

Abstract
In this manuscript, we report the full account of our efforts to couple the northern and the southern building blocks, whose synthesis were described in the preceding paper, along with the modifications required which ultimately lead to a successful synthesis of laulimalide. Key highlights include an exceptionally efficient and atom-economical intramolecular ruthenium-catalyzed alkene-alkyne coupling to build the macrocycle followed by a highly stereoselective 1,3-allylic isomerization promoted by a rhenium complex. Interestingly, the designed synthetic route also allowed us to prepare an analogue of the natural product that possesses significant cytotoxic activity. We also report in this paper a second generation route which provided a more concise synthesis of the natural product.
Keywords: total synthesis, laulimalide, ruthenium catalysis, rhodium catalysis, vinylidene complex, alkene-alkyne coupling
Introduction
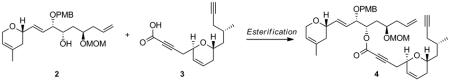
In the preceding paper, we described the synthesis of the two required fragments 2 and 3 for our synthesis of laulimalide (1). The preparation of these fragments highlights the use of our dinuclear-zinc catalyst I to install the two contiguous stereogenic centers bearing hydroxy groups in a syn-relationship on the northern fragment 2 and the utilization of a rhodium-catalyzed cycloisomerization to elaborate the dihydropyran moiety of the southern fragment 3 (Scheme 1). In this paper, we report efforts to utilize these building blocks for the synthesis of laulimalide and the modifications in the southern fragment needed in order to complete the synthesis. In addition, we report a second generation route that shortens the overall synthesis.
Scheme 1.

Completed Northern and Southern Fragments
Results and discussion
At this juncture, we were in a favorable position to explore the coupling between the two fragments 2 and 3 in order to obtain enyne 4, thus setting the stage for the crucial macrocyclisation step via an intramolecular ruthenium-catalyzed alkene-alkyne coupling (4→5; Scheme 2).
Scheme 2.

Proposed Esterification and Intramolecular Alkene-Alkyne Coupling
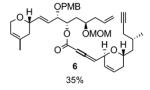
Although this esterification seemed to be a priori a trivial process, the transformation proved to be synthetically challenging as outlined in Table 1. Classical esterifications method such as Steglich conditions[i] (DCC/DMAP) and related protocols, or conversion of the carboxylic acid into an acyl chloride[ii] or acyl fluoride[iii] followed by addition of alcohol 2 all failed to give the desired ester 4. The use of the Ghosez reagent[iv] (entry 1) proved to be efficient in providing a coupling product, which in fact turned out to be exclusively allene 6 and no trace of the desired coupling product 4 was observed. We believed that acidic conditions would be best suited as it would prevent formation of the allene. We therefore attempted the esterification using Kita conditions (entry 2),[v] which unfortunately did not provide the desired ester 4. Transesterification between the methyl ester of 3 and alcohol 2 in the presence of Otera’s catalyst[vi] provided the desired ester 4, albeit in low yield (entry 3). The use of the Shiina mixed anhydride[vii] (entry 4) was disappointing since it only yielded traces of coupling product as judged by TLC. The best result was obtained by using the Yonemitsu modification[viii] of the Yamaguchi protocol[ix] (entry 5), which yielded 18% of ester 4.
Table 1.
Attempts at Esterification
| Entry | Conditions | Results |
|---|---|---|
| 1 |
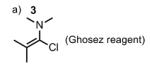
|
|
| b) 2, Et3N, CH2Cl2 | 35% | |
| 2 |

|
Ethoxyvinylester formed (TLC) No esterification |
| b) 2, CSA (10 mol%) DCE |
||
| 3 |
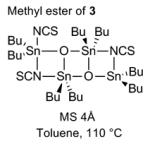
|
13% |
| 4 |
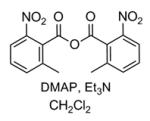
|
Traces of 4 by TLC |
| 5 | 2,4,6-trichlorobenzoylchloride Et3N, DMAP, THF |
18% |
Despite the lack of an efficient protocol to achieve this esterification, these disappointing attempts allowed us to collect sufficient material to investigate the crucial ruthenium-catalyzed alkene-alkyne coupling reaction. Gratifyingly, exposure of enyne 4 to 10 mol% of [CpRu(CH3CN)3]PF6 in acetone at 50 °C resulted in the stereo-, regio- and chemoselective formation of the desired branched 1,4-diene 5 as a single diastereoisomer in 36% yield (not optimized). According to 1H NMR analysis of the crude reaction mixture, which revealed the complete consumption of the starting material and the nearly exclusive formation of the desired product, we believed that this transformation occured in a much higher yield (Scheme 3).
Scheme 3.

Intramolecular Ru-Catalyzed Alkene-Alkyne Coupling
At this point of our synthesis, the frustrating esterification issue clearly needed to be addressed since our ruthenium-catalyzed strategy to form the macrocycle had proven to be viable. We reasoned that a slight modification of our strategy could help us reach our goal. Accordingly, the (Z)-alkene at C2-C3 could be installed by performing an intermolecular Still-Gennari olefination between the β-ketophosphonate 8 and aldehyde 10, both fragments in principle readily available from the previously synthesized compounds 2 and 9 respectively (Scheme 4).
Scheme 4.

Revised Retrosynthetic Analysis
The coupling between the previously obtained alcohol 2 and carboxylic acid 7 under Yamaguchi conditions proceeded uneventfully to give the required β-ketophosphonate 8 in near quantitative yield. It is believed that the facile acylation of sterically hindered alcohol 2 involves the in-situ formation of a more reactive ketene intermediate from phosphonoacetic acid 7 facilitated by the presence of a strongly electron-withdrawing substituent in the α-position of the carbocylic acid group.[x] The required aldehyde 10 was successfully obtained via a highly diastereoselective Ferrier-type addition of (tert-butyldimethylsilylvinyl)ether onto vinylogous acetal 9 in the presence of Montmorillonite K-10 (82% yield),[xi] with the required anti-relationship for the dihydropyran moiety as confirmed by the absence of an nOe between the two protons H5 and H9 (Scheme 5).
Scheme 5.

Synthesis of β-Ketophosphonate 8 and Aldehyde 10
With the two required fragments in hand, the Still-Gennari olefination could be implemented (Scheme 6). Condensation of the potassium salt of phosphonate 8 with aldehyde 10, in the presence of 18-crown-6, gave rise to the formation of the desired alkene 11 as a (1/5) mixture of (E/Z)-isomers, which could be easily separated by flash chromatography on silica gel. To our delight, the subsequent key intramolecular alkene-alkyne coupling proceeded extraordinarily well. Indeed, treatment of enyne 11 with 10 mol% of [CpRu(CH3CN)3]PF6 in acetone at 50 °C generated the desired branched 1,4-diene 12 in a spectacular 99% yield! More strikingly, only 15 minutes were required to transform enyne 11 into macrolactone 12 in a chemo-, regio- and stereoselective fashion. Importantly, exclusive formation of the E-stereoisomer of the newly formed C15-C16 allylic double bond within the macrocyle was observed according to 1H NMR analysis (JH15-H16 = 16 Hz) which would set the stage for the subsequent installation of the required trans-epoxide. Noteworthy, no isomerization of the (Z)-alkene at C2-C3 could be detected as judged by 1H NMR spectroscopy. The core reaction is in addition highly atom economical, and the solvent is easily recoverable and can be recycled since the work-up involves as the first step a simple removal of the solvent by evaporation which makes the process reasonably eco-friendly.
Scheme 6.

