

Abstract
A bicyclam-based biodegradable polycation with CXCR4 antagonistic activity was developed with potential for combined drug/gene cancer therapies. The dual-function polycation prevents cancer cell invasion by inhibiting CXCL12 stimulated CXCR4 activation, while at the same time efficiently and safely delivers plasmid DNA into cancer cells.
Keywords: CXCR4, Polycations, Transfection, DNA, Polyplexes
Synthetic polycations have been investigated widely as delivery vectors of nucleic acid therapeutics, such as plasmid DNA and small interfering RNA. Polycations form polyelectrolyte complexes (polyplexes) with the nucleic acids, thereby protecting them from degradation and facilitating transport across cellular membranes. Significant effort has been devoted to synthesizing polycations that have improved transfection activity and reduced toxicity.[1] Traditionally, polycations have been viewed as pharmacologically inert components of delivery systems.
Herein we propose a conceptually new approach that polycations can have both a delivery function as well as pharmacologic activity, to enhance the therapeutic outcome of gene and RNA interference therapies. We describe a novel class of polycations that not only deliver plasmid DNA but also function as CXCR4 antagonists that inhibit cancer cell invasion and thus could limit metastasis in various approaches to gene therapy for cancer (Scheme 1).
Scheme 1.

Mechanism of action of dual-function polycations as CXCR4 antagonists and gene delivery vectors.
CXCR4 is a highly conserved transmembrane G-protein-coupled receptor that exclusively binds its ligand CXCL12. Several studies established that cancer cells utilize the CXCL12/CXCR4 axis to metastasize to distant sites.[2] Consistent with the seed-and-soil hypothesis of metastatic dissemination,[3] high levels of CXCL12 are found in sites commonly affected by metastases in cancer types known to overexpress CXCR4.[4] CXCR4 expression is often associated with poor survival and aggressive types of cancer.[5]
Among the most widely investigated CXCR4 inhibitors are cyclam derivatives,[6] including AMD3100 (Figure 1a). It is a highly specific CXCR4 antagonist, as it inhibits binding and signaling of CXCL12.[7] The AMD3100 binding site on CXCR4 and its antimetastasis activity have been well characterized.[8] Not all eight amino groups of AMD3100 are required for that activity,[6a] making it a suitable building block of CXCR4 antagonizing polycations described in this study.
Figure 1.

Synthesis and characterization of RPA. (a) RPA synthesis by Michael addition polymerization (*any secondary amine of the ring could be substituted and there could be multiple substitutions per ring); (b) Size exclusion chromatogram of RPA; (c) Comparison of cytotoxicity of RPA and PEI 25 kDa in HepG2 cells (RPA: ○, PEI: ●) and CXCR4+ U2OS cells (RPA: △, PEI: ▲) determined by MTS.
Although several reports have described lipid-based delivery vectors that incorporated peptide-targeting ligands to increase delivery to CXCR4-overexpressing cells,[9] no attempts have been made to synthesize polymeric CXCR4 antagonists that take advantage of their antagonistic properties to enhance the outcomes of gene therapies. Here, we synthesized such polycations, named RPA, by direct Michael addition polymerization of AMD3100 with a disulfide-containing bisacrylamide CBA (Figure 1). Soluble RPAs were synthesized using a molar ratio of AMD3100 to CBA 1:1 in MeOH/water at 37°C. The reaction was terminated by addition of excess AMD3100 to assure consumption of all residual acrylamides and presence of the terminal cyclam residues in the prepared RPA. RPA was purified by precipitation and extensive dialysis. The weight-average molecular weight, Mw, of the RPA used in this study was 13.9 kDa, and the polydispersity index, Mw/Mn, was 1.26 (Figure 1b). A control bioreducible polycation that lacks CXCR4 activity, RHB, was synthesized by copolymerization of CBA with N,N-dimethylaminodipropylene-triamine as described previously[10] and had Mw = 11.3 kDa and Mw/Mn = 1.95. Cytotoxicity of RPA was measured by MTS assay in HepG2 liver cells and in U2OS osteosarcoma cells overexpressing CXCR4 (Figure 1c). In both cell lines RPA had remarkably low toxicity compared with 25-kDa poly(ethyleneimine) (PEI) control. The IC50 of RPA was almost 50 times higher than that of PEI in HepG2 cells (599 vs. 12 μg/mL) and 116 times higher in U2OS cells (464 vs. 4 μg/mL). The IC50 of RHB was 57 μg/mL in HepG2 (Figure S5 in Supporting Information). RPA and plasmid DNA formed polyplexes that were positively charged (25 mV, RPA/DNA w/w ratio 5) and had a relatively small size (56 nm at w/w 5) compared with PEI and RHB polyplexes (84 nm at w/w 1.2 and 157 nm at w/w 5, respectively) (Table S1 in Supporting Information). Glutathione treatment of the RPA/DNA polyplexes triggered DNA release due to depolymerization of RPA (Figure S4 in Supporting information).
