehyde, easily separable from 63 by flash chromatography on silica gel. We then needed to convert the TES ether into a MOM protecting group as suggested above. Accordingly, TES-ether 63 was treated with TBAF, which resulted in complete decomposition of the starting material. It seemed that the 2-ethyl-N-acylpyrrole moiety was not stable to the basic reaction conditions, based on the TLC of the reaction mixture indicating the release of 2-ethylpyrrole. Even though using milder conditions could certainly circumvent this issue, we decided to investigate the dinuclear zinc aldol chemistry applied directly to a β-OMOM aldehyde, thus avoiding this impractical and inelegant change of protecting groups.
Utilizing a similar route as outlined in Scheme 10, the required β-methoxymethyloxy aldehyde 65 was obtained in 63% yield from tosyl glycidol 52. Remarkably, using 15 mol% of (R,R)-ProPhenol 32 catalyst and 2-ethyl acylpyrrole 55 as donor provided the desired syn 1,2-diol 66 with a 10:1 dr, in 51% isolated yield (Scheme 14). Surprisingly and to our delight, the chelation properties of the MOM protecting group did not seem to affect the diastereoselectivity of the reaction in the case of the ethyl pyrrole. This was not true in the case of the parent acylpyrrole (See Table 2, above); evidently the additional steric bulk associated with the ethyl group overcomes any stereochemical bias of the chelating protecting group.
Scheme 14.

Completion of the Synthesis of the Northern Fragment
After protection of the resulting 1,2-diol as a PMP acetal and cleavage of the acylpyrrole upon exposure to sodium borohydride in THF, alcohol 67 could be isolated in 70% yield (over 2 steps). Oxidation of 67 with Dess-Martin periodinane gave rise to the corresponding aldehyde, which was treated with the lithium salt of sulfone 30 in a THF/HMPA mixture to give alkene 68 in 64% yield and as a single (E)-geometric isomer. Taking advantage of the presence of the MOM chelating group, the PMP acetal 68 could be opened in a regioselective fashion (3:1, C20:C19) upon treatment with DIBAL-H in dichloromethane at −78 °C, to give the desired fragment 7 as a single E-configured geometric isomer, completing the synthesis of the northern fragment. The PMB regioisomers were separable by column chromatography, and the undesired C19-PMB ether could be recycled by exposure to DDQ giving back the starting acetal 68, increasing the overall synthetic efficiency.
Rh(I) Cycloisomerization for the Synthesis of the Southern Fragment
As noted in the introduction, our retrosynthetic analysis for the southern fragment is based upon the notion that a Ferrier-type reaction would install the C1-C4 side chain of dihydropyran 8 stereoselectively via an axial delivery of a nucleophile onto the oxocarbenium ion generated from vinylogous acetal 12. The vinylogous acetal 12 in turn was envisioned to come from a key rhodium-catalyzed cycloisomerization. Cycloisomerization reactions are efficient and atom-economical[ 48 ] transformations since all of the atoms in the starting material are present in the product.[49] Previous work in our group had demonstrated the feasibility of Ru- and Rh-catalyzed cycloisomerization reactions to form dihydropyran rings.[50] For the synthesis of laulimalide, we chose to implement the Rh-catalyzed reaction to take advantage of its broader functional group tolerance. In particular, the Rh-catalyzed reaction is more tolerant of substituents at the propargylic position (such as the OBn in diyne 13). Recently, Morris and Shair demonstrated the utility of the Rh-catalyzed cycloisomerization in the formation of an intermediate glycal for the synthesis of lomaiviticin A and B.[51] In our synthesis of laulimalide, we were interested in exploring the scope of the reaction by employing a challenging diyne substrate 13. The success of this cycloisomerization step would rely on the reversibility of the Rh-vinylidene formation and the kinetically more facile production of the six-membered cyclic glycal product over a seven-membered ring. [52]
The synthesis of the key diyne substrate 13 is shown in Scheme 15. Starting with oxirane 69, (readily prepared from D-aspartic acid),[53] alkylation with the lithium salt of methyl propiolate provided alcohol 70. Conjugate addition of dimethylcuprate followed by acid-catalyzed lactonization gave lactone 71 in excellent yield for the two step procedure. Diastereoselective hydrogenation and concomitant benzyl deprotection furnished saturated lactone 72 as a single stereoisomer. Reduction of lactone 72 with DIBAL-H to the lactol, followed by protection as the mixed acetal gave alcohol 73. Dess-Martin periodinane oxidation to the aldehyde, followed by Grignard reaction with ethynylmagnesium bromide and then protection of the resulting alcohol as a benzyl ether proceeded in good yield (73%) for the three step procedure providing ether 74. Hydrolysis of the acetal gave lactol 75, which upon treatment with the Ohira-Bestmann reagent[54] provided diyne 13 in moderate yield.
Scheme 15.