Still-Gennari Olefination and Ru-Catalyzed Alkene-Alkyne Coupling
This intramolecular alkene-alkyne coupling deserves some attention as to its mechanistic rationale. It has been previously stipulated that coordination of the ruthenium catalyst to both the alkene and the alkyne (A) would lead to an oxidative cylisation giving rise to the ruthenacyclopentene B, which in turn would undergo a syn β-hydride elimination to yield the vinylruthenium species C (Scheme 7).[xii] Reductive elimination of the latter would ultimately furnish the branched 1,4-diene 12 with regeneration of the [CpRu(CH3CN)3]PF6 catalyst. It is worth mentioning that in this case, the coordination and subsequent cyclization occurs such that the group attached to the terminal alkyne is opposite to the ruthenium thus giving rise exclusively to the desired branched 1,4-diene 12. In the path to such branched products, steric interactions occur during the C-C bond-forming event and cyclisation leads to the kinetically formed ruthenacycle B. As it can be assumed that the ruthencyclopentene formation is fast and reversible,[xii] the product formation is only determined at the β-hydride elimination step, which is presumably slower than the ruthenacyclopentene formation. As β-hydride elimination is favored in the path toward branched product, the latter is generally favored with monosubstituted alkenes and terminal alkynes which explains the regioselectivity obtained in our case.[xii] The formation of highly functionalized macrocycles which highlights the chemoselectivity has also been illustrated in the case of the synthesis of amphidinolide A by our group, albeit with lower efficiency.[xiii] The synthesis of pinnatoxin A[xiv] through this method further illustrates the power of the Ru-catalyzed coupling reaction which wherein the alkene-alkyne macrocyclization proceeded in 79% yield represents a new opportunity to form macrocycles in an atom economical fashion.
Scheme 7.

Proposed Catalytic Cycle for the Ru-Catalyzed Alkene-Alkyne Coupling
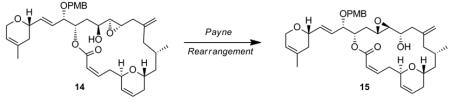
With macrocycle 12 in hand, we were in a favorable position to tackle the envisaged Payne rearrangement. To this end, deprotection of the MOM protecting group proceeded smoothly in tert-BuOH in the presence of PPTS, to furnish allylic alcohol 13 in 66% yield. A hydroxyl-directed diastereoselective Sharpless epoxidation[xv] of the latter in the presence of (+)-DET ultimately furnished the trans-epoxide 14 as a single diastereoisomer (Scheme 8). The stage was now set to probe the challenging Payne rearrangement, which should in principle, after deprotection of the PMB group, lead to the natural product laulimalide.
Scheme 8.

Synthesis of the Epoxyalcohol Precursor of Laulimalide
Various conditions were investigated to promote the formation of the Payne-rearranged product 15 as summarized in Table 2. In the initial report of Payne,[xvi] it is described that the epoxide migration works best using sodium hydroxide in an aqueous solution. Interestingly, under these conditions and after one hour at room temperature, neither saponification of the lactone nor isomerization of the (Z)-alkene were observed, but neither was any of the desired Payne rearrangement product. An alternative approach involved the formation of the trimethylsilyl ether of the starting epoxyalcohol 14 upon exposure to bis-(trimethylsilyl)trifluoroacetamide (BSTFA), followed by treatment with a fluoride source such as tris(dimethylamino)sulfonium difluorotrimethylsilicate (TAS-F) (Entry 3) also failed to generate any Payne rearranged product. Finally, epoxyalcohol 14 was exposed to sodium hydride in THF at 0 °C, which resulted in the formation of a new product by TLC. However spectroscopic analysis (1H NMR, COSY), indicated the formation of the ring-contracted lactone 16, in 50% yield (95% brsm), resulting from an intramolecular attack of the alkoxy group α to the epoxide onto the ester functionality (Table 2). This last result discouraged us from pursuing this transformation since the formation of the ring-contracted lactone seemed to be thermodynamically favored over the formation of the desired translocated epoxyalcohol 15 under basic conditions.
Table 2.
Attempts at Payne Rearrangement
| Entry | Conditions | Results |
|---|---|---|
| 1 |
tert-BuOk tert-BuOH, rt, 20min |
Decomposition |
| 2 | NaOH, H2O Et2O, rt, 1h |
No rearrangement |
| 3 |
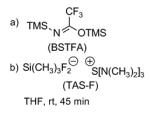
|
Starting Material + unidentified product |
| 4 | NaH, THF 0 °C, 30 min |
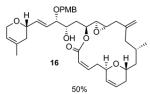
|
Being unable to perform the epoxide translocation as planned in our retrosynthetic analysis necessitated the developement of a new strategy that would allow us to obtain laulimalide. We anticipated that a selective 1,3-isomerization of the previously prepared allylic alcohol 13 would give rise to the rearranged allylic alcohol 17, allowing us to access laulimalide 1 (Scheme 9).
Scheme 9.

1,3-Allylic Transposition: A Revised Strategy
A number of chemical transformations that, in principle, can provide the rearranged allylic alcohol 17 from 13 formally through a 1,3-allylic transposition have been described in the literature.[xvii] We first resorted to a secondary allylic selenoxide to perform the above-mentioned transformation since, after exposure of the allylic alcohol 13 to a phenylselenol under Mitsunobu-type conditions, the allyl selenide 18 would be obtained with inversion of configuration (Scheme 10).[xviii] Oxidation of the phenyl selenide 18 to the corresponding selenoxide 19 should set the stage for a [2,3]-sigmatropic shift leading to the desired rearranged allylic alcohol 17. Unfortunately, using standard Mitsunobu-type conditions (2-nitrophenylselenol, trimethyl or tributylphosphine, THF) we were not able to convert allylic alcohol 13 into the corresponding selenide product 18 and the starting material was recovered.
Scheme 10.

1,3-Allylic Transposition Using an Allylic Selenoxide
It has been shown that the isomerization of allylic alcohols by 1,3-transposition of the hydroxy group could be catalyzed by a number of high oxidation state transition metal oxo complexes, such as vanadium, tungsten, molybdenum and rhenium.[xix] Rhenium oxo-catalysts display several advantages over the other metal complexes. They are active at low temperatures and do not undergo reduction at the metal center by the alcohol, which usually causes a loss of catalytic activity with time (case of Mo).[xix] Applying the rhenium oxo catalysis conditions developed by Osborn et al.[xx] and extensively utilized by Grubbs et al. to allylic alcohol 13,[xxi] which involve the highly active triphenylsilyl perrhenate complex O3ReOSiPh,[xxii] 3 resulted in the formation of the rearranged product 20 in good yield, with retention of configuration in the product (Scheme 11). We found that using one equivalent of the rhenium complex for 5 min in Et2O at -50 °C were the optimum conditions to obtain the rearranged allylic product 20 in 78% yield, easily separable by flash chromatography on silica gel from the starting material 13 (97% brsm). Inversion of the C15 stereogenic center using an oxidation/CBS[xxiii]-reduction sequence allowed us to obtain the desired epimeric allylic alcohol 17. Epoxidation of the allylic alcohol under Sharpless conditions followed by DDQ deprotection ultimately completes the synthesis of laulimalide 1. Spectral and physical data of the synthetic sample were in complete agreement with those reported in the literature for the natural product.[xxiv,xxv]
Scheme 11.

Completion of the Synthesis of Laulimalide
A potential improvement in the synthesis would be to use the epimer at C17 of allylic alcohol 13. This epimer should indeed directly provide intermediate 17 under the rhenium oxo-catalysis conditions described above and therefore provide a more efficient synthesis of laulimalide as it would avoid the oxidation/CBS-reduction sequence. To that end, the required β-ketophosphonate 28 was prepared using the same strategy as for the synthesis of its epimer 8, starting from the known homoallylic alcohol 21 (derived from commercially available (R)-tosylglycidol).[xxvi] Protection of the latter as a MOM-ether followed by cleavage of the 1,3-dithiane acetal under standard conditions furnished β-hydroxyaldehyde 22 (63% yield over 2 steps), setting the stage for the direct zinc-catalyzed aldol reaction using α-hydroxyacetyl 2-ethylpyrrole 23. Gratifyingly, the use of 15 mol% of (R,R)-dinuclear zinc catalyst I gave rise to the syn-1,2-diol 24 with a 10:1 dr, in 53% isolated yield. This result further demonstrates, as highlighted in the preceding paper, that the dinuclear zinc aldol reaction proceeds with catalyst control as the stereochemistry of the remote allylic alcohol protected as a MOM ether on substrate 22 has no influence on the stereochemical outcome of the obtained syn-1,2-diol 24. Protection of the latter 1,2-diol as a PMP acetal followed by cleavage of the acylpyrrole using sodium borohydride in THF afforded primary alcohol 25 in 70% yield (over 2 steps). Oxidation of 25 with Dess-Martin periodinane gave rise to the corresponding aldehyde, which was treated with the lithium salt of sulfone 26 in a THF/HMPA mixture to give the corresponding alkene as a single (E)-geometric isomer. Regioselective opening of the PMP acetal upon treatment with DIBAL-H provided the desired secondary alcohol 27. The coupling between alcohol 27 and carboxylic acid 7 under Yamaguchi conditions ultimately furnished the desired β-ketophosphonate 28 in excellent yield (Scheme 12).
Scheme 12.