When CXCL12 binds to CXCR4 it induces downstream signaling through multiple pathways, including Ras and PI3 kinase. Treatment with CXCR4 antagonists not only prevents the CXCL12-induced downstream signaling but it also inhibits endocytosis of the receptor. To evaluate CXCR4 antagonism by RPA and RPA/DNA, we used a receptor redistribution assay (Figure 2). The assay uses U2OS cells stably expressing human CXCR4 receptor fused to the N-terminus of enhanced green fluorescent protein (GFP). The assay monitors cellular translocation of GFP-CXCR4 upon stimulation with CXCL12. We observed internalization of the CXCR4 receptor into endosomes in CXCL12-stimulated cells, as suggested by the punctate fluorescence distribution (Figure 2b) away from the original diffuse pattern (Figure 2a). RPA inhibited the CXCL12-triggered CXCR4 internalization in a dose-dependent manner, and full inhibition was observed above 0.5 μg/mL (Figure S6 in Supporting Information). To exclude the possibility that the observed effect was caused by nonspecific electrostatic binding of RPA to the negatively charged binding site of the CXCR4 receptor, we used control RHB but observed no CXCR4 antagonism (Figure 2f).
Figure 2.

CXCR4 antagonism of RPA and RPA/DNA polyplexes. CXCR4 receptor redistribution assay was conducted in U2OS cells expressing GFP-tagged CXCR4 (a). Before stimulation with 10 nM CXCL12, the cells were treated for 30 min with (b) no drug; (c) 0.24 μg/mL AMD3100.8HCl; (d) 1.5 μg/ml RPA.HCl; (e) 2.5 μg/ml RPA.HCl; (f) 1.5 μg/ml RHB.HCl; (g) RPA/DNA polyplexes (w/w 5, total RPA conc. 2.5 μg/ml); (h) RPA/DNA polyplexes (w/w 1, total RPA conc. 0.5 μg/ml); and (i) RHB/DNA polyplexes (w/w 5, total RHB conc. 2.5 μg/ml). The scale bars for all the images are 200 μm.
Next, we examined whether polyplexes themselves exhibit CXCR4 antagonism. CXCR4 internalization was inhibited more efficiently by RPA/DNA prepared at w/w 5 (2.5 μg/mL total RPA) than at w/w 1 (Figure 2g and 2h). DNA is not fully condensed in polyplexes at w/w 1 (Figure S3 in Supporting Information); thus, the formulation contains only a minimum amount of free RPA, suggesting that the polyplexes themselves may inhibit CXCR4. Similar to RHB, no CXCR4 antagonism was observed with RHB/DNA polyplexes (Figure 2i), confirming the specific CXCR4 antagonism of RPA and RPA/DNA.
The CXCR4/CXCL12 axis plays a critical role in cancer metastasis due to its function in trafficking and homing of cancer cells to organs that express high levels of CXCL12. Blocking the CXCR4/CXCL12 interactions with small-molecule antagonists suppresses metastasis in a variety of cancers.[11] Using a Matrigel invasion assay, we show that RPA and RPA/DNA effectively block CXCL12-mediated invasion of CXCR4+ U2OS cells (Figure 3). Both free RPA and RPA/DNA blocked invasion of 71–77% of cells, similar to that of AMD3100 (75%). The DNA dose used in the experiment with the polyplexes (1 μg/mL DNA) was in the range of typical doses used in transfection experiments. The observed decrease in cell invasion with control RHB/DNA polyplexes was not statistically significant (Supporting Information).
Figure 3.
