

Aplyviolene (1) and macfarlandin E (2, Figure 1A) are representative of the more complex members of the rearranged spongian diterpene class of natural products.[1,2] These diterpenes are structurally defined by attached cis-perhydroazulene and 6-acetoxy-2,7-dioxabicyclo[3.2.1]octan-3-one fragments. The substantial challenge in assembling these structures centers on the construction of the sensitive bicyclic lactone subunit and the formation of the C8–C14 σ-bond joining the two ring systems, a challenge augmented by the quaternary nature of C8.[3] We previously reported preparation of the lactone subunits of 1 and 2 by the synthesis of truncated congeners 3 and 4 (Figure 1A),[4] as well as the first total synthesis of aplyviolene (outlined in Figure 1B).[5] In this latter effort, the key C8–C14 σ-bond was formed by Michael addition of tertiary enolate 5 to enone 6.[6] Subsequent elaboration of product 7 provided intermediate 8, which was converted to (−)-aplyviolene along the lines of our earlier synthesis of 3. Undesirable aspects of this first-generation synthesis are the lengthy preparation of the cis-perhydroazulene unit and the need to remove the extraneous ketone carbonyl group from the product of the fragment-coupling step.
Figure 1.
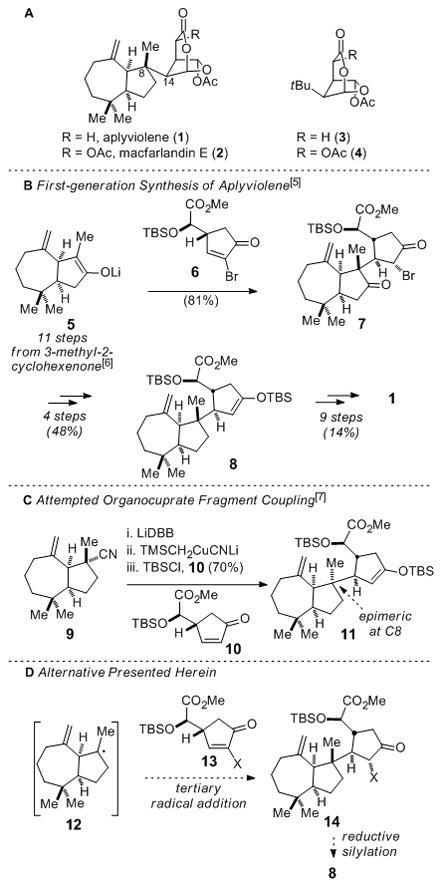
Structurally complex rearranged spongian diterpenes (A), previous synthetic efforts (B and C), and current synthetic approach (D). TMS = trimethylsilyl. TBS = tert-butyldimethylsilyl.
As described in the accompanying communication,[7] our initial attempt to streamline the synthesis by using a tertiary organocuprate derived from cis-perhydroazulene nitrile 9 in the key fragment coupling was prevented by exclusive formation of adduct 11, which is epimeric to aplyviolene at the critical quaternary carbon stereocenter (Figure 1C). This unexpected stereochemical outcome of the organocuprate conjugate addition reaction provoked consideration of an alternative strategy in which the C8–C14 σ-bond would be formed by the union of the tertiary radical 12 and enone 13 (Figure 1D, X = leaving group). Reductive silylation of the coupled product 14 would then provide enoxy silane 8, an advanced intermediate in the synthesis of (−)-aplyviolene. Despite the many advances in stereoselective radical chemistry,[ 8 ] bimolecular reactions that transform trialkyl-substituted tertiary radicals to stereogenic quaternary carbons are extremely rare.[9] Moreover, the use of tertiary radicals to couple two complex fragments in the context of target-oriented synthesis is also uncommon.[ 10 ] Encouraged by reports that achiral tertiary radicals undergo particularly diastereoselective additions to chiral alkenes, we chose to pursue this strategy.[ 11 ] Reported herein is a much-improved second-generation total synthesis of (−)-aplyviolene (1) that features the stereoselective coupling of a tertiary carbon radical and a carbon electrophile to combine chiral fragments with formation of a new quaternary carbon stereocenter.