Synthesis of β-ketophosphonate 28
Condensation of the potassium salt of phosphonate 28 with aldehyde 10, in the presence of 18-crown-6 gave rise to the formation of the desired alkene 29 as a (1/5) mixture of (E/Z)-isomers, which could be easily separated by flash chromatography on silica gel (Scheme 13). The subsequent intramolecular alkene-alkyne coupling proceeded again with exceptional efficiency, in the presence of 5 mol% of [CpRu(CH3CN)3]PF6 in acetone at 50 °C, to give almost instantaneously the desired branched 1,4-diene 30 in 98% yield. Deprotection of the MOM protecting group under mild acidic conditions finally provided the desired allylic alcohol 31. With the latter in hand, the crucial rhenium-catalyzed allylic transposition could be investigated. Allylic alcohol 31 was therefore exposed to the triphenylsilyl perrhenate catalyst O3-ReOSiPh3 under identical conditions as the ones used for its epimer 13 (Et2O at -50 °C for 5 min). To our dismay, the equilibrium between the two regioisomers 17 (product) and 31 (starting material) lay mainly towards the starting material 31 as judged 1H NMR analysis of the crude mixture (17/31 = 1/4) as prolonged reaction time did not improve this ratio further (Scheme 13). Furthermore, the two regioisomers 17 and 31 could not be separated by flash chromatography on silica gel. The separation was therefore performed using HPLC on an achiral column and successfully provided 16% of the desired allylic alcohol 17 (formal synthesis), along with 62% of recovered starting material 31 (50% brsm).
Scheme 13.

Completion of the Synthesis of Laulimalide via a Shorter Route
In the original isolation along with laulimalide 1 (IC50 = 7 nM), isolaulimalide 32 was also isolated but the latter possesses significantly diminished activity as compared to its congener (IC50 = 20 000 nM). It was shown that under mild acidic conditions laulimalide (1) was prone to furan formation through a SN2-type attack of the hydroxy group situated on the lateral chain at C20 onto the epoxide at C17, thus leading to isolaulimalide (32) (Scheme 14).
Scheme 14.

Acid-Catalyzed Conversion of Laulimalide into Isolaulimalide
This observation undoubtedly indicated that there is a need to synthesize new analogues designed in such a way that would prevent the furan formation, while at the same time retaining similar or even improved activity as compared with laulimalide. Our synthesis of laulimalide provided an opportunity to access a novel analogue. Accordingly, we performed the PMB-deprotection of compound 14 upon exposure to DDQ which led to the formation of laulimalide analogue 33 (Scheme 15). We were happy to see that our analogue displayed significant activity against Granta 519 and Jurkat cell lines with an IC of 200 nM and 182 nM respectively.[xxvii] 50 Even though this analogue displays inferior potency to that of laulimalide, it shows nevertheless a much better activity than isolaulimalide and possesses enhanced stability over the natural product laulimalide as furan formation is no longer possible.
Scheme 15.

