

Abstract
Small changes – big effect A new aureothin derivative, aureopyran, that features an unusual pyran backbone was generated by simply altering the enzymatic methylation topology. The α-pyrone ring hampers the correct placement of the polyketide backbone in the multifunctional cytochrome P450 monooxygenase AurH. Instead of a THF ring an oxo intermediate is formed that readily undergoes a rare electrocyclization reaction.
Keywords: Aureothin, AurH, Combinatorial biosynthesis, Cytochrome P450 monooxygenase, Polyketide
Cytochrome P450 (CYP) monooxygenases are known as multifarious biocatalysts that are able to transform a variety of substrates.[1–3] Members of this large group not only play an essential role in the degradation of biomolecules and xenobiotics in the liver, [4] but are also often components of biosynthetic assembly lines leading to complex natural products such as polyketides.[5, 6] In these pathways the high regio- and stereoselectivities by CYP monooxygenases is often striking and usually cannot be emulated by the synthetic reagents available.[7, 8] However, in light of the large number of known CYPs, the range of oxygenation reactions is surprisingly small. Apart from few exceptions, CYP monooxygenases generally function as hydroxylases or epoxidases. In this respect AurH is a rather exotic CYP as it is essential and sufficient to form the pharmacophoric THF moiety in aureothin (1) (Scheme 1), an antifungal and cytotoxic Streptomyces thioluteus metabolite.[9] Similarly, the homologue NorH mediates THF formation of neoaureothin (spectinabilin)[10] and the rearrangement products SNF4435C/D and orinocin.[11] Studies at the genetic, biochemical and chemical levels revealed that AurH is uniquely capable of stereoselectively hydroxylating the polyketide backbone of 2 at the C-7 position and subsequently mediating heterocyclization of the hydroxyl-intermediate 3.[12–14] For this unparalleled reaction sequence to happen, a finely tuned reactive site is crucial, as was shown by modeling studies using the AurH crystal structure and targeted point mutations.[15] Here, we report the unexpected discovery of a pyran analogue of aureothin that is generated by AurH using a non-natural α-pyrone substrate. Mechanistic considerations and a model using the x-ray structure of AurH support a four-electron oxidation yielding an oxo group with subsequent rearrangement to yield the 2H-pyran ring.
Scheme 1.
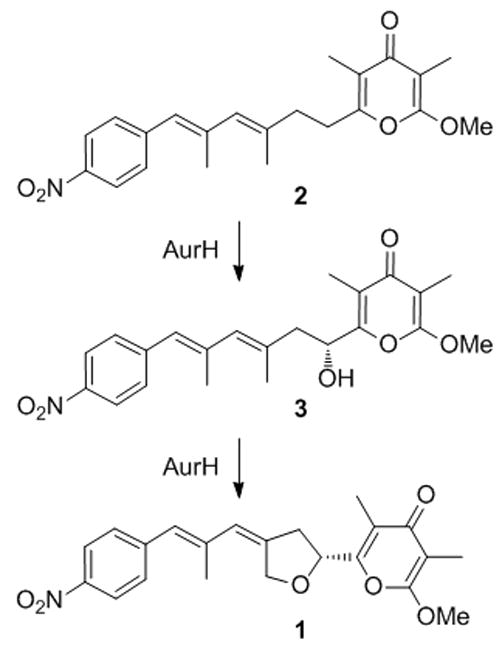
Sequential oxygenation of deoxyaureothin (2) to aureothin (1) via 3 by a multifunctional cytochrome P450 monooxygenase, AurH.
Our previous mutagenesis experiments and using variants of AurH for biotransformations revealed that the binding of the pyrone substrate is crucial for the proper course of the reaction. Tampering the binding site of AurH in same cases resulted in the aberrant multiple oxygenation of the allylic methyl group (C-9a) in lieu of the methylene (C-7).[15] We conceived setting up a similar scenario, yet confronting wild-type AurH with a slightly altered substrate. A promising strategy was to exchange the γ-pyrone for an α-pyrone ring. Whereas AurI specifically forms γ-pyrones, [16] the only known methyltransferase producing an O-methylated α-pyrone ring is EncK from the enterocin pathway.[17, 18] We succeeded in changing the methylation topology by swapping methyltransferase genes from the aureothin and enterocin pathways in a ΔaurH mutant, yielding deoxyisoaureothin (5) (Scheme 2).[19] To probe the fate of this compound in the presence of AurH in vivo we aimed at generating a mutant containing the all aur genes except for the MT gene aurI and complemented this mutant encK. To avoid possible side reactions by a peroxide shunt in vitro we chose to probe the biotransformation in vivo under natural conditions.