A critical consideration was the identity of the radical precursor. The common option of employing a bromide or iodide precursor was not viable, as preliminary studies showed that these tertiary halide intermediates were not accessible in satisfactory yield.[8] Barton esters were also unsuited for this application.[12] The use of such radical precursors would entail the coupling of 12 and enone 13 (X = H) to provide product 14 (X = 2-thiopyridine), whose reductive silylation to form enoxy silane 8 was expected to be unsuccessful.[13] By contrast, an (N-acyloxy)phthalimide precursor appeared distinctly advantageous. Over 20 years ago Okada demonstrated that, when exposed to visible light, Ru(bpy)3Cl2 (bpy = 2, 2′-bipyridine), and the hydrogen-donor 1-benzyl-1,4-dihydronicotinamide, such compounds are transformed in the presence of α,β-unsaturated ketones to products of conjugate addition in excellent yield.[14] We noted that in a single instance a tertiary radical (1-admantanyl) had been coupled effectively. Surprisingly, the use of (N-acyloxy)phthalimides as radical precursors in conjugate addition reactions has not been described since this initial disclosure, and their photosensitized cleavage has only rarely been reported in any form.[15] We hoped that realizing the proposed demanding application of the Okada chemistry would be assisted by insight gained from recent advances in photoredox catalysis.[16,17]
Prior to exploring the union of the complex fragments depicted in Figure 1D, we examined the addition of the tertiary radical generated from a simpler (N-acyloxy)phthalimide to methyl vinyl ketone. Using a slight modification of Okada’s conditions, a THF/H2O solution of (N-acyloxy)phthalimide 15[ 18 ] and methyl vinyl ketone (1.5 equiv) were coupled using 1 mol % of Ru(bpy)3Cl2, excess 1,4-dihydronicotinamide 17, and 1.5 h irradiation with blue light to give adduct 18 in 70% yield (Scheme 1).[19] We were gratified to observe that this coupling took place with high selectivity from the convex face (dr >20:1). This finding was particularly promising because the corresponding addition of an organocuprate intermediate provided 18 as the minor component of a mixture of epimeric adducts.[7]
Scheme 1.
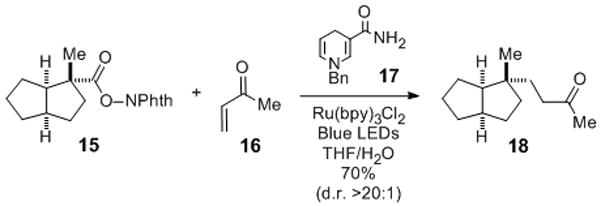
Synthesis of 18: a) 15, 16 (1.5 equiv), 17 (1.5 equiv), Ru(bpy)3Cl2 (0.01 equiv), THF/H2O (2:1), blue LEDs, RT, 90 min, 70%. NPhth = N-phthalimido. bpy = 2,2′-bipyridine
Encouraged by these initial studies, we developed a synthesis of the cis-perhydroazulene N-(acyloxy)phthalimide 27 (Scheme 2). The synthesis began with inexpensive (+)-fenchone (19), which was readily transformed by Beckmann fragmentation to tertiary nitrile 20.[20] We were pleased to find that heating the oxime intermediate in 4 M H2SO4 for 8 h provided 20 in 83% yield as a 2:1 mixture of alkene regioisomers favoring the desired Δ1,2 isomer, whereas halting the reaction after 30 min gave 20 as the 1:1 mixture of double bond isomers described by Kreiser in the original description of this transformation.[20] Reduction of nitrile 20 with diisobutylaluminum hydride and Z-selective Wittig reaction of the resulting crude aldehyde products with 3-hydroxypropyltriphenyl-phosphonium bromide yielded a 2:1 mixture of (Z)-allylic alcohol 21 and its Δ1,5 isomer.[21] Separation of the alkene isomers was possible at this stage by column chromatography on silver nitrate-embedded silica gel, which could be carried out on large scale to give pure 21 in 55% overall yield (up to 25 g of 21 isolated per batch) from the Beckman fragmentation product 20. Homoallylic alcohol 21 was converted to the corresponding nitro compound 22 by way of the primary iodide in 67% yield.[22] Intramolecular nitrile oxide cycloaddition of 22 proceeded efficiently at 90 °C under Mukaiyama conditions to provide the desired isoxazoline product, which did not require purification prior to further use.[23,24] After some experimentation, we found the most effective way to process the crude oxazoline intermediate to enone 24 was by initial hydrogenation using a mixture of 10% palladium on carbon and Raney-nickel in the presence of boric acid to provide the β-hydroxy ketone product 23.[25,26] Acid-catalyzed dehydration of 23 in toluene at elevated temperature provided enone 24 in 61% overall yield from nitro diene 22.[27]
Scheme 2.