Synthesis of a Potent Laulimalide Analogue
Conclusion
In conclusion, by using several atom-economic transformations such as a rhodium-catalyzed cycloisomerization reaction to form the endocyclic trans-dihydropyran, an enantio- and diastereoselective zinc-catalyzed glycolate aldol to prepare the syn-1,2-diol and an exceptionally efficient ruthenium-catalyzed alkene-alkyne coupling to build the macrocycle we were able to obtain a novel biologically active analogue of laulimalide 33. Remarkably, this analogue displayed nanomolar potency against some cancer cell lines. The application of a 1,3-allylic isomerization promoted by a rhenium complex within a complex setting ultimately allowed us to synthesize the natural product laulimalide 1. This work clearly demonstrates that the development of new methodologies within our laboratories allowed for a very efficient synthesis of laulimalide but also, conversely, that the inherent synthetic challenges arising from the natural product truly served as a springboard for the development of new methodologies. The latter point has been thoroughly illustrated in the preceding paper by the work conducted around the dinuclear zinc catalysis to form the syn-1,2 diol, using for the first time a donor partner at the carboxylic acid oxidation state.
Experimental Section
(3S,4S,6R,E)-3-(4-Methoxybenzyloxy)-6-(methoxymethoxy)-1-[(S)-4-methyl-3,6-dihydro-2H-pyran-2-yl]nona-1,8-dien-4-yl-2-[bis(2,2,2-trifluoroethoxy)phosphoryl]acetate (8)
Alcohol 2 (65 mg, 0.150 mmol) and 2-[bis(2,2,2-trifluoroethoxy)phosphoryl]acetic acid[xxviii] (105 mg, 0.346 mmol) were azeotroped together three times with toluene in a round bottom flask. THF (15 mL) was added and iPr2NEt (120 μL, 0.690 mmol) followed by 2,4,6-trichlorobenzoylchloride (68 μL, 0.435 mmol) were then sequentially added at rt. The resulting mixture was stirred for 10 min at rt. The solvent was removed in vacuo and the residue was dissolved in benzene (15 mL). 4-DMAP (126 mg, 1.035 mmol) was added in one portion at rt and the resulting white suspension was stirred for 2 hours. The mixture was then diluted with EtOAc and the resulting solution was successively washed with a saturated aqueous solution of sodium bicarbonate, a 1M solution of KHSO4 and brine. The organic layer was dried over MgSO4, filtered and concentrated under reduced pressure. Purification of the residue by flash chromatography on silica gel (petroleum ether/EtOAc : 80/20) provided 107 mg (99%) of the title compound 8 as a colorless oil. [α] 25 D = −22.7 (c 1.06, CHCl3); IR (neat) : 3076, 2931, 2852, 1741, 1641, 1613, 1514, 1445, 1421, 1383, 1301, 1268, 1174, 1101, 1071, 1037, 964, 917, 888, 845, 783 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.20 (m, 2H), 6.85 (m, 2H), 5.83 (dd, J = 15.5, 5.5 Hz, 1H), 5.75 (m, 1H), 5.59 (ddd, J = 15.5, 7.5, 1.0 Hz, 1H), 5.43 (br s, 1H), 5.24 (ddd, J = 10.5, 5.0, 2.5 Hz, 1H), 5.10-5.05 (m, 2H), 4.64 (d, J = 7.0 Hz, 1H), 4.56 (d, J = 7.0 Hz, 1H), 4.55 (d, J = 11.5 Hz, 1H), 4.48-4.38 (m, 4H), 4.27 (d, J = 11.5 Hz, 1H), 4.18 (br s, 2H), 4.06 (m, 1H), 3.84 (dd, J = 7.0, 5.5 Hz, 1H), 3.80 (s, 3H), 3.57 (m, 1H), 3.34 (s, 3H), 3.18-3.06 (m, 2H), 2.37-2.24 (m, 2H), 2.05 (m, 1H), 1.90 (m, 1H), 1.77 (ddd, J = 14.5, 10.0, 2.5 Hz, 1H), 1.71 (s, 3H), 1.68 (m, 1H); 13C NMR (125 MHz, CDCl ): δ 164.4 (d, 2 3 JPC = 4.0 Hz), 159.5, 136.3, 134.1, 131.5, 130.1, 129.8 (2C), 126.5, 119.9, 118.1, 114.0 (2C), 96.6, 79.5, 74.1, 73.9, 73.4, 70.4, 65.9, 63.4-62.2 (m, 2C), 56.1, 55.5, 39.9, 35.9, 35.4, 34.2 (d, 1JPC = 143.9 Hz), 23.2, CF3-signals missing (2C); HRMS (ESI): Calcd. for C31H41O10NaPF6 [M + Na]+: 741.2224. Found: 741.2239.
2-{(2R,6R)-6-[(S)-2-Methylpent-4-ynyl]-5,6-dihydro-2H-pyran-2-yl}acetaldehyde (10)
To a solution of dihydropyrane 9 (64 mg, 0.237 mmol) and tert-butyldimethyl(vinyloxy)silane[xxix] (75 mg, 0.473 mmol) in CH2Cl2 (1 mL) at 0 °C was added Montmorillonite K-10 (70 mg) in one portion. The cold bath was removed and the resulting slurry was stirred for 45 min at rt. The reaction mixture was then filtered over cotton and the resulting crude residue was purified by flash chromatography on silica gel (petroleum ether/EtOAc : 95/5 to 90/10) to provide 40 mg (82%) of the title aldehyde 10 as a colorless oil. IR (neat) : ψ 3294, 3034, 2959, 2924, 2727, 1725, 1459, 1431, 13.91, 1260, 1214, 1179, 1096, 804, 701 cm−1; 1H NMR (600 MHz, CDCl3): δ 9.81 (dd, J = 3.6, 1.8 Hz, 1H), 5.88 (ddt, J = 10.2, 14.8, 2.4 Hz, 1H), 5.69 (m, 1H), 4.79 (br m, 1H), 3.75 (m, 1H), 2.74 (ddd, J = 16.2, 9.0, 3.6 Hz, 1H), 2.54 (ddd, J = 16.2, 4.8, 1.8 Hz, 1H), 2.17 (ddd, J = 16.8, 6.0, 3.0 Hz, 1H), 2.12 (ddd, J = 16.8, 6.6, 3.0 Hz, 1H), 2.03 (m, 1H), 1.98-1.87 (m, 2H), 1.96 (t, J = 3.0 Hz, 1H), 1.72 (ddd, J = 14.4, 10.2, 4.2 Hz, 1H), 1.28 (ddd, J = 14.4, 9.6, 3.6 Hz, 1H), 0.99 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 198.6, 125.4, 123.2, 80.6, 67.1, 65.4, 63.4, 45.6, 38.8, 28.5, 28.4, 26.0, 24.2; HRMS (ESI): Calcd. for C13H18O2Na [M + Na]+: 229.1207. Found: 229.1204.
(Z)-{(3S,4S,6R,E)-3-(4-Methoxybenzyloxy)-6-(methoxymethoxy)-1-[(S)-4-methyl-3,6-dihydro-2H-pyran-2-yl]nona-1,8-dien-4-yl}-4-{(2R,6R)-6-[(S)-2-methylpent-4-ynyl]-5,6-dihydro-2H-pyran-2-yl}but-2-enoate (11)