Synthesis of the Diyne Cycloisomerization Substrate
With the diyne substrate 13 in hand, we explored the key cycloisomerization reaction. Employing chloro(1,5-cyclooctadiene)rhodium dimer as the precatalyst, we examined a series of electron-poor triarylphosphine ligands (76–79). To our delight, the desired dihydropyran was obtained in moderate yield with no noticeable 7-membered ring formation (Scheme 16). However, when the reaction time was too long, there was significant decomposition (entry 3). While tri(3-fluorophenyl)phosphine 76 was most effective in this series, the cyclization required up to 1.1 molar equivalents of ligand for acceptable reactivity.
Scheme 16.

Optimization of the Key Rh-Catalyzed Cycloisomerization
It is proposed that the use of an excess of the phosphine ligand is essential to suppress the coordination of the second alkyne to the metal center after vinylidene formation, which would serve as an entry to an undesired alkyne-alkyne coupling reaction leading to dimeric and oligomeric mixtures.[55] In our case, it was hypothesized that if a bidentate phosphine such as 79 was used, this would overcome the need for large excesses of phosphine, as it would require less phosphine to maintain a coordinatively saturated rhodium. The use of the bidentate phosphine 79 indeed allowed for a lower ligand loading, and at the same time, higher yield of the cyclized product. Surprisingly, using a large excess of the bidentate phosphine 79 (entry 5) led to almost complete suppression of the reaction, presumably due to full saturation of the rhodium and the lack of any available coordination sites to bind the alkyne. However, when the bidentate phosphine was used at significantly lower loadings (entries 6 and 7), a good yield of the product could be obtained. Under optimized conditions, only 5 mol % of the precatalyst and as low as 10 mol % of the bidentate phosphine was required for moderate to good reactivity (entry 7).
Electron poor ligands are crucial for the success of the cycloisomerization, which are thought to facilitate the rate-determining cyclization event in the catalytic cycle by stabilizing the ensuing Rh(III) to Rh(I) reduction step. The subsequent protoderhodation would then regenerate the catalyst. We attempted to further optimize the reaction by facilitating the protonation step with an exogenous acid, to no avail. For instance, the addition of CSA was detrimental, lowering the yield significantly (entry 8). As with the reaction with monodentate ligands, it was important to stop the reaction after about 5 h to prevent decomposition of the product, presumably via intermolecular alkyne dimerization. We also explored the use of alternative solvents. For instance, acetonitrile provided a 43% yield of the product at 100% conversion, while dioxane gave only a 21% yield. In both cases, a significant number of side products were obtained.
It is noteworthy that the reaction was completely chemoselective toward 6-membered ring formation. In addition, the fact that the alkyne at C13 remained intact appears to clearly indicate that the vinylidene-rhodium complex formation step was reversible. Although the elucidation of the exact mechanism awaits further study, the initial vinylidene rhodium complex formation (80 and 81, Scheme 17) is likely to be indiscriminant and reversible, and only the kinetically viable complex 81 undergoes subsequent cyclization to form the desired dihydropyran 12. The 7-membered ring cycloisomerization product 82 arising from vinylidene-rhodium 80 was not observed.
Scheme 17.

Rationale For the Selective Formation of a 6-Membered Ring
In order to complete the synthesis of the southern fragment, the obtained glycal 12 was activated with BF3•Et2O and underwent a highly diastereoselective Ferrier addition of a Marshall-type[56] with allenylstannane 83 at low temperature to furnish exclusively the trans-disubstituted dihydropyran 84 in 85% yield, as confirmed by the absence of an nOe between the C5 and C9 hydrogens (Scheme 18). This transformation allowed for the installation of the desired propargylic ester in one step from glycal 12. Williams[7h] and Nelson[7i] both successfully employed similar allenyl stannanes in their syntheses of laulimalide. Finally, saponification of the propargylic methyl ester 84 with lithium hydroxide (2 equiv) resulted in the formation of the desired acid 8 in 81% yield, completing the synthesis of the southern fragment.
Scheme 18.

Completion of the Southern Fragment
In summary, we have developed a novel synthesis of two similar sized fragments of laulimalide. The northern fragment was completed using a diastereoselective dinuclear zinc aldol reaction of a novel acylpyrrole aldol donor, followed by a Julia-Kocienski olefination reaction. The southern fragment was assembled via a key Rh(I)-catalyzed cycloisomerization reaction applied to a challenging diyne substrate. In the following paper, we detail the union of the two fragments and the completion of the total synthesis of laulimalide.