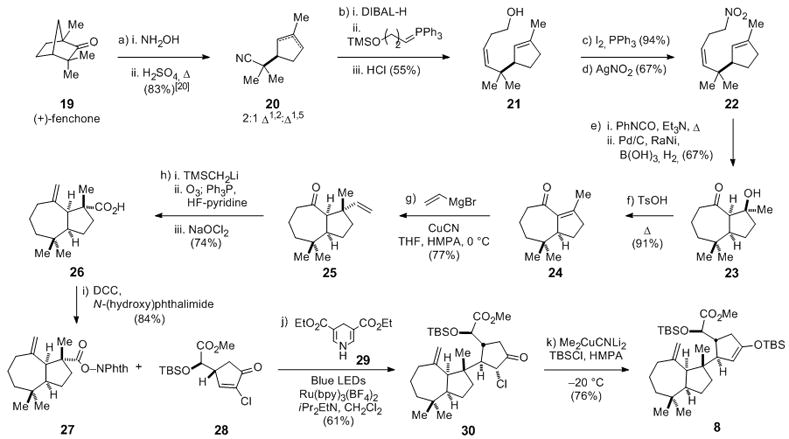
Formal total synthesis of (−)-aplyviolene: Synthesis of enoxysilane 8: a) i. 19, sodium acetate (2 equiv), NH2OH•HCl (1.75 equiv), EtOH, reflux, 36 h; ii. 4 M H2SO4, reflux, 8 h, 83% as a 2:1 mixture of Δ1,2:Δ1,5 isomers. b) i. 20, DIBAL-H (1.2 equiv), CH2Cl2, −78 °C, 1 h; ii. 3-hydroxypropyltriphenylphosphonium bromide (2 equiv), n-BuLi (4 equiv), TMSCl (2 equiv), THF, 0 °C, 20 min; + aldehyde from i., −78 °C, 1 h; 2 N H2SO4, RT, 18 h; 55%. c) 21, I2 (1.05 equiv), PPh3 (1.05 equiv), imidazole (1.1 equiv), benzene, 18 h, RT, 94%. d) alkyl iodide from c), AgNO2 (1.5 equiv), 18 h, RT, 67%. e) i. 22, PhNCO (3 equiv), Et3N (0.5 equiv), toluene, 18 h, 90 °C; ii. H2, 10% Pd/C (5 wt%), Raney-Ni (5 wt%), B(OH)3 (3 equiv), MeOH/H2O (5:1), 36 h, RT, 67% f) 23, TsOH (0.1 equiv), 5 h, 100 °C, 91%. g) 1 M vinylmagnesium bromide (5 equiv), CuCN (2.5 equiv), THF/HMPA (5:1), 10 min, 0 °C, + 24, 7 h, 0 °C, 77%. h) i. TMSCH2Li (5 equiv), pentane, −78 °C; + 25; ii. O3, CH2Cl2, −78 °C; Ph3P (1.5 equiv), HF−pyridine, 1 h, 0 °C; iii. NaClO2 (3 equiv), 2-methyl-2-butene (3 equiv), NaH2PO4 (1 equiv), acetone/H2O (30:1), RT, 1 h, 74%. i) 26, DCC (1.5 equiv), N-(hydroxy)phthalimide (1.7 equiv), DMAP (0.05 equiv), THF, 18 h, RT, 84%. j) 27, 28 (1.5 equiv), 29 (1.5 equiv), iPr2EtN (2.25 equiv), Ru(bpy)3(BF4)2 (0.01 equiv), CH2Cl2, blue LEDs, 2.5 h, RT, 61%. k) 31, Me2CuCNLi2 (2.0 equiv), TBSCl (5 equiv), Et2O/THF/HMPA (4:2:1), 1 h, −20 °C, 76%. DIBAL-H = Diisobutylaluminum hydride. TsOH = p-toulenesulfonic acid. DCC = N,N′ dicyclohexylcarbodiimide. DMAP = 4-dimethylaminopyridine. HMPA = hexamethylphosphoramide.