To a mixture of phosphonate 8 (146 mg, 0.204 mmol) and 18-crown-6 (247 mg, 0.934 mmol) in THF (7 mL) at -78 °C was added KHMDS (0.35 M in THF). After 45 min at this temperature, a solution of aldehyde 10 (35 mg, 0.1698 mmol) in THF (3.5 mL) was added dropwise and the mixture was stirred for 25 min. The reaction mixture was hydrolyzed by adding a saturated aqueous solution of ammonium chloride. Ethyl acetate was added and the layers were separated. The aqueous phase was extracted with EtOAc and the combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. 1H NMR spectroscopy of the crude residue indicated the quantitative formation of the desired alkene as a 1/5 mixture of E/Z geometric isomers. Purification of the crude residue by flash chromatography on silica gel (petroleum ether/EtOAc : 95/5 to 80/20) furnished 56 mg (50%) of the desired Z-isomer 11 along with 14 mg (12%) of the undesired E-isomer (combined yield = 62%). Excess phosphonate 8 was entirely recovered (m8 = 23 mg, 92% recovery). [α] 25 D = − 73.3 (c 1.18, CHCl3); IR (neat) : ψ 3294, 3032, 2927, 1717, 1643, 1613, 1514, 1439, 1380, 1364, 1300, 1248, 1212, 1168, 1093, 1037, 918, 819 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.21 (m, 2H), 6.84 (m, 2H), 6.42 (ddd, J = 11.5, 7.5, 6.5 Hz, 1H), 5.88 (dt, J = 11.5, 2.0 Hz, 1H), 5.81 (m, 1H), 5.76 (m, 1H), 5.67 (m, 1H), 5.59 (ddd, J = 16.0, 7.0, 1.8 Hz, 1H), 5.42 (br m, 1H), 5.24 (ddd, J = 10.0, 5.0, 2.5 Hz, 1H), 5.09-5.04 (m, 2H), 4.64 (d, J = 7.0 Hz, 1H), 4.58 (d, J = 7.0 Hz, 1H), 4.57 (d, J = 12.0 Hz, 1H), 4.31 (d, J = 12.0 Hz, 1H), 4.27 (br m, 1H), 4.18 (m, 2H), 4.05 (m, 1H), 3.87 (m, 1H), 3.80-3.74 (m, 1H), 3.79 (s, 3H), 3.55 (m, 1H), 3.34 (s, 3H), 2.97 (dddd, J = 16.5, 8.0, 4.5, 2.0 Hz, 1H), 2.88 (dddd, J = 16.0, 9.5, 6.5, 2.0 Hz, 1H), 2.33-2.28 (m, 2H), 2.21 (ddd, J = 16.5, 5.5, 2.5 Hz, 1H), 2.12 (ddd, J = 17.0, 7.0, 3.0 Hz, 1H), 2.07-1.85 (m, 5H), 1.96 (t, J = 3.5 Hz, 1H), 1.78-1.66 (m, 3H), 1.70 (s, 3H), 1.31-1.21 (m, 2H), 0.98 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 165.9, 159.3, 147.5, 135.4, 134.4, 131.5, 130.5, 129.7, 129.2 (2C), 127.1, 124.9, 121.0, 119.9, 117.8, 113.9 (2C), 96.7, 83.4, 79.5, 74.4, 73.6, 72.3, 71.2, 70.5, 69.6, 65.8, 65.6, 56.0, 55.5, 41.7, 40.1, 35.9, 35.6, 33.8, 31.4, 28.8, 26.8, 23.2, 19.1; HRMS (ESI): Calcd. for C40H54O8Na [M + Na]+: 685.3716, found: 685.3715.
(1R,3Z,7S,9S,10E,15S,17R)-7-{(S,E)-1-(4-Methoxybenzyloxy)-3-[(S)-4-methyl-3,6-dihydro-2H-pyran-2-yl]allyl}-9-(methoxymethoxy)-15-methyl-13-methylene-6,21-dioxabicyclo[15.3.1]henicosa-3,10,19-trien-5-one (12)
To a solution of enyne 11 (103 mg, 0.157 mmol) in freshly distilled acetone (160 mL) at 50 °C was added CpRu(CH3CN)3PF6 (3.4 mg, 7.85 μmol) in one portion. The resulting light brown solution was stirred at this temperature for 15 min, at which time TLC indicated complete consumption of the starting material. The mixture was filtered over a short plug of silica gel to remove the Ru-catalyst and was concentrated in vacuo. Purification of the residue by flash chromatography on silica gel (petroleum ether/EtOAc : 80/20) furnished 103 mg (99%) of the title 1,4-diene 12 as a colorless oil. [α] 25 D = − 145 (c 2.56, CHCl3); IR (neat) : ψ 2925, 1717, 1641, 1613, 1513, 1460, 1378, 1248, 1172, 1092, 1034, 975, 919, 820 cm−1; 1H NMR (500 MHz, CDCl3): δ 7.22 (m, 2H), 6.84 (m, 2H), 6.28 (ddd, J = 11.5, 9.5, 5.5 Hz, 1H), 5.88-5.80 (m, 3H), 5.69 (m, 1H), 5.61 (ddd, J = 16.0, 7.0, 1.5 Hz, 1H), 5.58 (dd, J = 14.5, 7.0 Hz, 1H), 5.42 (br m, 1H), 5.31 (br dd, J = 15.5, 8.5 Hz, 1H), 5.17 (ddd, J = 9.5, 5.0, 2.5 Hz, 1H), 4.80 (br s, 1H), 4.69 (br s, 1H), 4.67 (d, J = 6.5 Hz, 1H), 4.57 (d, J = 12.0 Hz, 1H), 4.45 (d, J = 7.0 Hz, 1H), 4.34 (d, J = 11.5 Hz, 1H), 4.30 (br m, 1H), 4.17 (m, 2H), 4.04 (m, 1H), 3.90 (m, 1H), 3.79 (s, 3H), 3.75 (br m, 1H), 3.42 (m, 1H), 3.32 (s, 3H), 2.81 (dd, J = 15.0, 7.0 Hz, 1H), 2.68 (dd, J = 15.0, 7.0, 1H), 2.35 (m, 1H), 2.13 (m, 1H), 2.09-1.98 (m, 3H), 1.92-1.72 (m, 6H), 1.70 (s, 3H), 1.49 (dt, J = 14.0, 7.0 Hz, 1H), 1.19 (m, 1H), 0.85 (d, J = 6.5 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 165.7, 159.3, 146.5 (2C), 135.8, 133.2, 131.6, 130.8, 130.5, 129.6 (2C), 129.0, 126.8, 125.2, 122.4, 120.0, 113.9 (2C), 113.3, 93.3, 79.5, 74.9, 73.6, 72.8, 71.7, 70.5, 67.1, 65.8, 55.7, 55.5, 43.5, 43.2, 40.3, 36.2, 35.9, 34.7, 31.5, 28.8, 23.2, 20.7; HRMS (ESI): Calcd. for C40H54O8Na [M + Na]+: 685.3716, found: 685.3720.
(1R,3Z,7S,9S,10E,15S,17R)-9-Hydroxy-7-{(S,E)-1-(4-methoxybenzyloxy)-3-[(S)-4-methyl-3,6-dihydro-2H-pyran-2-yl]allyl}-15-methyl-13-methylene-6,21-dioxabicyclo[15.3.1]henicosa-3,10,19-trien-5-one (13)
A solution of MOM-protected macrolactone 12 (103 mg, 0.156 mmol) in tert-BuOH (6.5 mL) was placed in a reaction vial containing a stir bar and PPTS (508 mg, 2.02 mmol) was added in one portion. The tube was sealed and subsequently immersed in a 85 °C oil bath. After stirring for 8 hours at this temperature, the reaction mixture was then allowed to cool to rt and was subsequently poured into water. The aqueous layer was extracted with EtOAc (x3) and the combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel (petroleum ether/EtOAc : 80/20 to 70/30) provided 63.6 mg (66%) of allylic alcohol 13 as a colorless oil. [α] 25 D = −118 (c 1.14, CHCl3); IR (neat) : ψ 3445, 3031, 2956, 2921, 2835, 1714, 1642, 1613, 1513, 1422, 1380, 1300, 1247, 1175, 1085, 1035, 973, 892, 819 cm−1; 1H NMR (600 MHz, CDCl3): δ 7.22 (m, 2H), 6.85 (m, 2H), 6.29 (ddd, J = 11.4, 9.5, 5.4 Hz, 1H), 5.88-5.81 (m, 3H), 5.69 (m, 1H), 5.65-5.55 (m, 2H), 5.50 (dd, J = 15.6, 7.2 Hz, 1H), 5.42 (br m, 1H), 5.15 (quintapp, J = 3.6 Hz, 1H), 4.81 (br s, 1H), 4.70 (br s, 1H), 4.58 (d, J = 11.4 Hz, 1H), 4.34 (d, J = 11.4 Hz, 1H), 4.28 (br m, 1H), 4.18 (m, 2H), 4.14 (m, 1H), 4.06 (m, 1H), 3.95 (tapp, J = 6.6 Hz, 1H), 3.80 (s, 3H), 3.75 (br m, 1H), 3.42 (dt, J = 15.0, 9.0 Hz, 1H), 2.79 (dd, J = 15.0, 7.2 Hz, 1H), 2.67 (dd, J = 15.0, 7.2 Hz, 1H), 2.36 (m, 1H), 2.12-1.96 (m, 4H+OH), 1.92-1.72 (m, 5H), 1.70 (s, 3H), 1.53 (dt, J = 13.6, 6.0 Hz, 1H), 1.16 (dt, J = 13.8, 6.6 Hz, 1H), 0.86 (d, J = 6.0 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 165.8, 159.4, 146.7, 146.5, 135.9, 133.7, 131.5, 130.4, 130.3, 129.6 (2C), 128.9, 126.6, 125.1, 122.0, 119.9, 114.0 (2C), 113.1, 79.3, 73.5, 72.6, 72.2, 71.3, 70.6, 67.0, 65.8, 55.5, 43.6, 43.1, 40.2, 37.9, 35.9, 34.6, 31.4, 28.4, 23.2, 20.6; HRMS (ESI): Calcd. for C38H50O7Na [M + Na]+: 641.3454, found: 641.3458.
(1R,3S,7S,9S,10S,12S,18R,Z)-10-Hydroxy-12-{(S,E)-1-[(4-methoxybenzyl)oxy]-3-[(S)-4-methyl-3,6-dihydro-2H-pyran-2-yl]allyl}-3-methyl-5-methylene-8,13,22-trioxatricyclo[16.3.1.07,9]docosa-15,19-dien-14-one (14)