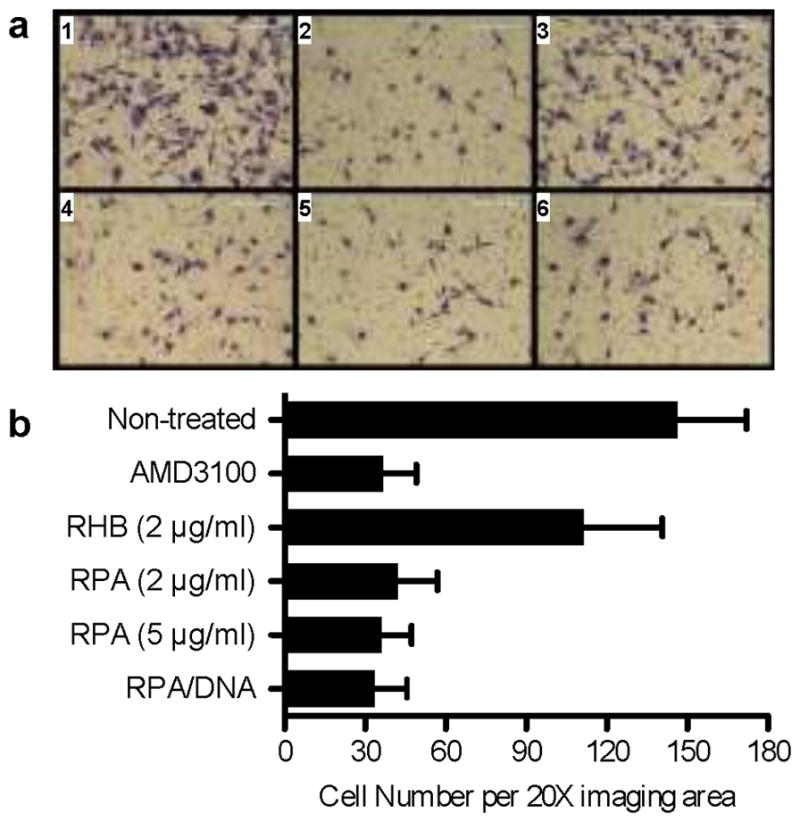
Inhibition of cancer cell invasion by RPA and RPA/DNA polyplexes. (a) Cell invasion assay with CXCR4+ U2OS cells treated with 1) no drug; 2) 0.24 μg/mL AMD3100.8HCl; 3) 2 μg/ml RHB.HCl; 4) 2 μg/ml RPA.HCl; 5) 5 μg/ml RPA.HCl and 6) RPA/DNA polyplexes (w/w 5, total RPA conc, 5 μg/ml). Cells were seeded in Matrigel-coated inserts and allowed to invade towards CXCL12-containing medium for 16 h before fixation and imaging. (b) Average number of invaded cells in 20× imaging area.
Having confirmed CXCR4 antagonism and inhibition of cancer cell invasion of the synthesized RPA, we then evaluated its gene delivery capability (Figure 4). RPA/DNA polyplexes mediated transfection that was fully comparable with that of control PEI/DNA polyplexes in B16F10 and U2OS cell lines at a DNA dose of 2.35 μg/mL.
Figure 4.
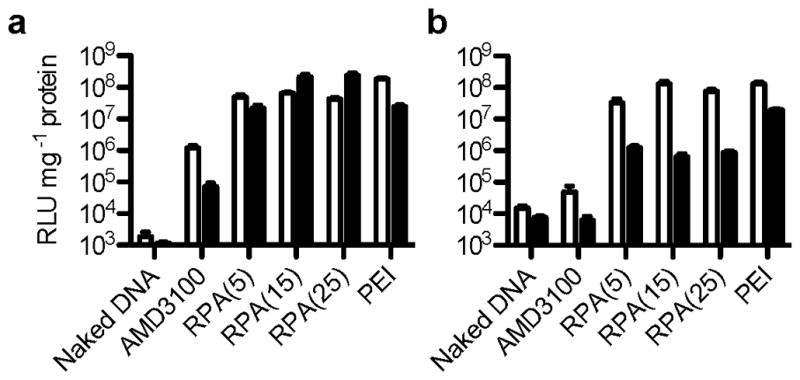
Transfection activity of RPA/DNA polyplexes prepared at w/w 5, 15 and 25 in the absence (white bars) and the presence (black bars) of 10% FBS in (a) B16F10 and (b) CXCR4+ U2OS cells. (Results are shown as mean luciferase expression in RLU/mg protein ± SD, n=3).