Experimental Section
(S,E)-1-((4R,5S)-5-(((S)-2,2-dimethyl-1,3-dioxolan-4-yl)methyl)-2,2-dimethyl-1,3-di-oxolan-4-yl)-7-hydroxy-5-methylenehept-2-enyl methyl carbonate (17)
A solution of 3-butyn-1-ol 10 (291 μl, 3.85 mmol) and alkene 9 (3.978 g, 11.55 mmol) in dry degassed acetone (15 mL) was cooled to 0 °C, and [CpRu(CH3CN)3]PF6 (139 mg, 0.39 mmol) was added in one portion. The resulting orange mixture was stirred at 0 °C for 10 min, and at rt for 18 h. The mixture was then concentrated in vacuo, and the residue was purified by column chromatography (petroleum ether/EtOAc = 4:1 to 2:1) to give the title compound 17 (1.165 g, 2.811 mmol, 73%). (c 1.06, CH2Cl2); IR (neat): ν 3496, 2987, 1752, 1443, 1380, 1269, 1069 cm−1; 1H NMR (500 MHz, CDCl3): δ 5.86 (ddd, J = 15.5, 7.1, 6.2 Hz, 1H), 5.59 (dd, J = 15.5, 7.4 Hz, 1H), 5.20 (dd, J = 7.0, 4.9 Hz, 1H), 4.90 (s, 1H), 4.89 (s, 1H), 4.26-4.21 (m, 1H), 4.11-4.08 (m, 1H), 4.06-4.02 (m, 1H), 3.81 (dd, J = 8.1, 4.5 Hz, 1H), 3.78 (s, 3H), 3.70 (t, J = 6.5 Hz, 2H), 3.60-3.57 (m, 1H), 2.83 (d, J = 6.8 Hz, 2H), 2.29 (t, J = 6.5 Hz, 2H), 1.94 (ddd, J = 13.8, 7.7, 1.7 Hz, 1H), 1.72 (ddd, J = 13.8, 9.9, 5.4 Hz, 1H), 1.40 (s, 3H), 1.39 (s, 3H), 1.37 (s, 3H), 1.36 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 154.9, 143.7, 134.5, 125.5, 113.2, 109.6, 108.7, 81.7, 77.7, 75.0, 73.6, 69.7, 60.3, 54.8, 39.1, 39.0, 38.6, 27.6, 27.3, 26.9, 26.6, 25.6. HRMS: Calc’d for C20H31O8 (M-CH3): 399.2020. Meas: 399.2023.
(S)-2-((E)-2-((4S,5S)-5-(((S)-2,2-dimethyl-1,3-dioxolan-4-yl)methyl)-2,2-dimethyl-1,3-dioxolan-4-yl)vinyl)-4-methylene-tetrahydro-2H-pyran (18)
A mixture of Pd2(dba)3·CHCl3 (0.175 g, 0.169 mmol), dppf (0.187 g, 0.338 mmol) and dry degassed dichloroethane (10 mL) was stirred under a N2 atmosphere at rt for 15 min, during which time it formed a clear deep red solution. This solution was transferred to a flask that contained a solution of 17 (2.800 g, 6.755 mmol) in dry degassed dichloroethane (120 mL), rinsing with 5 ml of dichloroethane to ensure complete transfer. The resulting mixture was placed in an oil bath, which was preheated to 70 °C, and stirred for 12 hr. After cooling to room temperature, the mixture was concentrated in vacuo, and the residue was purified by column chromatography (petroleum ether/EtOAc = 15:1 to 10:1) to give 18 (1.999 g, 5.907 mmol, 87%). (c 1.01, CH2Cl2); IR (neat): ν 2986, 1380, 1370, 1244, 1090, 1059, 890 cm−1; 1H NMR (500 MHz, CDCl3): δ 5.84 (ddd, J = 15.5, 5.5, 0.6 Hz, 1H), 5.70 (ddd, J = 15.5, 7.4, 1.3 Hz, 1H), 4.77-4.74 (m, 2H), 4.22 (ddd, J = 13.0, 7.1, 6.0 Hz, 1H), 4.00 (t, J = 7.9 Hz, 1H), 3.83-3.79 (m, 2H), 3.58 (dd, J = 8.2, 7.2 Hz, 1H), 3.42 (ddd, J = 12.1, 11.0, 2.8 Hz, 1H), 2.34-2.27 (m, 2H), 2.17-2.14 (m, 1H), 2.10-2.05 (m, 1H), 1.92 (ddd, J = 13.8, 7.1, 2.6 Hz, 1H), 1.65 (ddd, J = 13.8, 9.9, 5.8 Hz, 1H), 1.40 (s, 9H), 1.35 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 143.7, 135.5, 127.2, 109.0, 108.9, 108.6, 82.1, 78.0, 77.7, 73.7, 69.8, 68.4, 41.0, 36.2, 34.8, 27.2, 27.0, 25.7; HRMS: Calc’d for C19H30O5: 338.2093. Meas: 333.2087.