To a suspension of flame-dried 4 Å MS (100 mg) in CH2Cl2 (2 mL) at -20 °C were sequentially added (+)-diethyl-L-tartrate (28 μL, 0.134 mmol) and Ti(OiPr)4 (34 μL, 0.113 mmol). The resulting mixture was stirred for 15 min at this temperature and tert-butylhydroperoxide (5.5M in dodecane, 40 μL, 0.218 mmol) was then added dropwise. The mixture was stirred another 15 min and a solution of allylic alcohol 13 (10.9 mg, 0.0176 mmol) in CH2Cl2 (4 mL) was subsequently added dropwise. After stirring for 1 hour at -20 °C, the reaction mixture was hydrolyzed by adding a mixture of a 4 N solution of sodium hydroxide (2 mL) and brine (2 mL). The resulting mixture was stirred at 0 °C for 1 hour, and EtOAc was added. The layers were separated and the aqueous phase was extracted with EtOAc (3x). The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel (Hexanes/EtOAc : 80/20); yielded 9.6 mg (86%) of epoxide 14 as a colorless oil. [α] 25 D = −78.3 (c 0.49, CHCl3); IR (neat) : ψ 3435, 2922, 1715, 1643, 1612, 1513, 1445, 1379, 1247, 1213, 1172, 1086, 1034, 974, 896, 821 cm−1; 1H NMR (600 MHz, CDCl3): δ 7.22 (m, 2H), 6.85 (m, 2H), 6.34 (ddd, J = 11.4, 9.6, 6.6 Hz, 1H), 5.91 (d, J = 12.0 Hz, 1H), 5.85 (dd, J = 15.6, 5.4 Hz, 1H), 5.83 (m, 1H), 5.67 (dd, J = 10.2, 1.8 Hz, 1H), 5.64 (ddd, J = 15.6, 7.2, 0.6 Hz, 1H), 5.43 (br s, 1H), 5.24 (quintapp, J = 4.8 Hz, 1H), 4.91 (br s, 1H), 4.79 (br s, 1H), 4.58 (d, J = 12.0 Hz, 1H), 4.33 (d, J = 11.4 Hz, 1H), 4.24 (br m, 1H), 4.19 (br s, 2H), 4.06 (m, 1H), 3.93 (tapp, J = 6.0 Hz, 1H), 3.88 (m, 1H), 3.84 (m, 1H), 3.80 (s, 3H), 3.41 (dt, J = 13.8, 8.4 Hz, 1H), 2.99 (td, J = 6.0, 1.8 Hz, 1H), 2.87 (t, J = 2.4 Hz, 1H), 2.38 (m, 1H), 2.30-2.28 (m, 2H), 2.21-2.17 (m, 2H), 2.06 (m, 1H), 1.98 (ddd, J = 15.0, 6.0, 4.2 Hz, 1H), 1.92-1.72 (m, 5H), 1.71 (s, 3H), 1.68 (m, 1H), 1.57 (br s, OH), 1.11 (ddd, J = 13.8, 7.8, 6.0 Hz, 1H), 0.88 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 165.9, 159.5, 147.2, 144.0, 136.1, 131.5, 130.2, 129.6 (2C), 128.8, 126.5, 124.8, 122.0, 119.9, 114.5, 114.0 (2C), 79.4, 73.5, 71.9, 71.5, 70.7, 67.5, 67.4, 65.9, 60.6, 55.5, 54.6, 44.7, 42.5, 38.4, 35.9, 34.7, 33.9, 31.1, 27.8, 23.2, 20.3; HRMS (ESI): Calcd. for C38H50O8Na [M + Na]+: 657.3403, found: 657.3388
(1R,3Z,7S,9E,11R,15S,17R)-11-Hydroxy-7-{(S,E)-1-(4-methoxybenzyloxy)-3-[(S)-4-methyl-3,6-dihydro-2H-pyran-2-yl]allyl}-15-methyl-13-methylene-6,21-dioxabicyclo[15.3.1]henicosa-3,9,19-trien-5-one (20)
In the glove box, O3ReOSiPh3 (16.5 mg, 0.032 mmol, 1.0 equiv) was inserted into a flame-dried round bottom flask. Out of the glove box, Et2O (2.5 mL) was added under argon. The flask was cooled to -50 °C and the solution was stirred at this temperature for 10 min. A solution of the allylic alcohol 13 (19.8 mg, 0.032 mmol, 1.0 equiv) in Et2O (2.5 mL) was then added dropwise and the mixture was stirred for 5 min. The reaction mixture was quenched by successively adding silica gel and Et3N (200 μL) and was allowed to warm to rt. After removal of the solvents in vacuo, the crude reaction mixture was analyzed by 1H NMR which indicated the presence of the desired rearranged product 20 along with the starting material 13 in a 4:1 ratio in favor of the rearranged product 20. Purification of the residue by flash chromatography on silica gel (Hexanes/EtOAc : 90/10 to 85/15) furnished the desired compound 20 (15.4 mg, 78%) as a colorless oil. Moreover, 3.9 mg of the starting material 13 could be recovered (yield = 97% brsm). [α] 25 D = −85 (c 0.46, CHCl3); IR (neat) : ψ 3439, 2958, 2921, 2854, 1718, 1644, 1613, 1513, 1421, 1381, 1297, 1250, 1213, 1169, 1085, 1036, 971, 811, 756 cm−1; 1H NMR (600 MHz, CDCl3): δ 7.21 (m, 2H), 6.86 (m, 2H), 6.31 (td, J = 10.8, 4.8 Hz, 1H), 5.88 (br d, J = 12.0 Hz, 1H), 5.85 (dd, J = 15.6, 5.4 Hz, 1H), 5.85-5.79 (m, 1H), 5.70 (m, 1H), 5.63-5.54 (m, 2H), 5.49 (dd, J = 15.6, 7.2 Hz, 1H), 5.43 (br s, 1H), 5.09 (ddd, J = 7.8, 3.6, 2.4 Hz, 1H), 4.86 (s, 1H), 4.84 (s, 1H), 4.59 (d, J = 11.4 Hz, 2H), 4.31 (d, J = 12.0 Hz, 1H), 4.20 (br s, 2H), 4.15 (m, 1H), 4.12-4.06 (m, 2H), 3.83 (tapp, J = 6.6 Hz, 1H), 3.80 (s, 3H), 3.78 (sept, J = 4.2 Hz, 1H), 3.71 (m, 1H), 2.31 (m, 1H), 2.27-2.14 (m, 4H), 2.11-2.01 (m, 3H), 1.95-1.83 (m, 3H), 1.79-1.72 (m, 2H), 1.71 (s, 3H), 1.67 (ddd, J = 12.0, 7.8, 3.6 Hz, 1H), 0.93 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 165.6, 159.4, 147.6, 144.6, 136.1, 135.9, 131.5, 130.4, 129.6 (2C), 128.6, 127.9, 126.7, 125.0, 121.7, 119.9, 115.8, 113.9 (2C), 79.5, 73.5, 72.9, 71.8, 70.3, 70.2, 67.8, 65.8, 55.5, 43.9, 40.7, 36.0, 34.3, 34.0, 31.6, 28.5, 23.2, 21.2; HRMS (ESI): Calcd. for C38H50O7Na [M + Na]+: 641.3454, found: 641.3461.
(1R,3Z,7S,9E,11S,15S,17R)-11-Hydroxy-7-{(S,E)-1-(4-methoxybenzyloxy)-3-[(S)-4-methyl-3,6-dihydro-2H-pyran-2-yl]allyl}-15-methyl-13-methylene-6,21-dioxabicyclo[15.3.1]henicosa-3,9,19-trien-5-one (17)
Oxidation
To a solution of allylic alcohol 20 (10.1 mg, 0.0163 mmol, 1.0 equiv) in CH2Cl2 (1 mL) at 0 °C was added Dess-Martin periodinane (14 mg, 0.0327 mmol, 2.0 equiv) in one portion. The resulting mixture was stirred for 2 hours at rt. The reaction mixture was poured into a 1/1 mixture of a saturated aqueous solution of sodium bicarbonate and a saturated aqueous solution of sodium thiosulfate and Et2O (2 mL) was added. The layers were separated and the aqueous phase was extracted with Et2O (3x). The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. Purification of the residue by flash chromatography on silica gel (Hexanes/EtOAc : 80/20) furnished the desired title enone (9.6 mg, 96%) as a colorless oil. [α] 25 D = −71 (c 0.84, CHCl3); IR (neat) : ψ 3032, 2921, 1719, 1671, 1640, 1614, 1513, 1421, 1380, 1248, 1212, 1168, 1087, 1035, 976, 896, 818 cm−1; 1H NMR (600 MHz, CDCl3): δ 7.22 (m, 2H), 6.86 (m, 2H), 6.72 (ddd, J = 16.2, 7.8, 6.6 Hz, 1H), 6.37 (ddd, J= 11.4, 10.2, 4.8 Hz, 1H), 6.09 (d, J = 16.2 Hz, 1H), 6.88-6.83 (m, 3H), 5.69 (m, 1H), 5.63 (ddd, J = 15.6, 7.2, 1.2 Hz, 1H), 5.43 (br s, 1H), 4.90 (s, 1H), 4.82 (s, 1H), 4.59 (d, J = 12.0 Hz, 1H), 4.33 (d, J = 12.0 Hz, 1H), 4.26 (m, 1H), 4.20 (m, 2H), 4.08 (m, 1H), 3.92 (tapp, J = 6.0 Hz, 1H), 3.80 (s, 3H), 3.72 (m, 1H), 3.55 (dt, J = 15.6, 10.2 Hz, 1H), 3.25 (d, J = 15.6 Hz, 1H), 3.11 (d, J = 15.0 Hz, 1H), 2.54-2.44 (m, 2H), 2.34 (m, 1H), 2.09-2.00 (m, 3H), 1.95-1.86 (m, 2H), 1.78 (dd, J = 13.8, 10.2 Hz, 1H), 1.71 (s, 3H), 1.49 (ddd, J = 14.4, 8.4, 5.4 Hz, 1H), 1.19 (ddd, J = 14.4, 8.4, 2.0 Hz, 1H), 0.83 (d, J = 6.6 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ 198.4, 165.6, 159.4, 148.8, 143.4, 142.1, 136.2, 132.1, 131.4, 130.2, 129.6 (2C), 128.8, 126.2, 125.4, 120.9, 119.9, 116.1, 114.0 (2C), 79.2, 73.4, 72.8, 72.3, 70.5, 66.6, 65.8, 55.5, 46.5, 44.7, 43.7, 35.9, 34.3, 33.6, 31.8, 28.1, 23.2, 19.9; HRMS (ESI): Calcd. for C38H48O7Na [M + Na]+: 639.3298, found: 639.3316.