The conversion of enone 24 to the N-(acyloxy)phthalimide coupling partner 27 was ultimately achieved by way of only two isolated intermediates. Addition of the cuprate derived from copper cyanide and 2 equiv of vinylmagnesium bromide in THF containing hexamethylphosphoramide at 0 °C afforded vinyl addition product 25 as a 4.8:1 mixture of cis-perhydroazulene epimers in 77% yield.[28,29] Attempted elaboration of alkenyl ketone 25 to carboxylic acid 26 by initial methylenation of the ketone, followed by selective oxidative cleavage of the terminal vinyl group was prevented by competitive oxidation of the exomethylene functionality. As an alternative, slow addition of 25 to a mixture of (trimethylsilyl)methyllithium in pentane at −78 °C provided the β-silyl alcohol adduct. Without purification, this intermediate was exposed sequentially to ozone in CH2Cl2 at −78 °C, triphenylphosphine and HF-pyridine to cleanly generate the exomethylene aldehyde product. Sodium chlorite oxidation of this unpurified intermediate delivered carboxylic acid 26 in 74% overall yield from 25. The desired radical-coupling partner 27 was then secured in 84% yield by carbodiimide coupling of carboxylic acid 26 with N-hydroxyphthalimide.
With convenient access to cis-perhydroazulene N-(acyloxy)phthalimide 27 in hand, we investigated the pivotal fragment-coupling reaction. To our delight, initial efforts to implement the radical coupling of N-(acyloxy)phthalimide 27 and α-chlorocyclopentenone 28[30,31] using only a minor modification (Scheme 2) of the conditions of Okada provided the crystalline addition product 30 as a single diastereomer, albeit in low and variable yields (20–40%).[32] The low yield is best attributed to the sensitivity of 30, as chloride reduction and hydrolysis byproducts were observed.[33] Seeking to minimize the latter, we were attracted to a recent report by Gangé disclosing the photoredox-mediated reaction of glycosyl halides and electron-deficient alkenes under anhydrous conditions.[34] In this way, irradiation of a 1.5:1 mixture of enone 28 and N-(acyloxy)phthalimide 27, Ru(bpy)3(BF4)2, Hantzsch ester 29, N,N-diisopropylethylamine and dichloromethane with blue light provided coupled product 30, containing <5% of the reductively dechlorinated analogue as an inseparable byproduct, in a 61% yield.[35] Reductive enol silylation of 30 using dilithium-dimethyl(cyano)cuprate in the presence of tert-butyldimethylsilyl chloride at −20 °C delivered tricyclic enoxysilane 8, which we had previously carried on to (−)-aplyviolene.[5]
In summary, we report an improved second-generation total synthesis of aplyviolene (1) that is more efficient and proceeds by way of five fewer isolated intermediates than our original synthesis. The defining feature of this synthesis is the addition of the tertiary radical generated by photoredox-mediated fragmentation of N-(acyloxy)phthalimide 27 to α-chlorocyclopentenone 28, a reaction that combines two fragments of significant complexity and fashions adjacent quaternary and tertiary carbon stereocenters with high stereoselectivity. This transformation highlights the largely untapped utility of addition reactions of tertiary carbon radicals in the construction of quaternary carbon stereocenters and in stereoselectively fashioning demanding σ-bonds that join two rings. Furthermore, the results of this study, and those reported in the accompanying communication,[7] show that the creation of quaternary carbon stereocenters by the union of trialkyl-tertiary carbon radicals and related organometallic intermediates with carbon electrophiles can take place with complimentary stereoselection (Scheme 3). The generality of this observation and its exploitation in fragment coupling steps in stereoselective synthesis are under active investigation in our laboratories.