(2R,3S,5R)-2,3-Dihydroxy-5-(4-methoxybenzyl hydroxy)-(2-methoxyphenyl)oct-7-en-1-one (48)
(R,R)-Prophenol ligand (32) (4.0 mg, 0.00625 mmol) was placed under nitrogen and 320 μL of THF was added. Diethylzinc (13 μL, 0.0125 mmol, 1.0 M in THF) was added dropwise and stirred for 15 min. Separately, powdered molecular sieves (4Å, 50 mg) and 2-hydroxy-2′-methoxyacetophenone (33) (54.0 mg, 0.325 mmol) were placed into a vial and placed under nitrogen. The aldehyde (57.0 mg, 0.250 mmol) in 320 μL of THF was added in one portion. The mixture was cooled to 0 °C and the catalyst solution was added via syringe. The reaction mixture was then stirred at room temperature for 12 h. 1M HCl was added, and the mixture was extracted with DCM twice, the combined organic extracts were washed with brine, dried (MgSO4) and evaporated. Column chromatography eluting with 4:1 to 2:1 petroleum ether/EtOAc provided 53 mg (52%) of the title compound as a white solid. mp = 70–71 °C; (c 1.0 CHCl3); IR (neat): ν 3452, 3075, 3008, 2936, 2915, 2839, 1667, 1599, 1533, 1514, 1487, 1465, 1438, 1395, 1293, 1246, 1179, 1076, 1035; 1H NMR (400 MHz, CDCl3) δ 7.88 (dd, J = 1.8, 5.9, 1H), 7.53–7.58 (m, 1H), 7.06–7.10 (m, 3H), 6.91 (d, J = 7.9, 1H), 5.79–5.89 (m, 2H), 5.07–5.14 (m, 2H), 4.96 (d, J = 1.8, 1H), 4.53 (d, J = 10.8, 1H), 4.31 (d, J = 10.8, 1H), 4.20 (m, 1H), 4.09–4.13 (m, 1H), 3.89 (d, J = 8.7, 1H), 3.78 (s, 3H), 3.73 (s, 3H), 2.29–2.45 (m, 2H), 2.06 (d, J = 9.8, 1H), 1.73 (dddd, J = 1.7, 2.5, 2.5, 7.2, 1H), 1.25 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 200.9, 158.7, 134.5, 131.5, 130.7, 129.4, 123.8, 121.2, 117.4, 113.6, 111.6, 79.9, 75.5, 71.3, 68.4, 55.6, 55.2, 40.3, 38.7; HRMS (EI, M++Na) Calcd. for C23H28O6 423.1784, found 423.1780; Elemental Anal. calc. C23H28O6, C 68.98, H 7.05, found C 68.90, H 7.08.
(2R,3S,5R)-1-(2-ethyl-1H-pyrrol-1-yl)-2,3-dihydroxy-5-(methoxymethoxy)oct-7-en-1-one (66)
(R,R)-Prophenol ligand 33 (30 mg, 0.047 mmol) was placed under argon and THF (500 μL) was added. Diethylzinc (1.0 M in hexanes, 95 μL, 0.095 mmol) was added dropwise at rt and the reaction mixture was stirred for 20 min to give the dinuclear zinc catalyst as a yellow solution. Separately, powdered molecular sieves (4Å, 50 mg) were placed into a flame-dried flask followed by acyl pyrrole 55 (63 mg, 0.411 mmol) and the flask was placed under argon. Aldehyde 65 (50 mg, 0.316 mmol) in THF (500 μL) was then added to the acyl pyrrole/molecular sieves mixture in one portion. The resulting mixture was stirred vigorously and the catalyst was added dropwise via syringe. After stirring at rt for 12 hours, the reaction mixture was hydrolyzed by adding a 1M aqueous solution of HCl (2 mL) and the mixture was extracted with CH2Cl2 (3x). The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. Purification of the residue by chromatography on silica gel (petroleum ether/EtOAc: 9:1 to 7:3) provided 1,2-diol 66 as a mixture of two inseparable diastereoisomers (52.1 mg, 51%) in an 10:1 ratio as determined by the ratio of the NMR peaks at δ 6.97 and 7.06 ppm (brown solid). mp = 54 °C; (c 0.75, CHCl3); IR (neat): ν 3375, 2928, 1726, 1641, 1502, 1450, 1420, 1377, 1322, 1252, 1147, 1125, 1094, 1040, 914, 872, 813, 704 cm−1; 1H NMR (400 MHz, CDCl3): δ 6.97 (dd, J = 3.6, 1.6 Hz, 1H), 6.20 (t, J = 3.6 Hz, 1H), 6.04 (m, 1H), 5.76 (ddt, J = 16.8, 10.4, 7.2 Hz, 1H), 5.12-5.06 (m, 2H), 4.66 (dd, J = 7.6, 1.6 Hz, 1H), 4.63 (d, J = 6.8 Hz, 1H), 4.61 (d, J = 6.8 Hz, 1H), 4.23 (m, 1H), 3.85 (m, 1H), 3.69 (d, J = 7.6 Hz, OH), 3.27 (s, 3H), 2.97-2.90 (m, 2H+OH), 2.41-2.27 (m, 2H), 1.97 (ddd, J = 14.8, 10.8, 2.8 Hz, 1H), 1.70 (ddd, J = 14.8, 9.6, 2.8 Hz, 1H), 1.20 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 172.5, 140.1, 134.1, 119.1, 118.2, 112.9, 111.4, 96.8, 75.3, 74.0, 69.4, 56.0, 39.8, 38.5, 22.8, 13.1; HRMS (ESI): Calcd. for C16H25NO5Na[M + Na]+: 334.1620, found: 334.1630.