Scheme 2.
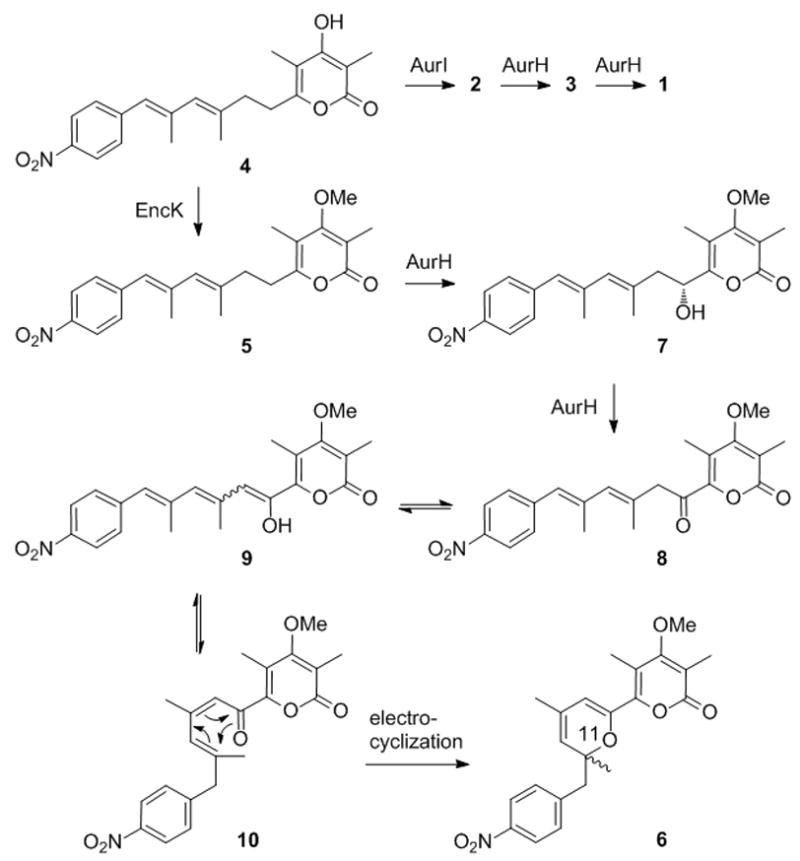
Model for four-electron oxidation of deoxyisoaureothin (5) to oxo intermediate 8 and electrocyclic (non-enzymatic) rearrangement to yield pyran (6)
Specifically, a ΔaurI mutant was created by excising the aurI gene from an integrative Streptomyces vector (pHJ48) containing the aur gene cluster, [20] yielding pBU172. For heterologous expression plasmid pBU172 was introduced into Streptomyces albus via tri-parental mating, and exo-conjugants were selected with appropriate antibiotics. Subsequently, expression plasmids pHJ72 and pHJ95 harboring the encK and aurI genes, respectively, were individually introduced into S. albus::pBU172 by PEG-mediated protoplast transformation. The metabolic profiles of the resulting strains, S. albus::pBU172/pHJ72 and S. albus::pBU172/pHJ95, as well as S. albus::pBU172 were monitored by HPLC-MS. As expected, aureothin production was abolished in S. albus::pBU172. Instead of aureothin nordeoxyaureothin (4) was detected.[16] In the complemented ΔaurI mutant strain, S. albus::pBU172/pHJ95, aureothin production was fully restored. In the ΔaurI mutant complemented with encK (S. albus::pBU172/pHJ72) a more complex metabolic pattern was observed. Besides the immediate product of the modular aur PKS, nordeoxyaureothin (4), a new compound (6) was detected. High-resolution mass spectrometry (calcd. for [M+H]+: 398.1600 Da, found: 398.1567 Da) suggested that 6 has a molecular formula of C22H23NO6, which is actually identical with the one for aureothin. However, the retention times of both compounds and the UV spectra differed starkly.