CXCR4 antagonism by RPA does not require the polymer to be internalized, which is advantageous because no intracellular barriers need to be overcome.[13] However, because the CXCR4 receptor is not internalized in the presence of an antagonist like RPA, binding of RPA/DNA to this receptor would be unproductive in mediating transfection. Polyplex transfection by CXCR4-mediated uptake was clearly unsuitable in experiments that attempted to use AMD3100 to target PEI polyplexes to CXCR4-overexpressing cells. The authors found no significant increase in transfection unless phorbol myristate acetate (PMA) was used to trigger CXCR4 receptor endocytosis.[12] Here we show that RPA/DNA polyplexes use an alternative uptake pathway that does not require CXCR4. This is documented by the lack of signal from RPA/DNA polyplexes with fluorescently labeled DNA colocalized with the membrane-present CXCR4 receptor (Figure S9 in Supporting Information). Further support for the lack of involvement of the CXCR4 receptor in transfection activity of RPA/DNA comes from experiments showing that PMA did not enhance transfection despite its ability to trigger internalization of the CXCR4 receptor by an alternative pathway from CXCL12 (Figures S7 and S8 in Supporting Information). Importantly for practical utility of the dual-function polycations, we also show concurrent CXCR4 inhibition and transfection with RPA/DNA polyplexes (Figure S10 in Supporting Information). These findings support our proposed mechanism of action in which a small part of the free RPA inhibits CXCR4 while the rest of the RPA/DNA polyplex formulation participates in transfection, most likely through nonspecific charge-mediated uptake.
In summary, we have described the preparation of a synthetic polycation that functions as a CXCR4 antagonist capable of blocking cancer cell invasion while simultaneously effectively delivering plasmid DNA and mediating transfection. Such dual-function delivery vectors can enhance antimetastatic efficacy of a variety of cancer gene therapy protocols.
Supplementary Material
Footnotes
This work was supported in part by a grant from the Office of Vice President for Research, Wayne State University and in part by NIH grant EB014570.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Jing Li, Department of Pharmaceutical Sciences, Wayne State University, 259 Mack Avenue, Detroit, Michigan 48202, USA.
Yu Zhu, Department of Pharmaceutical Sciences, Wayne State University, 259 Mack Avenue, Detroit, Michigan 48202, USA.
Dr. Stuart T. Hazeldine, Department of Pharmaceutical Sciences, Wayne State University, 259 Mack Avenue, Detroit, Michigan 48202, USA
Prof. Chunying Li, Department of Biochemistry and Molecular Biology, Wayne State University, 540 E. Canfield, Detroit, Michigan 48202, USA
David Oupický, Email: oupicky@wayne.edu, Department of Pharmaceutical Sciences, Wayne State University, 259 Mack Avenue, Detroit, Michigan 48202, USA.
References
- 1.Schaffert D, Troiber C, Salcher EE, Fröhlich T, Martin I, Badgujar N, Dohmen C, Edinger D, Kläger R, Maiwald G, Farkasova K, Seeber S, Jahn-Hofmann K, Hadwiger P, Wagner E. Angew Chem Int Ed. 2011;50:8986–8989. doi: 10.1002/anie.201102165. [DOI] [PubMed] [Google Scholar]
- 2.Balkwill F. Nat Rev Cancer. 2004;4:540–550. doi: 10.1038/nrc1388. [DOI] [PubMed] [Google Scholar]