(R)-4-(benzyloxy)-2-((S)-2-methylpent-4-ynyl)-3,4-dihydro-2H-pyran (12)
To a solution of 13 (438 mg, 1.62 mmol) in dry, degassed DMF (15 mL) in a Schlenk flask was added a mixture of [Rh(COD)Cl]2 (64 mg, 0.081 mmol) and bidentate phosphine 79 (142 mg, 0.324 mmol). Then, the Schlenk flask was purged with argon, closed and was immersed in a preheated oil bath (85 °C). After 5 hours, the reaction mixture was cooled to rt, diluted with diethyl ether, and washed with water. The aqueous layer was extracted with ether, and the combined organic layers were dried over MgSO4. After filtration, the filtrate was concentrated under vacuum, and the residue was purified by flash column chromatography on deactivated silica (petroleum ether/EtOAc: 30/1) to give 243 mg (55%) of 12 as a colorless oil. IR (neat): ν 3301, 2926, 1640, 1243, 1081 cm−1; 1H NMR (400 MHz, CDCl3): δ 7.37-7.27 (m, 5H), 6.54 (d, J = 6.1 Hz, 0.9H), 6.39 (d, J = 6.3 Hz, 0.1H), 4.99 (ddd, J = 6.1, 5.3, 2.0 Hz, 0.9H), 4.86 (dt, J = 6.3, 1.9 Hz, 0.1H), 4.76 (ABq, J = 11.7, Δν = 41.8 Hz, 0.2H), 4.57 (ABq, J = 11.9, Δν = 31.3 Hz, 1.8H), 4.09-4.02 (m, 1H), 3.85 (deformed ddd, J = 5.3, 4.1, 1.8 Hz, 1.8H), 2.22 (ddd, J = 16.6. 6.1. 2.7 Hz, 0.9H), 2.16 (ddd, J = 16.6, 6.3, 2.7 Hz, 0.9H), 2.07-2.00 (m, 1H), 1.98( t, J = 2.7 Hz, 0.9H), 1.95 (ddd, J = 14.3, 3.7, 1.8 Hz, 0.9H), 1.79 (ddd, J = 14.0, 9.8, 4.7 Hz, 0.9H), 1.58 (ddd, J = 14.3, 12.0, 4.1 Hz, 0.9H), 1.36 (ddd, J = 14.0, 9.5, 3.5 Hz, 0.9H), 1.05 (d, J = 6.6 Hz, 2.7H), 1.02(d, J = 6.7 Hz, 0.3H); 13C NMR (100 MHz, CDCl3): δ 147.3, 138.8, 128.4, 127.7, 127.6, 127.5, 100.2, 82.9, 69.4, 69.4, 66.4, 41.3, 34.8, 28.6, 26.5, 18.9; HRMS (ESI): Calcd. for C11H15O2 [M–PhCH2]: 179.1072. Found: 179.1071.
Acknowledgments
We thank the General Medical Sciences Institute of NIH (GM 33049) for their generous support of our programs. We thank Johnson-Matthey for their gifts of palladium and ruthenium salts and Aldrich for a generous supply of the ProPhenol ligand. W. M. S. thanks the National Institutes of Health for a post-doctoral fellowship. C. K. C. thanks Eli Lilly & Co. for a graduate fellowship.
References
- 1.Trost BM, Amans D, Seganish WM, Chung CK. J Am Chem Soc. 2009;131:17087. doi: 10.1021/ja907924j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Quinoa E, Kakou Y, Crews P. J Org Chem. 1988;53:3642. [Google Scholar]; (b) Corley DG, Herb R, Moore RE, Scheuer PJ, Paul VJ. J Org Chem. 1988;53:3644. [Google Scholar]
- 3.Jefford CW, Bernardinelli G, Tanaka JI, Higa T. Tetrahedron Lett. 1996;37:159. [Google Scholar]
- 4.Mooberry SL, Tien G, Hernandez AH, Plubrukarn A, Davidson BS. Cancer Res. 1999;59:653.for a review see: Gallagher BM. Curr Med Chem. 2007;14:2959. doi: 10.2174/092986707782794014.