From a 400 mL culture of S. albus::pBU172/pHJ72 supplemented with Amberlite XAD16 resin, 0.4 mg of a new nitro compound 6 were isolated and purified by open column chromatography and preparative HPLC. This small amount was sufficient to allow for a full structural characterization by 1D and 2D NMR experiments as well as by HRESI-MS and IR measurements. Although NMR data of p-nitro benzoate and pyrone moieties (γ-pyrone ring in 1) in 6 were almost identical to those of 1, NMR data from position 7 to 12 for 6 differed substantially from those for 1 (Table 1). The diagnostic signals for the aureothin THF ring were different or absent 1H NMR spectrum of 6. In particular, a triplet for the oxymethine proton (δ 5.17, H-7), two doublets for the methylene protons (δ 4.89 and 4.77, H-9a), a singlet methine proton (δ 6.42, H-12) were lacking. On the other hand, additional signals appeared in 1H NMR spectrum of 6, one methyl proton (δ 1.78, H-9a) and two doublets for methylene protons (δ 3.10 and 3.03, H-12). The HMBC correlations from H-8 (δ 6.00) to C-7 and C-8, H-9a to C-8, C-9, and C-10, H-11a to C-10, C-11, and C-12 corroborated the C-6 to C-12 stretch. Finally the chemical shifts δ 145.3 (C-7) and δ 79.9 (C-11) indicated that these carbons adjacent to an oxygen functionality (Figure 2) Because of the unusual pyran moiety the new compound was named aureopyran. Notably, whereas 6 features a chiral center at C-11, the CD spectrum revealed that 6 lacks optical activity and appears to be formed as a racemate.
Table 1.
Comparison of NMR data of aureothin (1) and aureopyran (6)
| Position | Aureopyran (6) | Aureothin (1) | ||
|---|---|---|---|---|
|
| ||||
| 1H (mult, J Hz) | 13C (mult) | 1H (mult, J Hz) | 13C (mult) | |
| 2 | 164.5 (s) | 162.0 (s) | ||
| 2a | – | 3.97 (s) | 55.2 (q) | |
| 3 | 112.5 (s) | 99.9 (s) | ||
| 3a | 2.06 (s) | 10.5 (q) | 1.90 (s) | 6.8 (q) |
| 4 | 168.3 (s) | 180.5 (s) | ||
| 4a | 3.78 (s) | 60.4 (q) | – | |
| 5 | 112.2 (s) | 120.1 (s) | ||
| 5a | 2.03 (s) | 10.6 (q) | 2.04 (s) | 9.4 (q) |
| 6 | 148.4 (s) | 154.6 (s) | ||
| 7 | 145.3 (s) | 5.17 (t 6.5) | 73.2 (d) | |
| 8 | 6.00 (s) | 107.5 (d) | 3.05 (m) | 38.2 (t) |
| 9 | 130.2 (s) | 140.5 (s) | ||
| 9a | 1.78 (d 1.5) | 20.2 (q) | 4.89 (d 14.2) 4.77 (d 14.2) |
70.0 (t) |
| 10 | 4.97 (t 1.5) | 120.1 (d) | 6.22 (s) | 128.3 (d) |
| 11 | 79.9 (s) | 138.5 (s) | ||
| 11a | 1.37 (s) | 25.4 (q) | 2.08 (s) | 17.6 (q) |
| 12 | 3.10 (d 13.4) 3.03 (d 13.4) |
45.2 (t) | 6.42 (s) | 125.9 (d) |
| 13 | 144.4 (s) | 144.1 (s) | ||
| 14, 18 | 7.32 (d 8.8) | 131.5 (d) | 7.43 (d 8.5) | 129.5 (d) |
| 15, 17 | 8.11 (d 8.8) | 123.0 (d) | 8.20 (d 8.5) | 123.5 (d) |
| 16 | 146.7 (s) | 146.0 (s) | ||
Figure 2.
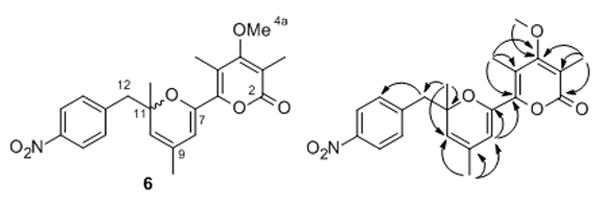
Structure of aureopyran (6) and key 2D NMR correlations.