Scheme 3.
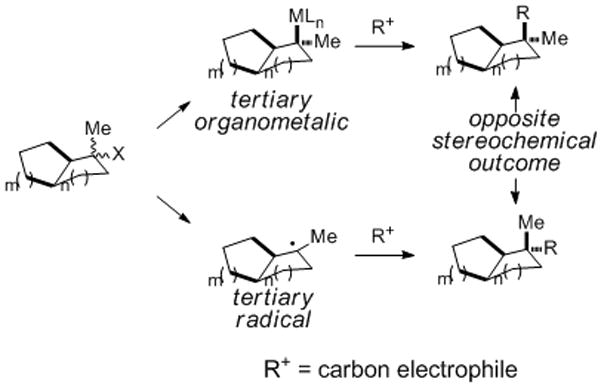
Complimentary stereoselection in forming quaternary carbon stereocenters by the reaction of tertiary carbon radicals and tertiary organometallic intermediates with carbon electrophiles.
Supplementary Material
Footnotes
We thank Dr. Joe Ziller, University of California, Irvine, for the single-crystal X-ray analyses and Dr. John Greaves, University of California, Irvine, for mass spectrometric analyses. This research was supported by the NIH Neurological Disorders & Stroke Institute (Grant NS-12389), the NIH National Institutes of General Medical Sciences (Grant GM-098601), and NIH a postdoctoral fellowship for M.J.S. (CA-138084). NMR spectra, mass spectra, and the X-ray analyses were obtained at UC Irvine using instrumentation acquired with the assistance of NSF and NIH Shared Instrumentation grants. Unrestricted funds from Amgen and Merck are also gratefully acknowledged.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.a) Keyzers RA, Northcote PT, Davies-Coleman MT. Nat Prod Rep. 2006;23:321. doi: 10.1039/b503531g. [DOI] [PubMed] [Google Scholar]; (b) Gonzalez M. Curr Bioact Comp. 2007;3:1. [Google Scholar]
- 2.a) Hambley TW, Poiner A, Taylor WC. Tetrahedron Lett. 1986;27:3281. [Google Scholar]; b) Buckleton JS, Bergquist PR, Cambie RC, Clark GR, Karuso P, Rickard CEF. Acta Crystallogr. 1986;C42:1846. [Google Scholar]; c) Molinski TF, Faulkner DJ, He CH, Van Duyne GD, Clardy JJ. J Org Chem. 1986;51:4564. [Google Scholar]
- 3.For a discussion on the challenge of assembling attached-ring stereocenters and examples, see: Overman LE, Velthuisen EJ. J Org Chem. 2006;71:1581. doi: 10.1021/jo0522862.
- 4.a) Schnermann MJ, Beaudry CM, Egorova AV, Polishchuk RS, Sutterlin C, Overman LE. Proc Natl Acad Sci USA. 2010;107:6158. doi: 10.1073/pnas.1001421107. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schnermann MJ, Beaudry CM, Genung NE, Canham SM, Untiedt NL, Karanikolas BDW, Sutterlin C, Overman LE. J Am Chem Soc. 2011;133:17494. doi: 10.1021/ja207727h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schnermann MJ, Overman LE. J Am Chem Soc. 2011;133:16425. doi: 10.1021/ja208018s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For the initial development of this strategy in the context of a total synthesis of a structurally less complex spongian diterpene, see: Lebsack AD, Overman LE, Valentekovich RJ. J Am Chem Soc. 2001;123:4851. doi: 10.1021/ja015802o.