- 5.(a) Pryor DE, O’Brate A, Bilcer G, Diaz JF, Wang YF, Wang Y, Kabaki M, Jung MK, Andreu JM, Ghosh AK, Giannakakou P, Hamel E. Biochemistry. 2002;41:9109. doi: 10.1021/bi020211b. [DOI] [PubMed] [Google Scholar]; (b) Gaitanos TN, Buey RM, Diaz FJ, Northcote PT, Teesdale-Spittle P, Andreu JM, Miller JH. Cancer Res. 2004;64:5063. doi: 10.1158/0008-5472.CAN-04-0771. [DOI] [PubMed] [Google Scholar]; (c) Hamel E, Day BW, Miller JH, Jung KM, Northcote PT, Ghosh AK, Curran PD, Cushman M, Nicolaou KC, Paterson I, Sorensen EJ. Mol Pharmacol. 2006;70:1555. doi: 10.1124/mol.106.027847. [DOI] [PubMed] [Google Scholar]; (d) Gapud EJ, Bai R, Ghosh AK, Hamel E. Pharmacology. 2004;66:113. doi: 10.1124/mol.66.1.113. [DOI] [PubMed] [Google Scholar]
- 6.Bennett MJ, Barakat K, Huzil T, Tuszynski J, Schriemer DC. Chem Bio. 2010;17:725. doi: 10.1016/j.chembiol.2010.05.019. [DOI] [PubMed] [Google Scholar]
- 7.Ghosh AK, Wang Y. J Am Chem Soc. 2000;122:11027. doi: 10.1021/ja0027416.Ghosh AK, Wang Y, Kim JT. J Org Chem. 2001;66:8973. doi: 10.1021/jo010854h.Mulzer J, Öhler E. Angew Chem Int Ed. 2001;40:3842.Paterson I, De Savi C, Tudge M. Org Lett. 2001;3:3149. doi: 10.1021/ol010150u.Enev VS, Kaehlig H, Mulzer J. J Am Chem Soc. 2001;123:10764. doi: 10.1021/ja016752q.Wender PA, Hedge SG, Hubbard RD, Zhang L. J Am Chem Soc. 2002;124:4956. doi: 10.1021/ja0258428.Crimmins MT, Stanton MG, Allen SP. J Am Chem Soc. 2002;124:5958. doi: 10.1021/ja026269v.Williams DR, Liang M, Mullins RJ, Stites RE. Tetrahedron Lett. 2002;43:4841.Nelson SG, Cheung WS, Kassick AJ, Hilfiker MA. J Am Chem Soc. 2002;124:13654. doi: 10.1021/ja028019k.Ahmed A, Hoegenauer EK, Enev VS, Hanbauer M, Kaehlig H, Öhler E, Mulzer J. J Org Chem. 2003;68:3026. doi: 10.1021/jo026743f.Gallagher BM, Fang FG, Johannes CW, Pesant M, Tremblay MR, Zhao H, Akasaka K, Li XY, Liu J, Littlefield BA. Bioorg Med Chem Lett. 2004;14:575. doi: 10.1016/j.bmcl.2003.12.001.Uenishi J, Ohmi M. Angew Chem Int Ed. 2005;44:2756. doi: 10.1002/anie.200500029.Gollner A, Altmann KH, Gertsch J, Mulzer J. Chem Eur J. 2009;15:5979. doi: 10.1002/chem.200802605.Gollner A, Altmann KH, Gertsch J, Mulzer J. Tetrahedron Lett. 2009;50:5790.For a review see: Mulzer J, Ohler E. Chem Rev. 2003;103:3753. doi: 10.1021/cr940368c.
- 8.Payne GB. J Org Chem. 1962;27:3819. [Google Scholar]
- 9.Trost BM, Machacek MR, Faulk BD. J Am Chem Soc. 2006;128:6745. doi: 10.1021/ja060812g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trost BM, Rhee YH. J Am Chem Soc. 2003;125:7482. doi: 10.1021/ja0344258. [DOI] [PubMed] [Google Scholar]
- 11.Ferrier RJ. Glyoscience: Epimerisation, Isomerisation and Rearrangement Reactions of Carbohydrates. Vol. 215. Springer-Verlag Berlin; Berlin: 2001. p. 153. [Google Scholar]
- 12.Barton DHR, McCombie SW. J Chem Soc Perkin Trans 1. 1975:1574. [Google Scholar]
- 13.Both allylzinc and allylindium reagents gave the same diastereoselectivity. Chattopadhyay A. J Org Chem. 1996;61:6104. doi: 10.1021/jo9604696.Binder WH, Prenner RH, Schmid W. Tetrahedron. 1994;50:749.
- 14.For a review, see: Trost BM, Toste FD, Pinkerton AB. Chem Rev. 2001;101:2067. doi: 10.1021/cr000666b.
- 15.Trost BM, Papillon JPN, Nussbaumer T. J Am Chem Soc. 2005;127:17921. doi: 10.1021/ja055967n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Trost BM, Older CM. Organometallics. 2002;21:2544. [Google Scholar]
- 17.Disfavoring β-hydride elimination by a structural constraint or coordinative saturation has been studied. For examples see Zhang L, Zetterberg K. Organometallics. 1991;10:3806.Kayaki Y, Shimizu I, Yamamoto A. Organometallics. 1994;13:3517.
- 18.Trost BM, VanVranken DL. Chem Rev. 1996;96:395. doi: 10.1021/cr9409804.A similar stereospecific palladium-catalyzed cyclization was reported by Lee and Hansen: Hansen EC, Lee D. Tetrahedron Lett. 2004;45:7151.