The structure of aureopyran (6), specifically its enol ether moiety, suggests that this metabolite could be derived from a 7-oxo-functionalized deoxyisoaureothin intermediate (Scheme 2). A plausible scheme would be that deoxyisoaureothin was first hydroxylated at C-7 in analogy to the natural substrate. However, because of the different pyrone docking the non-natural substrate is likely misplaced at the active site. As a consequence, a second oxidation would take place at C-7, thus yielding an oxo functionality. By tautomerism a thermodynamically favored enol can be formed, with concomitant delocalization of the π-electron system throughout the entire backbone. This tautomerism may also facilitate an E/Z-isomerization, which is a prerequisite for the following heterocyclization. For the pyran ring closure, two scenarios are conceivable. Promoted by the electron-withdrawing nitroaryl moiety, the enol could add to the double bond adjacent to the aromatic ring. Alternatively, the polyketide backbone could cyclize by an electrocyclic rearrangement. In either way, the net outcome is a 2H-pyran moiety and formation of a new chiral centre. The finding that 6 is formed as a racemate is in accord with the model of a non-enzymatic reaction. The most plausible scenario would be an electrocyclization, which is often a key reaction in the rearrangement of polyunsaturated systems, as evidenced by biomimetic total synthesis.[21] The closed 2H-pyran would be thermodynamically preferred due to the electron-withdawing properties of the α–pyron ring in the 2-position of the 2H-pyran.[21] These findings are in line with the observed spontaneous cyclization of a synthetic oxo derivative of deoxyaureothin.[22]
In order to gain more insight into the potential course of the oxygenation step we aimed at modeling since co-crystallization and soaking experiments using substrate analogues into AurH crystals were unsuccessful. We first modeled deoxyisoaureothin (5) into both x-ray crystal structures of AurH that account for the two sequential reaction steps leading to the naturally occurring furan ring. The PDB entries 3P3X and 3P3L served as models for the first and the second step, respectively, of the natural reaction sequence. For each of the protein structures we observed a ligand orientation that renders a reaction at C7 possible. The binding energy of the 3P3X-based model is higher, and the distance between heme iron and C7 is smaller in the 3P3X-based model (−8.89 kcal/mol and 4.91 Å in 3P3X; −8.79 kcal/mol and 5.53 Å in 3P3L). Thus, the 3P3X-based model is more appropriate, and interestingly the (7R)-proton is directed towards the heme iron (Fe-C7-(7R)H-angle = 21.9°; Fe-C7-(7S)H-angle = 113.6°). This strongly supports the model in which (7R)-7-hydroxydeoxyisoaureothin (7) results from the first oxidation reaction (Scheme 2). In fact, this scenario is in full agreement with the first reaction of the natural substrate of AurH, deoxyaureothin (2), which is oxidized to yield the (7R)-hydroxylated intermediate (3, Scheme 1). Next, we modeled the tentative hydroxyl intermediate 7 into the templates 3P3X and 3P3L to obtain two models for downstream reactions. Again, the 3P3X-based model seems more reasonable because of the vicinity of C7 to the heme iron (5.58 Å in 3P3X; 6.85 Å in 3P3L). The binding energy for this model was calculated as −8.96 kcal/mol. The hydroxyl moiety is located orthogonally above the heme-iron (SCys355-Fe-O-angle = 174.1°), which places the functional group in a region, where a formation of an oxo-group (as in 8, Scheme 2) would be conceivable. These results strongly support the model for a four-electron oxidation that sets the stage for a rearrangement cascade that ultimately leads to pyran formation.
In recent years a growing number of microbial, fungal and plant CYPs have been identified and characterized that generate aldehyde or even carboxylate moieties by four- and six-electron oxidations of methyl groups. Such unusual biocatalysts have been found to catalyze key steps in terpene[23–28] and polyketide pathways.[29–31] However, to the best of our knowledge a scenario where a CYP initiates an electrocyclization by two consecutive oxidation reactions is fully unprecedented.
In summary, we have generated a new aureothin derivative, aureopyran (6), featuring an unusual pyran backbone by simply swapping the methylation topology. The α-pyrone methyltransferase EncK in lieu of the native γ-pyrone methyltransferase AurI generated deoxyisoaureothin (5), which was subject to a complex downstream processing. The results from biotransformations and modeling experiments clearly demonstrate that AurH accepts the non-natural substrate, deoxyisoaureothin (5). However, the α-pyrone ring hampers the correct placement of the polyketide backbone that is a prerequisite for the second oxygenation step. As a consequence, instead of a THF ring an oxo intermediate (8) is formed that can readily undergo a rare electrocyclization reaction, that is, to our knowledge, unprecedented in biosynthetic pathways.[21] These findings not only provide new insights into the action of a finely tuned cytochrome P450 monooxygenase, but also reveal a mechanistically intriguing reaction cascade. On combinatorial biosynthesis grounds it is remarkable that the exchange of a single methyl transferase gene opened an avenue to a new natural product skeleton. This result encourages future approaches to generate structural diversity by pathway engineering.