Diastereoselective reduction
To a solution of the previously obtained α,β-unsaturated ketone (7.5 mg, 0.0122 mmol, 1.0 equiv) in THF (1.5 mL) were successively added (R)-2-methyl-CBS-oxazaborolidine (1.0 M in toluene, 61 μL, 0.0609 mmol, 5.0 equiv) followed by BH3•THF (1.0 M in THF, 43 μL, 0.0426 mmol, 3.5 equiv) slowly via syringe at 0 °C. The reaction mixture was stirred for 5 min at this temperature, and was hydrolyzed by adding H2O (1.5 mL). The resulting mixture was warmed to rt, Et2O was added and the organic phase was washed with a 1.0 M aqueous solution of HCl. The aqueous phase was extracted with Et2O (3x) and the organic layer was washed with brine, dried over MgSO4, filtered and concentrated in vacuo. 1H NMR of the crude residue indicated the presence of only one diastereoisomer. Purification of the residue by flash column chromatography on silica gel (Hexanes/EtOAc : 85/15) furnished the titled allylic alcohol 17 (7.3 mg, 97%) as a colorless oil. [α] 25 D = − 105 (c 0.59, CHCl3); IR (neat): 3433, 2924, 2854, 1720, 1644, 1612, 1513, 1450, 1379, 1248, 1166, 1087, 1037, 974, 815 ψ cm−1; 1H NMR (600 MHz, CDCl3): δ 7.21 (m, 2H), 6.85 (m, 2H), 6.32 (ddd, J = 11.4, 9.6, 5.4 Hz, 1H), 5.90 (d, J = 11.4 Hz, 1H), 5.84 (dd, J = 15.6, 5.4 Hz, 1H), 5.84-5.80 (m, 1H), 5.70 (m, 1H), 5.63-5.58 (m, 3H), 5.42 (br s, 1H), 5.07 (ddd, J = 10.2, 5.4, 3.0 Hz, 1H), 4.84 (s, 2H), 5.59 (d, J = 12.0 Hz, 1H), 4.31 (d, J = 11.4 Hz, 1H), 4.19 (br s, 2H), 4.18-4.10 (m, 2H), 4.07 (m, 1H), 3.89-3.83 (m, 2H), 3.80 (s, 3H), 3.53 (m, 1H), 2.35-2.18 (m, 5H), 2.15 (dd, J = 13.2, 4.2 Hz, 1H), 2.11 (dd, J = 14.4, 9.6 Hz, 1H), 2.07-2.01 (m, 1H), 1.90 (br d, J = 16.8 Hz, 1H), 1.85-1.71 (m, 3H), 1.70 (s, 3H), 1.64 (ddd, J = 13.8, 8.4, 4.8 Hz, 1H), 1.35-1.26 (m, 1H), 1.12 (ddd, J = 12.0, 7.8, 4.2 Hz, 1H), 0.85 (d, J = 6.0 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 165.6, 159.4, 146.9, 145.1, 135.8, 135.3, 131.5, 130.4, 129.7 (2C), 128.6, 127.2, 126.7, 124.9, 121.9, 119.9, 114.6, 114.0 (2C), 79.5, 74.1, 73.5, 71.4, 70.3, 70.0, 67.8, 65.9, 55.5, 45.0, 43.5, 42.4, 36.0, 34.5, 33.6, 31.1, 28.4, 23.2, 19.9; HRMS (ESI): Calcd. for C38H50O7Na [M + Na]+: 641.3454, found: 641.3464.
Laulimalide (1)
Sharpless Epoxidation
To a suspension of flame-dried 4 Å MS (85 mg) in CH2Cl2 (1.5 mL) at -20 °C were sequentially added (+)-diethyl-L-tartrate (18 μL, 0.0860 mmol) and Ti(OiPr)4 (21 μL, 0.0725 mmol). The resulting mixture was stirred for 15 min at this temperature and tert-butylhydroperoxide (5.5M in dodecane, 25 μL, 0.140 mmol) was then added dropwise. The mixture was stirred another 15 min and a solution of the previously obtained allylic alcohol 17 (7.0 mg, 0.0113 mmol) in CH2Cl2 (3 mL) was subsequently added dropwise. After stirring for 1 hour at -20 °C, the reaction mixture was hydrolyzed by adding a mixture of a 4 N solution of sodium hydroxide (1.5 mL) and brine (1.5 mL). The resulting mixture was stirred at 0 °C for 1 hour, and EtOAc was added. The layers were separated and the aqueous phase was extracted with EtOAc (3x). The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel (Hexanes/EtOAc 80:20) yielded 6.4 mg (88%) of the desired epoxide as a colorless oil. [α] 25 D = −141 (c 0.41, CHCl3); IR (neat): ψ 3452, 2918, 1720, 1643, 1612, 1513, 1421, 1378, 1300, 1247, 1213, 1171, 1115, 1084, 1033, 977, 891, 814 cm−1; 1H NMR (600 MHz, CDCl3): δ 7.21 (m, 2H), 6.86 (m, 2H), 6.42 (dt, J = 10.8, 3.6Hz, 1H), 5.89 (br d, J = 11.4 Hz, 1H), 5.84 (dd, J = 15.6, 5.4 Hz, 1H), 5.83 (m, 1H), 5.68 (m, 1H), 5.59 (dd, J = 15.6, 6.6 Hz, 1H), 5.43 (br s, 1H), 5.21 (dd, J = 10.8, 4.8 Hz, 1H), 4.85 (s, 1H), 4.83 (s, 1H), 4.58 (d, J = 11.4 Hz, 1H), 4.32 (d, J = 12.0 Hz, 1H), 4.32-4.28 (m, 1H), 4.19 (br s, 2H), 4.06 (m, 2H), 3.88 (tapp, J = 6.0 Hz, 1H), 3.80 (s, 3H), 3.79-3.75 (m, 1H+OH), 3.03 (m, 1H), 2.87 (m, 1H), 2.37 (dd, J = 13.8, 4.8 Hz, 1H), 2.30 (m, 1H), 2.20 (m, 1H), 2.11-1.86 (m, 8H), 1.77 (dd, J = 13.2, 10.2 Hz, 1H), 1.71 (s, 3H), 1.47-1.40 (m, 2H), 1.35-1.30 (m, 1H), 0.82 (d, J= 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 166.1, 159.4, 150.3, 145.2, 136.0, 131.5, 130.2, 129.7 (2C), 128.8, 126.3, 125.4, 121.0, 120.0, 114.0 (2C), 112.6, 79.3, 73.4, 71.1, 70.6, 68.1, 66.7, 65.9, 60.8, 55.5, 52.4, 45.9, 43.7, 37.3, 35.9, 33.9, 33.3, 31.9, 30.0, 23.2, 21.1; HRMS (ESI): Calcd. for C38H50O8Na [M + Na]+: 657.3403, found: 657.3411.
Final deprotection of the PMB group