- 3.a) Burger JA, Kipps TJ. Blood. 2006;107:1761–1767. doi: 10.1182/blood-2005-08-3182. [DOI] [PubMed] [Google Scholar]; b) Hanahan D, Weinberg RA. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 4.a) Rhodes LV, Short SP, Neel NF, Salvo VA, Zhu Y, Elliott S, Wei YK, Yu DH, Sun MH, Muir SE, et al. Cancer Res. 2011;71:603–613. doi: 10.1158/0008-5472.CAN-10-3185. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, et al. Nature. 2001;410:50–56. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]; c) Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Cancer Res. 2002;62:1832–1837. [PubMed] [Google Scholar]; d) Chinni SR, Sivalogan S, Dong Z, Filho JC, Deng X, Bonfil RD, Cher ML. Prostate. 2006;6(6):32–48. doi: 10.1002/pros.20318. [DOI] [PubMed] [Google Scholar]; e) Liang ZX, Yoon YH, Votaw J, Goodman MM, Williams L, Shim H. Cancer Res. 2005;65:967–971. [PMC free article] [PubMed] [Google Scholar]
- 5.a) Akashi T, Koizumi K, Tsuneyama K, Saiki I, Takano Y, Fuse H. Cancer Sci. 2008;99:539–542. doi: 10.1111/j.1349-7006.2007.00712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wallace TA, Prueitt RL, Yi M, Howe TM, Gillespie JW, Yfantis HG, Stephens RM, Caporaso NE, Loffredo CA, Ambs S. Cancer Res. 2008;6(8):927–936. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 6.a) Bridger GJ, Skerlj RT, Hernandez-Abad PE, Bogucki DE, Wang Z, Zhou Y, Nan S, Boehringer EM, Wilson T, Crawford J, Metz M, Hatse S, Princen K, De Clercq E, Schols D. J Med Chem. 2010;53:1250–1260. doi: 10.1021/jm901530b. [DOI] [PubMed] [Google Scholar]; b) Dessolin J, Galea P, Vlieghe P, Chermann JC, Kraus JL. J Med Chem. 1999;4(2):229–241. doi: 10.1021/jm980358u. [DOI] [PubMed] [Google Scholar]; c) Khan A, Greenman J, Archibald SJ. Curr Med Chem. 2007;14:2257–2277. doi: 10.2174/092986707781696618. [DOI] [PubMed] [Google Scholar]; d) Khan A, Nicholson G, Greenman J, Madden L, McRobbie G, Pannecouque C, De Clercq E, Ullom R, Maples DL, Maples RD, et al. J Am Chem Soc. 2009;131:3416–3417. doi: 10.1021/ja807921k. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Lam AR, Bhattacharya S, Patel K, Hall SE, Mao A, Vaidehi N. J Chem Inf Model. 2010;51:139–147. doi: 10.1021/ci1003027. [DOI] [PubMed] [Google Scholar]; f) Rosenkilde MM, Gerlach LO, Hatse S, Skerlj RT, Schols D, Bridger GJ, Schwartz TW. J Biol Chem. 2007;282:27354–27365. doi: 10.1074/jbc.M704739200. [DOI] [PubMed] [Google Scholar]
- 7.De Clercq E. Nat Rev Drug Discov. 2003;2:581–587. doi: 10.1038/nrd1134. [DOI] [PubMed] [Google Scholar]
- 8.Hatse S, Princen K, Bridger G, De Clercq E, Schols D. FEBS Lett. 2002;527:255–262. doi: 10.1016/s0014-5793(02)03143-5. [DOI] [PubMed] [Google Scholar]
- 9.a) Driessen WHP, Fujii N, Tamamura H, Sullivan SM. Mol Ther. 2008;16:516–524. doi: 10.1038/sj.mt.6300388. [DOI] [PubMed] [Google Scholar]; b) Egorova A, Kiselev A, Hakli M, Ruponen M, Baranov V, Urtti A. J Gene Med. 2009;1(1):772–781. doi: 10.1002/jgm.1366. [DOI] [PubMed] [Google Scholar]
- 10.Chen J, Wu C, Oupicky D. Biomacromolecules. 2009;10:2921–2927. doi: 10.1021/bm900724c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Liang Z, Zhan W, Zhu A, Yoon Y, Lin S, Sasaki M, Klapproth JMA, Yang H, Grossniklaus HE, Xu J, et al. PLoS One. 2012;7:e34038. doi: 10.1371/journal.pone.0034038. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yoon Y, Liang Z, Zhang X, Choe M, Zhu A, Cho HT, Shin DM, Goodman MM, Chen Z, Shim H. Cancer Res. 2007;67:7518–7524. doi: 10.1158/0008-5472.CAN-06-2263. [DOI] [PubMed] [Google Scholar]
- 12.Le Bon B, Craynest NV, Daoudi JM, Giorgio CD, Domb AJ, Vierling P. Bioconjugate Chem. 2004;1(5):413–423. doi: 10.1021/bc034220o. [DOI] [PubMed] [Google Scholar]
- 13.Wu K, Liu J, Johnson RN, Yang J, Kopeček J. Angew Chem Int Ed. 2010;49:1451–1455. doi: 10.1002/anie.200906232. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.