Experimental Section
(See Supplemental Material.)
Supplementary Material
Figure 1.
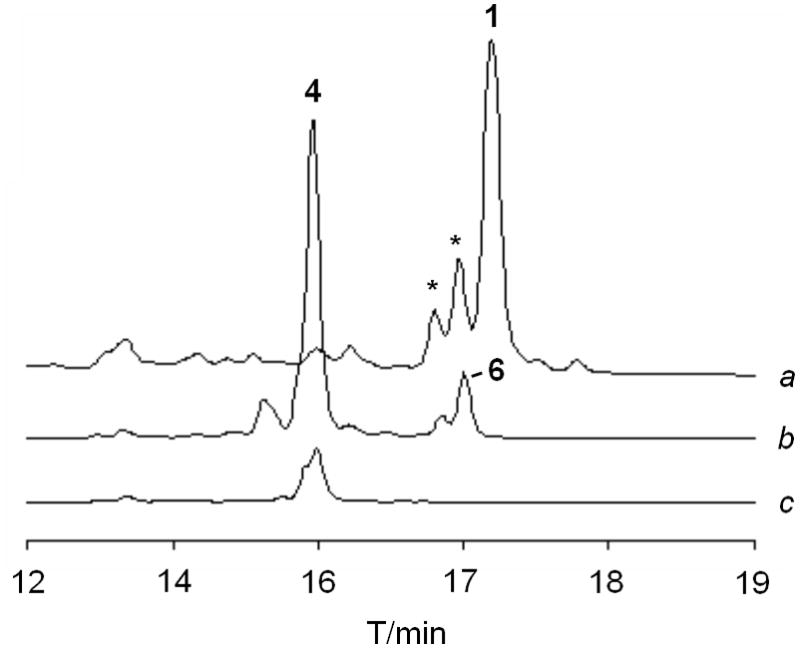
HPLC profiles of extracts from cultures of a) ΔaurI mutant complemented with aurI (peaks marked with asterisk designate E/Z-isomers of 1); b) ΔaurI mutant coexpressing encK; c) ΔaurI mutant.
Figure 3.


Models of deoxyisoaureothin (5, top) and (7R)-hydroxy intermediate (7, bottom) bound to the active site of AurH, supporting the proposed aberrant four-electron oxidation sequence.
Acknowledgments
(We thank A. Perner and F. Rhein for MS and NMR measurements, respectively. Financial support by the BMBF (GenoMik/GenBioCom) and the NIH (AI47818) is gratefully acknowledged.
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Werck-Reichhart D, Feyereisen R. Genome Biol. 2000;1:3003. doi: 10.1186/gb-2000-1-6-reviews3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong LL. Curr Opin Chem Biol. 1998;2:263–268. doi: 10.1016/s1367-5931(98)80068-9. [DOI] [PubMed] [Google Scholar]
- 3.Omura T. J Biochem. 2010;147:287–306. doi: 10.1093/jb/mvq001. [DOI] [PubMed] [Google Scholar]
- 4.Kumar S. Expert Opin Drug Metab Toxicol. 2010;6:115–131. doi: 10.1517/17425250903431040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hertweck C. Angew Chem Int Ed. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- 6.Rix U, Fischer C, Remsing LL, Rohr J. Nat Prod Rep. 2002;19:542–580. doi: 10.1039/b103920m. [DOI] [PubMed] [Google Scholar]
- 7.Ortiz de Montellano PR. Chem Rev. 2010;110:932–948. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernhardt R. J Biotechnol. 2006;124:128–145. doi: 10.1016/j.jbiotec.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 9.He J, Hertweck C. Chem Biol. 2003;10:1225–1232. doi: 10.1016/j.chembiol.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 10.Traitcheva N, Jenke-Kodama H, He J, Dittmann E, Hertweck C. ChemBioChem. 2007;8:1841–1849. doi: 10.1002/cbic.200700309. [DOI] [PubMed] [Google Scholar]
- 11.Müller M, Kusebauch B, Liang G, Beaudry CM, Trauner D, Hertweck C. Angew Chem Int Ed. 2006;45:7835–7838. doi: 10.1002/anie.200602840. [DOI] [PubMed] [Google Scholar]
- 12.He J, Müller M, Hertweck C. J Am Chem Soc. 2004;126:16742–16743. doi: 10.1021/ja046104h. [DOI] [PubMed] [Google Scholar]
- 13.Richter MEA, Traitcheva N, Knüpfer U, Hertweck C. Angew Chem Int Ed. 2008;47:8872–8875. doi: 10.1002/anie.200803714. [DOI] [PubMed] [Google Scholar]
- 14.Werneburg M, Hertweck C. Chembiochem. 2008;9:2064–2066. doi: 10.1002/cbic.200800301. [DOI] [PubMed] [Google Scholar]
- 15.Zocher G, Richter ME, Mueller U, Hertweck C. J Am Chem Soc. 2011;133:2292–2302. doi: 10.1021/ja110146z. [DOI] [PubMed] [Google Scholar]