To a solution of the previously obtained PMB-ether (5.4 mg, 8.52 μmol) in CH2Cl2 (1.5 mL), pH7 buffer (75 μL) and tert-BuOH (75 μL) was added DDQ (5.8 mg, 25.6 μmol) in one portion. The resulting green mixture was stirred at rt for 30 min. Another portion of DDQ (5.8 mg, 25.6 μmol) was added. After stirring for one hour, some more DDQ (5.8 mg, 25.6 μmol) was added and the mixture was stirred for another hour. The resulting orange suspension was then washed with a saturated aqueous solution of sodium bicarbonate and the aqueous phase was extracted three times with CH2Cl2. The combined organic layers were washed with brine, dried over MgSO4, filtered and concentrated in vacuo. Purification of the residue by flash chromatography on silica gel (Hexanes/EtOAc : 80/20 to 50/50) furnished 3.9 mg (89%) of laulimalide (1) as a colorless oil. The analytical and spectroscopic data perfectly matched those reported in the literature.xxiv [α] 25 D = −193 (c 0.15, CHCl ); 1 3 H NMR (600 MHz, CDCl3): δ 6.44 (ddd, J = 11.4, 9.6, 3.6 Hz, 1H), 3.91 (m, 1H), 5.88 (ddd, J = 15.6, 6.4, 1.2 Hz, 1H), 5.86-5.82 (m, 1H), 5.75 (ddd, J = 15.6, 6.0, 1.2 Hz, 1H), 5.69 (m, 1H), 5.42 (br s, 1H), 5.16 (ddd, J = 11.4, 5.4, 1.8 Hz, 1H), 4.86 (s, 1H), 4.85 (s, 1H), 4.31 (m, 1H), 4.22 (qapp, J = 5.4 Hz, 1H), 4.20-4.16 (m, 2H), 4.08 (m, 1H), 4.03 (m, 1H), 3.79-3.69 (m, 2H), 3.07 (m, 1H), 2.90 (t, J = 2.4 Hz, 1H), 2.40-2.35 (m, 2H), 2.40-2.35 (m, 2H), 2.22 (m, 1H), 2.12 (br d, J = 15.6 Hz, 1H), 2.05-1.84 (m, 6H), 1.78 (dd, J = 13.2, 10.2 Hz, 1H), 1.70 (s, 3H), 1.49 (ddd, J = 14.4, 11.4, 9.6 Hz, 1H), 1.45 (m, 1H), 1.33 (ddd, J = 14.4, 4.2, 3.0 Hz, 1H), 0.83 (d, J = 6.6 Hz, 3H); 13C NMR (125 MHz, CDCl3): δ 166.0, 150.4, 144.8, 133.9, 131.2, 128.7, 128.5, 125.2, 120.5, 119.7, 112.5, 73.5, 73.1 (2C), 72.2, 67.9, 66.5, 65.6, 60.6, 52.0, 45.5, 43.4, 37.0, 35.6, 33.7, 33.4, 31.6, 29.5, 22.9, 20.7; HRMS (ESI): Calcd. for C30H42O7Na [M + Na]+: 537.2828, found: 537.2825.
Supplementary Material
Acknowledgement
We gratefully acknowledge P. Crews (UC Santa Cruz) for providing an authentic sample of laulimalide. We also thank J. Flygare, J.-P. Stephan and P. Chan at Genentech for biological testings of our laulimalide analogue. We thank the General Medical Sciences Institute of NIH (GM 33 049) for their generous support of our programs. We thank Johnson-Matthey for ther gifts of palladium and ruthenium and Aldrich for generous supply of the ProPhenol ligand. W. M. S. thanks the National Institutes of Health for a post-doctoral fellowship. C. K. C. thanks Eli Lilly & Co. for a graduate fellowship.
Footnotes
Supporting information Available: Detailed experimental procedures and full characterization of compounds 4, 5, 16, 21-31, 33.
References and Footnotes
- [i].Neises B, Steglich W. Angew. Chem. Int. Ed. 1978;17:522. [Google Scholar]
- [ii].Von Stadler PA. Helv. Chim. Acta. 1978;61:1675. [Google Scholar]
- [iii].Pittelkow M, Kamounah FS, Boas U, Pedersen B, Christensen JB. Synthesis. 2004:2485. [Google Scholar]
- [iv].Haveaux B, Dekoker A, Rens M, Sidani AR, Toye J, Ghosez L. Org. Synth. 1980;59:26. [Google Scholar]
- [5].Kita Y, Maeda H, Omori K, Okuno T, Tamura Y. Synlett. 1993:273. [Google Scholar]
- [vi]a).Otera J, Ioka S, Nozaki H. J. Org. Chem. 1989;54:4013. [Google Scholar]; b) Otera J, Dan-oh N, Nozaki H. J. Org. Chem. 1991;56:5307. [Google Scholar]
- [vii]a).Shiina I, Ibuka R, Kubota M. Chem. Lett. 2002:286. [Google Scholar]; b) Shiina I, Kubota M, Oshiumi H, Hashizume M. J. Org. Chem. 2004;69:1822. doi: 10.1021/jo030367x. [DOI] [PubMed] [Google Scholar]
- [viii].Hikota M, Sakurai Y, Horita K, Yonemitsu O. Tetrahedron Lett. 1990;31:6367. [Google Scholar]
- [ix].Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989. [Google Scholar]
- [x].Nahmany M, Melman A. Org. Lett. 2001;3:3733. doi: 10.1021/ol0166855. [DOI] [PubMed] [Google Scholar]
- [xi].Toshima K, Miyamoto N, Matsuo G, Nakata M, Matsumura S. Chem. Commun. 1996:1379. [Google Scholar]
- [xii].For a review on Ruthenium-catalyzed reaction featuring the Ru-catalyzed alkene-alkyne coupling, see: Trost BM, Frederiksen MU, Rudd MT. Angew. Chem. Int. Ed. 2005;44:6630. doi: 10.1002/anie.200500136.
- [xiii]a).Trost BM, Harrington PE. J. Am. Chem. Soc. 2004;126:5028. doi: 10.1021/ja049292k. [DOI] [PubMed] [Google Scholar]; b) Trost BM, Harrington PE, Chisholm JD, Wrobleski ST. J. Am. Chem. Soc. 2005;127:13598. doi: 10.1021/ja053365y. [DOI] [PubMed] [Google Scholar]
- [xiv].Nakamura S, Kikuchi F, Hashimoto S. Angew. Chem. Int. Ed. 2008;47:7091. doi: 10.1002/anie.200802729. [DOI] [PubMed] [Google Scholar]
- [xv].Johnson RA, Sharpless KB. In: Catalytic Asymmetric Synthesis. Ojima I, editor. VCH Publishers; New York: 1993. pp. 103–158. [Google Scholar]
- [xvi].Payne GB. J. Org. Chem. 1962;27:3819. [Google Scholar]
- [xvii].Hodgson DM, Humphreys PG. In: Science of Synthesis: Houben-Weyl Methods of Molecular Transformations. Clayden JP, editor. Vol. 36. Thieme; Stuttgart: 2007. pp. 583–665. [Google Scholar]
- [xviii].Grieco PA, Gilman S, Nishizawa M. J. Org. Chem. 1976;41:1485. [Google Scholar]
- [xix].For a review of 1,3-isomerizations of allylic alcohols, see: Bellemon-Laponnaz S, Le Ny JP. C. R. Chim. 2002;5:217.
- [xx].Bellemin-Laponnaz S, Gisie H, Le Ny JP, Osborn JA. Angew. Chem., Int. Ed. Engl. 1997;36:976. [Google Scholar]
- [xxi].Morrill C, Grubbs RH. J. Am. Chem. Soc. 2005;127:2842. doi: 10.1021/ja044054a. [DOI] [PubMed] [Google Scholar]
- [xxii].Schoop T, Roesky HW, Noltemeyer M, Schmidt H-G. Organometallics. 1993;12:571. [Google Scholar]
- [xxiii].Corey EJ, Bakshi RK, Shibata S. J. Am. Chem. Soc. 1987;109:5551. [Google Scholar]; For a review on oxazaborolidine-mediated asymmetric reductions, see: Cho BT. Tetrahedron. 2006;62:7621.
- [xxiv]a).Quinoa E, Kakou Y, Crews P. J. Org. Chem. 1988;53:3642. [Google Scholar]; (b) Corley DG, Herb R, Moore RE, Scheuer PJ, Paul VJ. J. Org. Chem. 1988;53:3644. [Google Scholar]; (c) Ghosh AK, Wang Y. J. Am. Chem. Soc. 2000;122:11027. doi: 10.1021/ja0027416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [xxv].A 1H NMR spectrum (Varian, 600 MHz) of an authentic sample of laulimalide provided by P. Crews (UC Santa Cruz) was identical to the spectrum we obtained for the synthetic sample using the same NMR instrument (Varian, 600 MHz) – see experimental section for details and comparison of spectra.
- [xxvi].Yokokawa F, Asano T, Shioiri T. Tetrahedron. 2001;57:6311. [Google Scholar]
- [xxvii].The evaluation of the biological activity was conducted by a team at Genentech South San Francisco.
- [xxviii].Prepared by enzymatic reduction of the corresponding commercially available methyl ester according to: Sano S, Takemoto Y, Nagao Y. ARKIVOC. 2003;8:93.
- [xxix].Prepared according to: Srisiri W, Buyle Padias A, Hall HK. J. Org. Chem. 1994;59:5424.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.