- 7.Schnermann MJ, Untiedt NL, Jiménez–Osés G, Houk KN, Overman LE. previous communication in this issue.
- 8.For select recent reviews, see: Bar G, Parsons AF. Chem Soc Rev. 2003;32:251. doi: 10.1039/b111414j.Srikanth GSC, Castle SL. Tetrahedron. 2005;61:10377.
- 9.No examples of the stereoselective formation of quaternary carbon stereocenters from intermolecular additions of alkyl-substituted tertiary radicals to alkenes are mentioned in a recent comprehensive treatise, see Christoffers J, Baro A, editors. Quaternary Stereocenters–Challenges and Solutions for Organic Synthesis. Wiley–VCH; Weinheim: 2005. pp. 287–314.. We are aware of only one report of such reactions occurring with high stereoselectivity, see: Gansäuer A, Bluhm H, Rinker B, Narayan S, Schick M, Lauterbach T, Pierobon M. Chem Eur J. 2003;9:531. doi: 10.1002/chem.200390056.. For examples of such reactions that take place with poor to moderate diastereoselectivity, see: Damm W, Giese B, Hartung J, Hasskerl T, Houk KN, Zipse H. J Am Chem Soc. 1992;114:4067. and Srikrishna A, Hemamalini P, Sharma GVR. J Org Chem. 1993;58:2509.
- 10.For one example, see; Cossy J, Bouzbouza S, Hakiki A. Tetrahedron. 1999;55:11289.
- 11.The addition of achiral tertiary radicals to chiral alkenes often take place with higher diastereoselectivity than the corresponding reactions of secondary and primary radicals; see, for example: Giese B, Witzel T. Tetrahedron Lett. 1987;28:2571.Toru T, Watanabe Y, Tsusaka M, Ueno Y. J Am Chem Soc. 1993;115:10464.Hayen A, Koch R, Saak W, Haase D, Metzger JO. J Am Chem Soc. 2000;122:12458.
- 12.There are many examples of the use of tertiary radicals generated from Barton esters to form achiral quaternary carbons, see: Barton DHR, Crich D, Kretzschmar G. Tetrahedron Lett. 1984;25:1055.Barton DHR, Crich D, Kretzschmar G. J Chem Soc, Perkin Trans 1. 1986:39.
- 13.Lebsack A. PhD Dissertation. University of California; Irvine (USA): 2002. [Google Scholar]
- 14.Okada K, Okamoto K, Morita N, Okubo K, Oda M. J Am Chem Soc. 1990;113:9401. [Google Scholar]
- 15.Okada also reported light-promoted decarboxylative reduction, chlorination, and selenylation of N-(acyloxy)phthalimides, see: Okada K, Okamoto K, Oda M. J Am Chem Soc. 1988;110:8736.Okada K, Okamoto K, Oda M. J Chem Soc-Chem Comm. 1989:1636.Okada K, Okubo K, Morita N, Oda M. Chemistry Letters. 1993:2021.Okada K, Okubo K, Morita N, Oda M. Tetrahedron Lett. 1992;33:7377.(b) Two other groups have used this decarboxylative reduction, see: Fisher MJ, Myers CD, Joglar J, Chen SH, Danishefsky SJ. J Org Chem. 1991;56:5826.Cano M, Fabrias G, Camps F, Joglar J. Tetrahedron Lett. 1998;39:1079.
- 16.For important recent contributions, see: Nicewicz DA, MacMillan DWC. Science. 2008;322:77. doi: 10.1126/science.1161976.Ischay MA, Anzovino ME, Du J, Yoon TP. J Am Chem Soc. 2008;130:12886. doi: 10.1021/ja805387f.Nagib DA, Scott ME, MacMillan DWC. J Am Chem Soc. 2009;131:10875. doi: 10.1021/ja9053338.Narayanam JMR, Tucker JW, Stephenson CRJ. J Am Chem Soc. 2009;131:8756. doi: 10.1021/ja9033582.