- 16.Müller M, He J, Hertweck C. ChemBioChem. 2006;7:37–39. doi: 10.1002/cbic.200500161. [DOI] [PubMed] [Google Scholar]
- 17.Piel J, Hertweck C, Shipley P, Hunt DS, Newman MS, Moore BS. Chem Biol. 2000;7:943–955. doi: 10.1016/s1074-5521(00)00044-2. [DOI] [PubMed] [Google Scholar]
- 18.Cheng Q, Xiang L, Izumikawa M, Meluzzi D, Moore BS. Nat Chem Biol. 2007;3:557–558. doi: 10.1038/nchembio.2007.22. [DOI] [PubMed] [Google Scholar]
- 19.Werneburg M, Busch B, He J, Richter ME, Xiang L, Moore BS, Roth M, Dahse HM, Hertweck C. J Am Chem Soc. 2010;132:10407–10413. doi: 10.1021/ja102751h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.He J, Hertweck C. ChemBioChem. 2005;6:908–912. doi: 10.1002/cbic.200400333. [DOI] [PubMed] [Google Scholar]
- 21.Beaudry CM, Malerich JP, Trauner D. Chem Rev. 2005;105:4757–4778. doi: 10.1021/cr0406110. [DOI] [PubMed] [Google Scholar]
- 22.Henrot M, Richter ME, Maddaluno J, Hertweck C, De Paolis M. Angew Chem Int Ed. 2012:51. doi: 10.1002/anie.201204259. [DOI] [PubMed] [Google Scholar]
- 23.Helliwell CA, Poole A, Peacock WJ, Dennis ES. Plant Physiol. 1999;119:507–510. doi: 10.1104/pp.119.2.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rojas MC, Hedden P, Gaskin P, Tudzynski B. Proc Nat Acad Sci USA. 2001;98:5838–5843. doi: 10.1073/pnas.091096298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teoh KH, Polichuk DR, Reed DW, Nowak G, Covello PS. FEBS Lett. 2006;580:1411–1416. doi: 10.1016/j.febslet.2006.01.065. [DOI] [PubMed] [Google Scholar]
- 26.Ro DK, Paradise EM, Ouellet M, Fisher KJ, Newman KL, Ndungu JM, Ho KA, Eachus RA, Ham TS, Kirby J, Chang MC, Withers ST, Shiba Y, Sarpong R, Keasling JD. Science. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]
- 27.Ro DK, Arimura G, Lau SY, Piers E, Bohlmann J. Proc Nat Acad Sci USA. 2005;102:8060–8065. doi: 10.1073/pnas.0500825102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quaderer R, Omura S, Ikeda H, Cane DE. J Am Chem Soc. 2006;128:13036–13037. doi: 10.1021/ja0639214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen S, Mao X, Shen Y, Zhou Y, Li J, Wang L, Tao X, Yang L, Wang Y, Zhou X, Deng Z, Wei D. Appl Environ Microbiol. 2009;75:1778–1781. doi: 10.1128/AEM.00859-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olano C, Wilkinson B, Sánchez C, Moss SJ, Sheridan R, Math V, Weston AJ, Bran AF. Chem Biol. 2004;11:87–97. doi: 10.1016/j.chembiol.2003.12.018. [DOI] [PubMed] [Google Scholar]
- 31.Moebius N, Ross C, Scherlach K, Rohm B, Roth M, Hertweck C. Chem Biol. 2012;19 doi: 10.1016/j.chembiol.2012.07.022. in press. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.