- 19.For a selected set of conditions that were explored, see: Kaneko S, Yoshino T, Katoh T, Terashima S. Tetrahedron. 1998;54:5471.Chen YJ, Chang WH. J Org Chem. 1996;61:2536.Schostarez H, Paquette LA. J Am Chem Soc. 1981;103:722.Kitano Y, Chiba K, Tada M. Synlett. 1999:288.Grieco PA, Marinovic N. Tetrahedron Lett. 1978:2545.Corey EJ, Suggs JW. J Org Chem. 1973;38:3224.Chen MS, Prabagaran N, Labenz NA, White MC. J Am Chem Soc. 2005;127:6970. doi: 10.1021/ja0500198.Carless HAJ, Haywood DJ. J Chem Soc Chem Comm. 1980:980.Sen A, Lai TW. Inorg Chem. 1984;23:3257.Oltvoort JJ, Vanboeckel CAA, Dekoning JH, Vanboom JH. Synthesis. 1981:305.Frauenrath H, Runsink J. J Org Chem. 1987;52:2707.Masilamani D, Manahan EH, Vitrone J, Rogic MM. J Org Chem. 1983;48:4918.Markovic D, Vogel P. Angew Chem Int Ed. 2004;43:2928. doi: 10.1002/anie.200453897.
- 20.Examples include: PPTS, CSA, TfOH, HI, LDA, Rh(PPh3)2Cl, Pd/C, Ru(PPh3)3H2, [Ir(COD)Cl]2.
- 21.Pappo R, Allen DS, Lemieux RU, Johnson WS. J Org Chem. 1956;21:478. [Google Scholar]
- 22.Abad A, Agullo C, Arno M, Domingo LR, Zaragoza RJ. Tetrahedron Lett. 1986;27:3289. [Google Scholar]
- 23.(a) Herbert JM. Tetrahedron Lett. 2004;45:817. [Google Scholar]; (b) Marshall JA, Van Devender EA. J Org Chem. 2001;66:8037. doi: 10.1021/jo016048s. [DOI] [PubMed] [Google Scholar]
- 24.Martinelli MJ, Nayyar NK, Moher ED, Dhokte UP, Pawlak JM, Vaidyanathan R. Org Lett. 1999;1:447. [Google Scholar]
- 25.Huynh C, Derguiniboumechal F, Linstrumelle G. Tetrahedron Lett. 1979:1503. [Google Scholar]
- 26.(a) Julia M, Paris JM. Tetrahedron Lett. 1973:4833. [Google Scholar]; (b) Blakemore PR, Cole WJ, Kocieński PJ, Morley A. Synlett. 1998:26. [Google Scholar]
- 27.Leading reference: Trost BM, Ito H. J Am Chem Soc. 2000;122:12003.
- 28.(a) Sasmal S, Geyefr A, Maier ME. J Org Chem. 2002;67:6260. doi: 10.1021/jo025889b. [DOI] [PubMed] [Google Scholar]; (b) Honda T, Namiki H, Kaneda K, Mizutani H. Org Lett. 2004;6:87. doi: 10.1021/ol0361251. [DOI] [PubMed] [Google Scholar]
- 29.Molander GA, Harris CR. J Org Chem. 1997;62:2944. doi: 10.1021/jo9700235. [DOI] [PubMed] [Google Scholar]
- 30.(a) Marmsăter FP, Vanecko JA, West FG. Org Lett. 2004;6:1657. doi: 10.1021/ol049493t. [DOI] [PubMed] [Google Scholar]; (b) Maiti KK, Jeon OY, Lee WS, Chung SK. Chem Eur J. 2007;13:762. doi: 10.1002/chem.200600898. [DOI] [PubMed] [Google Scholar]
- 31.Kim H, Lee C. Org Lett. 2002;4:4369. doi: 10.1021/ol027104u. and references therein. [DOI] [PubMed] [Google Scholar]
- 32.Takacs JM. In: Comprehensive Organometallic Chemistry II. Abel EW, Stone FGA, Wilkinson G, Hegedus LS, editors. Vol. 12. Elsevier Science; New York: 1995. pp. 814–817.For early examples, see: Takahashi K, Miyake A, Hata G. Bull Chem Soc Jpn. 1972;45:230.Stork G, Poirier JM. J Am Chem Soc. 1983;105:1073.Stanton SA, Felman SW, Parkhurst CS, Godleski SA. J Am Chem Soc. 1983;105:1964.Keinan E, Roth Z. J Org Chem. 1983;48:1172.Keinan E, Sahai M, Roth Z, Nudelman A, Herzig J. J Org Chem. 1985;50:3558.
- 33.For reviews see: Nolan SP, Clavier H. Chem Soc Rev. 2010;39:3305. doi: 10.1039/b912410c.Vougioukalakis GC, Grubbs RH. Chem Rev. 2010;110:1746. doi: 10.1021/cr9002424. and references therein.
- 34.For a recent review, see: Trost BM, Brindle CS. Chem Soc Rev. 2010;39:1600. doi: 10.1039/b923537j.
- 35.Trost BM, Ito H, Silcoff E. J Am Chem Soc. 2001;123:3367. doi: 10.1021/ja003871h. [DOI] [PubMed] [Google Scholar]
- 36.Yoshikawa N, Kumagai N, Matsunaga S, Moll G, Ohshima T, Suzuki T, Shibasaki M. J Am Chem Soc. 2001;123:2466. doi: 10.1021/ja015580u.Kumagai N, Matsunaga S, Yoshikawa N, Ohshima T, Shibasaki M. Org Lett. 2001;3:1539. doi: 10.1021/ol015878p.Yoshikawa N, Suzuki T, Shibasaki M. J Org Chem. 2002;67:2556. doi: 10.1021/jo0162538.Kumagai N, Matsunaga S, Kinoshita T, Harada S, Okada S, Sakamoto S, Yamaguchi K, Shibasaki M. J Am Chem Soc. 2003;125:2169. doi: 10.1021/ja028926p.for a review see: Matsunaga S, Shibasaki M. Chem Soc Rev. 2006;35:269. doi: 10.1039/b506346a.