- 17.For recent reviews, see: Zeitler K. Angew Chem, Int Ed. 2009;48:9785. doi: 10.1002/anie.200904056.Angew Chem. 2009;121:9969.Yoon TP, Ischay MA, Du J. Nat Chem. 2010;2:527. doi: 10.1038/nchem.687.Narayanam JMR, Stephenson CRJ. Chem Soc Rev. 2011;40:102. doi: 10.1039/b913880n.Telpy F. Collect Czech Chem Commun. 2011;76:859.
- 18.Available in five steps from cyclooctene oxide, see supporting information for details.
- 19.An excess of (1.5 equiv) of the hydrogen donor was required to suppress over-addition of methyl vinyl ketone. The use of blue LEDs strips was experimentally convenient; the reaction took place in similar yield using a compact fluorescent bulb.
- 20.Kreiser W, Below P. Liebigs Ann Chem. 1985:203. [Google Scholar]
- 21.The hydroxyl group of the ylide was protected in situ as a trimethylsilyl ether and then cleaved during workup using 2 N hydrochloric acid, see: Couturier M, Dory YL, Rouillard F, Deslongchamps P. Tetrahedron. 1998;54:1529.
- 22.a) The displacement of the primary iodide with silver nitrite was most effective in the absence of solvent, in which case only a slight excess of AgNO2 was required. This reaction is typically carried out in ethereal solvents using 3–5 equiv of AgNO2, see, for example: Hewson AT, Macpherson DT. J Chem Soc, Perkin Trans 1. 1985:2625.
- 23.For the use of this approach in forming a perhydroazulene ring system, see: Kozikowski AP, Mugrage BB, Wang BC, Xu ZB. Tetrahedron Lett. 1983;24:3705.
- 24.Mukaiyama T, Hoshino T. J Am Chem Soc. 1960;82:5339. [Google Scholar]
- 25.Curran DP. J Am Chem Soc. 1983;105:5826. [Google Scholar]
- 26.We found that Raney-nickel was most effective for cleaving the N-O bond and Pd/C for hydrogenation of the alkene. Elevated temperature with either catalyst delivered the β-hydroxy ketone, albeit in reduced yield.
- 27.This sequence was readily scaled; more than 25 g of 24 has been prepared by this method.
- 28.This mixture of isomers is inconsequential; it can be carried forward to the coupling reaction or separated at this stage.
- 29.The use of HMPA is required in this reaction; in its absence 25 was not observed and only complex mixtures of products were obtained.
- 30.Available in 83% yield by chlorination of the corresponding enone[4] with the Mioskowski reagent (Et4NCl3), at 0 °C.[31]
- 31.Schlama T, Gabriel K, Gouverneur V, Mioskowski C. Angew Chem, Int Ed. 1997;36:2342. [Google Scholar]; Angew Chem. 1997;109:2440. [Google Scholar]
- 32.The relative configuration of 30 was secured by crystallographic analysis: CCDC 885482.
- 33.The use of the corresponding α-bromoenone (13, X = Br) in the radical coupling reaction resulted in extensive reductive debromination of coupled product.
- 34.a) Andrews RS, Becker JJ, Gagné MR. Angew Chem, Int Ed. 2010;49:7274. doi: 10.1002/anie.201004311. [DOI] [PubMed] [Google Scholar]; Angew Chem. 2010;122:7432. [Google Scholar]; b) Andrews RS, Becker JJ, Gagné MR. Org Lett. 2011;13:2406. doi: 10.1021/ol200644w. [DOI] [PubMed] [Google Scholar]
- 35.In line with previous observations,[34] we found that the use of 29 in conjunction with N,N-disopropylethylamine was essential for realizing high yields and short reaction times.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.