- 37.For a review see: Renz M, Meunier B. Eur J Org Chem. 1999:737.
- 38.Tatsumi T. In: Beckmann Rearrangement. Sheldon RA, Bekkum H, editors. Weinheim, Wiley-VCH; New York: 2001. p. 185. [Google Scholar]
- 39.Racherla US, Brown HC. J Org Chem. 1991;56:401. [Google Scholar]
- 40.Ellison RA, Lukenbach ER, Chiu CW. Tetrahedron Lett. 1975:499. [Google Scholar]
- 41.For studies on the reactivity of acyl pyrroles, see: Evans DA, Borg G, Scheidt K. Angew Chem Int Ed. 2002;41:3188. doi: 10.1002/1521-3773(20020902)41:17<3188::AID-ANIE3188>3.0.CO;2-H.
- 42.(a) Kinoshita T, Okada S, Park SR, Matsunaga S, Shibasaki M. Angew Chem Int Ed. 2003;42:4680. doi: 10.1002/anie.200352509. [DOI] [PubMed] [Google Scholar]; (b) Matsunaga S, Kinoshita T, Okada S, Harada S, Shibasaki M. J Am Chem Soc. 2004;126:7559. doi: 10.1021/ja0485917. [DOI] [PubMed] [Google Scholar]; (c) Mita T, Sasaki K, Kanai M, Shibasaki M. J Am Chem Soc. 2005;127:514. doi: 10.1021/ja043424s. [DOI] [PubMed] [Google Scholar]
- 43.Evans DA, Johnson DS. Org Lett. 1999;1:595. doi: 10.1021/ol990113r. [DOI] [PubMed] [Google Scholar]
- 44.Evans DA, Scheidt KA, Johnson JN, Willis MC. J Am Chem Soc. 2001;123:4480. doi: 10.1021/ja010302g. [DOI] [PubMed] [Google Scholar]
- 45.Harada S, Handa S, Matsunaga S, Shibasaki M. Angew Chem Int Ed. 2005;44:4365. doi: 10.1002/anie.200501180. [DOI] [PubMed] [Google Scholar]
- 46.Jin M, Taylor RE. Org Lett. 2005;7:1303. doi: 10.1021/ol050070g. [DOI] [PubMed] [Google Scholar]
- 47.Tinsley JM, Roush WR. J Am Chem Soc. 2005;127:10818. doi: 10.1021/ja051986l. [DOI] [PubMed] [Google Scholar]
- 48.Trost BM. Science. 1991;254:1417. doi: 10.1126/science.1962206. [DOI] [PubMed] [Google Scholar]
- 49.For several recent reviews of metal-catalyzed methods for the formation of 5- and 6-membered rings through carbon-heteroatom bond forming reactions: Majumdar KC, Roy B, Debnath P, Taher A. Curr Org Chem. 2010;14:846.Larrosa I, Romea P, Urpi F. Tetrahedron. 2008;64:2683.
- 50.(a) Trost BM, Rhee YH. J Am Chem Soc. 2003;125:7482. doi: 10.1021/ja0344258. [DOI] [PubMed] [Google Scholar]; (b) Trost BM, Rhee YH. J Am Chem Soc. 2002;124:2528. doi: 10.1021/ja011840w. [DOI] [PubMed] [Google Scholar]; (c) Trost BM, Rhee YH. J Am Chem Soc. 1999;121:11680. [Google Scholar]
- 51.Morris WJ, Shair MD. Tetrahedron Lett. 2010;51:4310. doi: 10.1016/j.tetlet.2010.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.For recent reviews of metal vinylidenes in catalysis: Weidermann SH, Chulbom L. Metal Vinylidenes and Allenylidenes in Catalysis. 2008:279.Trost BM, McClory A. Chem Asian J. 2008;3:164. doi: 10.1002/asia.200.For information on the mechanism of alkyne to vinylidene transformation: Wakasuki Y. J Organomet Chem. 2004;689:4092.
- 53.Frick JA, Klassen JB, Bathe A, Abramson JM, Rapoport H. Synthesis. 1992:621. [Google Scholar]
- 54.(a) Mueller S, Liepold B, Roth GJ, Bestmann HJ. Synlett. 1996:521. [Google Scholar]; (b) Ohira S. Synth Commun. 1989;19:561. [Google Scholar]
- 55.For Rh(I)-catalyzed dimerization of terminal alkynes, see: Schmidt HJ, Singer H. J Organomet Chem. 1978;153:165.
- 56.For the preparation and use of these types of stannanes, see: Marshall JA. Chem Rev. 1996;96:31. doi: 10.1021/cr950037f.